
Abstract
This paper reports the results of the experimental study of parameters for a DC oxygen discharge with water cathode in the pressure range of 0.1–1 bar and the discharge current of 40 mA. The radius of positive column, the cathode voltage drop, the cathode current density and the electric field strength were measured. Rotational temperatures of N2 (C3Πu, V = 0) and OH (A2Σ, V = 0) and absolute line intensities of atomic oxygen with wave length of 845 and 777 nm were determined as well. Plasma composition modeling was carried out by the combined solution of the Boltzmann equation for electrons, the equations of vibrational kinetics for ground states of N2, O2, H2O molecules, and the equations of chemical kinetics, and the plasma conductivity equation. Calculations were carried out taking into consideration the discharge radial heterogeneity and using experimental values of E/N and gas temperatures. The main particles being formed in plasma were shown to be ·OH, H2O2, O(3P), O2(a1Δg), O2(b1Σ +g ), H(1S). On the basis of this calculation and experimental values of line intensities, the populating mechanism of (3p 3P) level of atomic oxygen was discussed. The comparison of some properties of discharges in O2, N2 and air was done.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Recently, the large number of works on gas discharges with aqueous electrodes was published. One of the reasons is prospects of these discharges application for some practical tasks. Among of them, is the wastewater purification from organic pollutants. To date, many devices were designed for these purposes (see Reviews [1, 2]). Independently on the discharge type the discharges using molecular oxygen as a plasma-forming gas provide the highest efficiency with respect to organics decomposition [2–5]. For explaining the processes taking place in a solution, it is necessary to know the active species concentrations in plasma. Even more important is to know the reaction rates rather than concentrations since these rates determine the limit fluxes of active species from discharge into liquid phase. Unfortunately, the possibilities of experimental methods for concentration determination in non-equilibrium plasma of higher pressure are rather limited. Moreover, available methods can not provide the measurements of process rates. For this reason, the models describing the chemical composition are intensively developed for different types of discharges in various plasma-forming gases: Ref. [6]—a zero-dimension Global model for the helium RF atmospheric pressure discharge with admixture of water molecules; Ref. [7]—model of an atmospheric pressure RF discharge (13.56 MHz) in helium containing admixtures of O2 and humid air; Ref. [8]—model of an argon plasma jet in humid air; Ref. [9]—model of an air plasma of DC discharge with water cathode and some others. If the model describes the processes of formation and loss of emitting states, it gives the basis for determining the concentrations of ground states of appropriate molecules and atoms since emission spectroscopy is a most available method for atmospheric pressure discharges. Emission lines of atomic oxygen (845, 777 nm, transitions 3p 3P → 3s 3S, 3p 5P → 3s 5S, respectively) are recorded for any plasma-forming gas and for any discharge type burning both over and under water. We know only one study where mechanism of populating-depopulating of the 3p 3P, 3p 5P state was analyzed for the conditions of oxygen low pressure DC discharge (30–300 Pa, 30–120 mA) [10]. The population of 3p 3P, 3p 5P states was shown to be due to the direct electron impact from ground state of O(3P) whereas the main channel of depopulation is radiation transitions to 3s 3S, 3p 5S states. That mechanism relatively easy allows to determine the O(3P) state concentration using absolute emission intensities of 845 or 777 lines or argon actinometry [11]. It is clear that under the conditions of higher pressure discharges, that mechanism can be changed since the reduced electric field strengths (E/N) are lower, electron densities and gas temperatures are higher, and process set is more complex. For this reasons, the mechanism analysis must include the plasma composition modeling as well as the measurements of plasma parameters which are necessary for that modeling. As far as we know, plasma chemistry of higher pressure DC discharge in oxygen containing water admixtures has not been studied yet. Also, our data are compared with the appropriate data obtained for other plasma-forming gases in the same discharge type.
Experimental
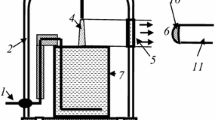
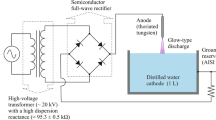
The experiments were carried out in a metal pin-water electrode system the sketch of which is represented in Fig. 1. The setup is similar to that in [12]. A cone stainless steel anode was movable for measuring the volt–distance characteristic of discharge. The digital voltmeter Fluke 289© (USA) was used for this purpose. The electric field strength in plasma was determined from the slope of linear part of this characteristic, whereas extrapolation to a zero distance gave the value of the cathode voltage drop. The geometrical sizes of discharge were determined with a digital camera. The AvaSpec-2048FT-2 monochromator (grating of 600 line/mm, wave length range is 200–900 nm) was applied for the emission spectra digital registration. In this case the anode–cathode distance was 10 mm. The light entered the monochromator entrance slit through a quartz optical fiber. The optical fiber-monochromator system was calibrated by the monochromator manufacturer on a radiation power. The emission from cathode and anode discharge parts was cut off by the size of the quartz window. Since the discharge radius (R D ≤ 0.25 mm) and discharge length (L D = 10 mm) were much less than the distance between the light fiber entrance lens and the discharge (L = 75 mm), the total amount of quanta, Ф, which is registered by monochromator per second is connected with line or band intensity, \({\bar{I}}\)(quantum/(cm3 \(\times\) s)), by expression
where R L is the radius of entrance lens (2.5 mm), \({\bar{I}}\) is the average on the discharge volume line or band intensity, \(V_{D}\) is the discharge volume.

Schematic diagram of the experimental set-up. 1 cathode, 2 glass bell-jar, 3 anode, 4 discharge, 5 quartz window, 6 radiation output to entrance lens of light fiber, 7 glass cell with distilled water, 8, 9 gas outlet and inlet, 10 entrance lens of light fiber, 11- light fiber
The Pyrex cell with distilled water of 80 ml volume was placed into a 5 l glass bell-jar. Molecular oxygen of the ultra high purity grade was used as a plasma-forming gas. The gas flow rate was 300 cm3/s. The pressure range was 0.1–1 (±0.01) bar, and discharge current, I, was 40 mA. In some cases experiments were carried out with molecular nitrogen as well.
The N2 admixture was estimated by the following way. The exhaust gas was passed through degassed glass vessel. After that the gas composition from vessel was analyzed by MX 7304 (Russia) monopole mass-spectrometer.
The main contributions to the emission spectrum of O2 discharge were the bands of OH (A2Σ+→X2Π), atomic hydrogen (Balmer lines) and the lines of atomic oxygen. The most intensive oxygen lines were 845 and 777 nm (3p 3P → 3s 3S, 3p 5P → 3s 5S transitions, respectively). A slight emission from N2s positive system (C3Πu → B3Πg) was also observed, due to the presence of air impurity in the system. According to our estimate, the N2 concentration was less than 0.2 %. The rotational temperature was determined from non-resolved rotational structure of the band for the C3Πu → B3Πg (0–2) transition as it was described elsewhere [13]. In the present case, the rotational temperature is equal to the translational one. In addition, the rotational temperature was measured on the OH A2Σ+ → X2 Π (0–0) band. As it will be shown below, the results of temperature determination by both methods were practically the same.
The reproducibility errors for all measured values were calculated on five and more measurements using the confidence probability of 0.95. The total errors are shown in appropriate Figures as the bars.
Calculations of the Plasma Composition
For the calculation of plasma composition it is convenient to divide the single plasma system into several subsystems according to community of processes taking place in each subsystem, and, as the result, according to community of mathematical description. Every subsystem is interconnected with the others. And input parameters necessary for the description of a subsystem are the output parameters of the others. If the gas temperature, reduced electric field, E/N, and discharge current density, j, are known from the experiment, the three subsystems are the most important for non-equilibrium plasma in molecular gases: (1) the subsystem of electron gas, (2) the subsystem of vibrational kinetics of molecules in ground states, (3) the subsystem of chemical kinetics of neutral species.
The subsystem of electron gas was described by the solution of the homogeneous Boltzmann equation using two-term expansion in spherical harmonics. In spite of low concentration of N2 molecules, we took into consideration the appropriate collisions. The collisions of second kind with vibrationally excited molecules as well as the e–e collisions were taken into consideration. For N2, O2, H2O, NO molecules and O(3P) atoms, the sets of cross-sections from studies [14–18] were used, respectively. The detailed description of appropriate equations was given elsewhere [19, 20]. The input parameters of this subsystem were gas temperatures, E/N, j, and plasma composition. At the first step, it was supposed that plasma-forming gas consists of molecular oxygen, 0.2 % of N2 and H2O molecules. The mole fraction of H2O molecules was used as a fitting parameter. According to data of various studies we assumed that H2O content cannot be more than 8 % at all pressures. The output parameters of this subsystem were the electron energy distribution function (EEDF) and electron gas characteristics including electron density and rate constants of processes with electron participation.
The subsystem of vibrational kinetics included the ground states of N2, O2, H2O, NO molecules. The populating of vibrational levels was described by the equations system of quasi- stationary kinetics. The kinetic equation for the concentration, [N (n) (V)], of molecules of kind n in vibrational state V was written as
where the summation on n is carried out on amount of components, K V,V−1 and K V−1,V are the rate constants of V–T exchange, \(K_{V - 1,V}^{W + 1,W}\) and \(K_{{V , {\text{V}} + 1}}^{W,W - 1}\) are the rate constants of V–V exchange, K V0 and K 0V are the rate constants of electron excitation and deactivation of level V, V and W are the vibrational quantum numbers, \([N^{(n)} (W)]\) is the concentration of molecule n in vibrational state W, Q V is the rate of other reactions, W* is the vibrational level corresponding to the dissociation limit. W* = 14, 13, 23, 40, 36, and 46 for H2O(001), H2O(100), H2O(010), NO, O2, and N2 molecules, respectively.
This system takes into account the single-quantum V–V, and V–T exchange between molecules, e-V excitation-de-excitation and some other events. The complete processes list is given in study [9]. For the dependence of rate constants on V we did not use any approximations of translation factors. They were calculated by the SSH generalized theory [21] and the data obtained were normalized on the experimental rate constants of studies [22–27] for the 0 ↔ 1 transitions. The parameters of vibrational states were taken from [28, 29] whereas values of reverse radius in exponential repulsive potential of interaction were taken from [30].
The subsystem of chemical kinetics of neutral species consists of 222 reactions describing the processes of formation and loss of the following neutral species: O2(X), O2(a1Δ), O2(b1Σ), O2(A3Σ), O(3P), O(1D), O(1S), O3, O(3p 3P), O(3s 3S), O(3s 5S), H2O, H, OH, H2O2, HO2, H2, N2(X), N2O, NO, NO2, NO3, HNO, HNO2, HNO3, N2(A3Σ +u ), N2(B3Πg), N2(C3Πu), and N2(a’1Σ+). The reaction set was based by us in study [9] where it was used for the modeling of an atmospheric pressure DC discharge in air. The list of processes and data on the rate constants with references is given in Table 1. Unlike the data of [9] some reactions for excited states of atomic oxygen were added.
Calculated concentrations were returned to the subsystem of electron gas and the calculation procedure was repeated again until the particle concentrations change did not exceed the error limit of 1 %.
Results and Discussion
Measured cathode voltage drops are represented in Fig. 2 together with the data of some other studies. The data of different authors agree quite satisfactorily. The cathode voltage drops are increased in a series of O2, air, and N2 whereas for the low pressure gas discharges with metallic cathode the opposite dependence is observed [31].

Figure 3 shows the electric field strength changes with pressure. The N2 discharge requires higher electric field strengths as comparing with air and oxygen discharges.

The data on the discharge radii are represented Fig. 4. The discharge in O2 is more contracted.

Measured gas temperatures are represented in Fig. 5. The discharge in O2 demonstrates the highest temperatures whereas in N2—the lowest ones. Air discharge occupies the intermediate position. As it can be seen, the temperatures obtained on the N2(C3Πu → B3Πg (0–2)) and OH [A2Σ+ → X2Π (0–0)] bands are practically the same. Therefore, unlike discharges in N2, He, Ar, N2O and CO2 [33] there is no specific processes for the O2 discharge resulting in an overpopulation of higher rotational levels of OH (A2Σ+, V = 0) and rotational population can be characterized with a single value of temperature.

The averaged gas temperature (T g ) and effective vibrational temperature (T V ) of O2(X) as a function of pressure. 1 Ar from study [12], 2 N2 from study [34], 3, 4, 7, 8 O2, 5 ambient air from study [9]. 1, 2, 3 were obtained on the rotational temperature of N2. 4 was obtained on the rotational temperature of OH. 6 the TV calculation at averaged gas temperature, 7 the temperature at discharge axis, T 0, 8 the temperature at discharge boarder, T R
It is interesting to compare the temperatures for N2 (study [34]) and O2 discharges. As it can be seen from Fig. 4, the gas temperature for N2 discharge is essentially lower than for the O2 one in spite of large difference in inputted power density, j × E (see Fig. 3, O2—~3.5 × 102 W/cm3, N2—~2.1 × 103 W/cm3 at 1 bar). The difference can be explained by the following way. Formally, the gas temperature depends on the heat conductivity coefficient, the power density inputted into discharge and fraction of power density, δ, transmitted to the gas heating. Molecular N2 and O2 have very close heat conductivity coefficients [33]. For both N2 [34] and O2 the EEDF calculations showed that more than 95 % of electron energy is inputted to the vibrational degrees of freedom of N2 and O2 ground states. The direct gas heating over elastic electron collisions with molecules gives δ of ~5 × 10−3. Therefore, the main source of gas heating is V–T exchange reactions transforming the energy of vibrational excitation into heat. If vibrationally excited molecules have no time to relax within discharge zone, it may be expected that the part of vibrational energy transforms to heat outside the glowing part of discharge. It must lead to gas cooling at the same inputted power density. Let us estimate the characteristic times of the diffusion and V–T relaxation. The diffusion coefficient of N2 in N2 at atmospheric pressure \(D(N_{2} ) = 0.17 \times (T/273)^{1.92}\), and O2 in O2—\(D(O_{2} ) = 0.19 \times (T/273)^{1.9}\)[35]. At the temperature of T = 1067 and 2600 K and the pressure of 1 bar (Fig. 4) it gives D(N 2 ) = 2.27 cm2/s and D(O 2 ) = 14.67 cm2/s. The characteristic time of the diffusion can be calculated by \(\tau_{D} \approx \frac{1}{D} \times \left( {\frac{R}{2.405}} \right)^{2}\), where R is the discharge radius (Fig. 5). Calculation gives the \(\tau_{D}\) value of 1.4 × 10−4 and 4 × 10−4 s for O2 and N2 molecules, respectively. The characteristic time of the V-T relaxation can be calculated as follows:
where K 10 is the rate constant and N is the concentration of appropriate molecules. The rate constant for O2-O2 is \(K_{10} = 4.67 \times 10^{ - 13} \times T_{g} \times \exp [ - 126/(T_{g} )^{1/3} ]\) cm3/s and for N2–N2 it is \(K_{10} = 4.15 \times 10^{ - 29} \times \exp [2.3 \times (T_{g} )^{0.357} ]\) cm3/s. For the temperatures of T g = 1067 and 2600 K, respectively for N2 and O2 at pressure of 1 bar \(K_{10} (O_{2} )\) = 1.48 × 10−13 cm3/s and \(K_{10} (N_{2} )\) = 4.53 × 10−17 cm3/s. And the characteristic time of the V–T relaxation for N2 is 3.2 × 10−3 s, whereas for O2 it is 2.5 × 10−6 s. The comparison of these times with the diffusion times shows that for O2 discharge the vibrational energy is transformed into heat in discharge zone, whereas for N2 discharge the essential part of vibrational energy (\(1 - \tau_{D} /\tau_{VT} \approx 0.88\)) is dissipated outside of the discharge. In spite of the large difference in the inputted power density, the power density transforming into heat for oxygen discharge is higher.
Using the data obtained we estimated the radial profile of temperature by the following way. At Bessel distribution of heat sources density on radius, r, the heat conductivity equation has the form:
where oxygen heat conductivity coefficient is equal to \(\lambda = \alpha + \beta \times T_{g} = 0.00507 + 6.615 \times 10^{ - 5} \times T_{g}\) W/(m K) [36], the power density (W/m3), F, at the discharge axis (r = 0) is \(F = \frac{I \times E}{{0.43 \times \pi \times R^{2} }}\), J 0 is the Bessel function of the first kind of the zero orderThe solution of this equation is
where T go is the temperature at the discharge axis (r = 0).The Eq. (5) allows calculating the temperature profile by means of variation T g0 to obtain the average value of temperature, \(\bar{T}_{g} = \frac{2}{{R^{2} }} \times \int\limits_{0}^{R} {T_{g} (r) \times r} \times dr\), which is measured experimentally. The results of such calculation of T g0 and T gR (temperature at the discharge border, r = R) are shown in Fig. 5. The values of reduced electric field strengths, E/N, calculated on the total concentration of particles (P = N×kT) and average temperature are shown in Fig. 6.

The averaged reduced electric field strength as a function of pressure. 1 O2, 2 N2 [34]
High values of temperature radial gradient result in a high gradient of E/N and in high gradients of rate constants of electron impact. Due to a non-linear dependence of rate constants on E/N the rate constant calculated on averaged E/N can differ strongly from the rate constant calculated by means of averaging the appropriate radial distribution. Also, the high temperature gradient leads to radial changes of concentrations of plasma forming gas. To describe this situation, we acted by the following way.
The EEDF characteristic time of relaxation \(\tau_{r}\) can be estimated as follows
where \(\bar{\varepsilon } \approx 1\) eV—electron average energy, \(m_{e}\)—electron mass, \(\sigma^{m} \approx 10^{ - 15}\) cm2—momentum transfer cross section for electron collision with O2 molecule, \(N = 4 \times 10^{18}\) cm−3—total particle concentration, \(\delta = (1.2 \times V_{D} /\bar{V})\)—average energy fraction losing with electron per one collision, \(\bar{V} = \sqrt {2\bar{\varepsilon } \times e/m_{e} }\) cm/c—average electron velocity, \(V_{D} \approx 5 \times 10^{6}\) cm/s—electron drift velocity.
The calculation gives the\(\tau_{r}\) ≈ 8 \(\times\) 10−9 s and \(\bar{V} \gg V_{D}\). Therefore, the EEDF can be really determined by means of solving the stationary Boltzmann equation in the two term expansion.
The EEDF characteristic length of relaxation l r is determined as
The calculation gives the \(l_{r} = 4 \times 10^{ - 3}\) cm. Comparison of this value with the discharge radius, R, (Fig. 4) shows that the EEDF is determined with the local value of E/N.
At the given experimental conditions the characteristic times of the most part of main chemical reactions (typically less than 10−6 s) are shorter than diffusion time. Therefore, the balance of particles formation-loss has to be fulfilled locally as well.
The radial distribution of electron density, N e , was described by Bessel function of the first kind of the zero order
where \(N_{e}^{0}\) is the electron density at r = 0.
The E/N and N were calculated in every point, r, using the temperature profile (1), pressure and E. And the plasma composition was calculated for every spatial discharge point at given pressure and current by means of procedure described in “Calculations of the Plasma Composition” section.
It was rather difficult to match the measured line intensities for wave lengths of 845 with calculations (Fig. 7). The threshold energies of O(3p 3P) excitation from O(3P) are rather high (10.9 eV). The dissociative excitation from O2 ground state (reaction 17 of Table 1) still requires more energy. For this reason, the excitation with electron impact from O(3P) including the emission transition from upper states gives the excitation rates of ~1010 cm−3/s at the pressure of 1 bar in pure oxygen. The experimental value is ~1012 cm−3/s (Fig. 7). The presence of H2O molecules makes the situation even worse. H2O additives result in the sharp dropping of electron impact rate coefficients due to the decrease in EEDF “tail” (Fig. 8). That effect depends on pressure. And it is the most significant at high pressure. In addition, the quenching rate coefficients of O(3p 3P) state with H2O are greater than of O2 (processes (18), (20) of Table 1). Molecular oxygen and water molecule due to the specific feature of its potential curves does not have any excited states the collisions of which could lead to the formation of O(3p 3P) state. Therefore, it is necessary to examine the possibilities of step-wise processes of electron impact with participation of electron-excited states of atomic oxygen.

Lines emission intensities of 845 nm (squares, line) and (777) nm (circles) versus gas pressure. Points are the experiment. The line is the calculation

Calculated electron energy function distributions at E/N = 1 × 10−16 V/cm2 and different content of water molecules. 1, 2, 3, 4 water molecules content is 0, 3, 5, and 10 %, respectively
Atomic oxygen has three states lying below O(3p 3P): 3s 3S (9.52 eV), 1S (4.19 eV), and1D (1.97 eV). Two last of them are metastable ones.
First of all, we added in a calculation the process of O(3p 3P) excitation from O(3s 3S) state (processes (19) of Table 1, thresholds energy is 1.47 eV). This transition is the allowed one. Therefore, the cross-sections, \(\sigma_{ik}\), can be estimated by Drawin’s semi-empirical expression [37] which is the version of Bethe approximation:
where \(a_{0} = 0.5292 \times 10^{ - 8} \;{\text{cm}}\), Ry = 13.606 eV, \(\varepsilon_{i} = E_{i} - E_{k}\)—the threshold energy, \(f_{ik}\)—the oscillator strength for i → k transition, \(u = (\varepsilon - \varepsilon_{i} )/\varepsilon_{i}\)—the dimensionless electron energy, \(\varepsilon\)—the electron energy. The oscillator strengths were calculated from radiation transition probabilities taken from [38]. The appropriate cross-section is shown in Fig. 9.

Cross-sections of O(3p 3P) excitation by electron impact from O(3s 3S) (1), O(1S) (2), and O(1D) (3) states
But we did not attain desired result since the threshold energy of O(3s 3S) excitation from O(3P) is too high For this reason the population rates are too low to provide the measured intensity of lines.
Unfortunately, the data on the excitation cross-sections of O(3p 3P) by electron impact from O(1 D,1S) are unknown. These transitions are the spin-forbidden ones. It is well known that the cross-sections for allowed and forbidden transitions are close on absolute value. But unlike cross-sections for allowed transition the cross-sections for forbidden transitions are pressed to threshold energy [62]. For this reason, for the estimate of these cross-sections we used the cross-section value of O(3p 3P) excitation from O(3s 3S) as initial approximation. Further, the cross-section absolute values and its shapes were fitted to agree with the experiment. In this case, the agreement was achieved. The appropriate data are shown in Figs. 7, 9.
The discharge was really heterogeneous (Fig. 10). The course of average electron energy reflects clearly the E/N increase in radial direction. The radial distribution of O(3P) concentration is essentially broader than emission of 845 nm line due to the sharp dependence of excitation rate constants on E/N. The similar behavior was experimentally observed in study [39].

The normalized radial distributions of 845 nm line emission (1), concentrations of O2(a1Δg) (2), O2(b1Σ +g ) (3), O(3P) (4), OH (5), and average electron energy (6). The pressure is 1 bar
Calculations showed that step-wise processes [(19), (21, 22)] provide up to 90 % of total population rate, whereas the direct excitation from O(3P) gives 10 % only. The main processes of loss are the emission (processes (23)) and quenching with O2 molecules (processes (18)). The both processes have approximately the same rates. We did not analyze the mechanism of formation-loss of (3p 5P) state. However, taking into account the structure of energy levels of O(I) atom, close intensities of 845 and 777 nm lines and the their dependence on the pressure we can assume that the mechanism of O(3p 5P) population has to be similar to the mechanism for O(3p 3P) state.
The main oxygen species were two lower metastable states of molecular oxygen O2(a1Δg) and O2(b1Σ +g ), atomic oxygen O(3P), O(1D) and O(1S) (Figs. 11, 12). All particle concentrations were essentially greater than for DC air discharge at the same parameters. The calculated O(3P) concentration is close to those measured for atmospheric pressure RF discharge burning in a mixture Ar-air (~1016 cm−3) [39]. The pressure increase leads to the increase in concentrations for all atomic oxygen species. Therefore, the emission intensities cannot be used as indicator of changes of oxygen atom concentration.

The concentration of species averaged over the discharge cross-section vs gas pressure. 1, 5 O2(a1Δg), 2, 6 O2(b1Σ +g ), 3, 7, 8 O(3P), 4 H2O, respectively. 5, 6, 7 calculated data of study [9] for air discharge. 8 experimental data of study [39] for atmospheric pressure RF plasma jet (Ar + 2 % air)

The concentration of species averaged over the discharge cross-section versus gas pressure. 1, 6 OH, 2, 7 H2O2, 3 H2, 4, 8 O(1D), 5 O(1S), 9 H(1S), respectively. 6, 7, 8 calculated data of study [9] for air discharge
Concentration dependences on pressure are defined by the following main reasons: results in the growth of electron densities (Fig. 13), in the increase in O2 concentration, and in the decrease in E/N and, as the result, electron average energy (Fig. 13). The two first factors promote a growth of excitation rate by electron impact. The third leads to the decrease in the rate constants of electron impact. The degree of the constants decrease depends on the process threshold energy. For processes with the relatively low threshold energy the decrease in the rate constants is slight and the first two factors are the predominate ones. For this reason, the concentrations of O2(a1Δg), O2(b1Σ +g ), O(3P) are increased with the pressure while the O(3p 3P) concentration is decreased.

Electron densities (1, 2) and average energy (3, 4) vs pressure. 1, 3 O2. 2, 4 N2 [34]. The data 1, 3 were averaged over the discharge cross-section
It is interesting to point out that ozone concentration in the discharge under study is essentially less (~1011–1012 cm−3) than for DBD discharges [107] or for impulse streamer corona [4] in oxygen (~1015–1016 cm−3). The mechanism of ozone formation and decay is different as well. For DBD and streamer corona the main reactions of ozone formation and decay are the reaction (161) and (162) of Table 1, that is
For the given discharge the main reactions are the reactions (32) and (167) of Table 1
The main oxygen-hydrogen particles were ·OH radicals and hydrogen peroxide. Their concentrations are shown in Fig. 11. In is interesting to point out that in spite of the difference in plasma-forming gases (He, Ar, N2) the orders of magnitude of ·OH radical concentrations are close [32, 40] (1015–1016 cm−3).
Furthermore, it is noteworthy to highlight that the main way of OH formation is not the reaction of water dissociation by electron impact. Calculations showed that at atmospheric pressure the OH concentration depends on a balance of only two reactions—(157) and (141). It gives the following expression for the OH concentration
where K 157, K 141—the appropriate rate constants.
The pressure decrease leads to the mechanism change. Thus, at the pressure of 0.1 bar the main process of OH formation is the reaction (173), whereas the lose reactions are (147) and (153).
In spite of many processes with H2O2 participation only few of them are essential. For the pressure of 1 bar, the H2O2 concentration is defined with the formation reaction (133) and loss (205). And the H2O2 concentration can be estimated as
For the pressure of 0.1 bar the H2O2 concentration is defined by the formation reactions (133) and (132) whereas loss reaction is (144). And the H2O2 concentration can be estimated as
Calculations showed that O2(X) vibrational distributions are generally not the equilibrium ones (Fig. 14). Nevertheless, the populations of low vibrational states can be described by the Boltzmann law, while the effective vibrational temperatures are almost equal to the gas temperatures (Fig. 5). This is due to the fast V–T relaxation rate of these molecules on O2 in comparison with the V–V relaxation rates. However, faster increase in V–V rate constants with V than V–T rate constants results in the overpopulation of higher vibrational levels as compared with equilibrium. The same situation was observed for O2 vibrational distributions in air plasma [9].

The O2(X) molecule distributions on vibrational levels at the averaged gas temperature. V—number of level. 1, 2 pressures are 0.1 and 1 bar, respectively
It is necessary to point out that ion-molecular reactions are not considered under calculations. The formation rates of positive ions particularly water cluster ions are limited by the primary ionization rate of molecules with electron impact. Under our experimental values of E/N the ionization rates are less than rates of excitation and dissociation more than several orders of magnitude. For example, at E/N = 10 Td the ionization rate constant of H2O with electron impact is ~1.2 × 10−24 cm3/s, the ionization rate constant of O2 is ~1.4 × 10−24 cm3/s, whereas the dissociation rate constant of O2(X) is ~3×10−14 cm3/s, the rate constant of O2(a1Δ) excitation is ~8×10−11 cm3/s. Thereby, ion-molecular reactions cannot influence on the concentrations of neutral species. Of course, these reactions necessary to take into account if the E/N values are unknown because E/N is determined from the equations of charge balance. But in our case the E/N was taken from experiment. Also, these reactions are important if we want to known the ion composition of plasma. But it was not the aim of given study.
Conclusions
On the basis of experimental data on parameters of an oxygen DC discharge with water cathode, the modeling chemical composition of plasma was carried out at the pressure range of 0.1–1 bar at the discharge current of 40 mA. The main particles being formed in plasma were shown to be ·OH, H2O2, O(3P), O2(a1Δg), O2(b1Σ +g ), H(1S). The dependencies of these particles concentrations on the pressure are defined with the change in an electron and O2(X) concentrations and in an EEDF. The specific feature of the discharge is a higher gas temperature as compared with the same discharges in air and nitrogen. This is due to the high rates of V-T relaxation of vibrationally-excited O2 molecules. Due to high temperature gradient the discharge is heterogeneous in the radial direction. The O(1S,1D) metastable states give essential input to population of OI(3p 3P) level. For this reason the behavior of radiation emission from this state (845 nm) does not reflect a course of atomic oxygen concentration.
References
Tatarova E, Bundaleska N, Sarette J, Ferreira C (2014) Plasma Sources Sci Technol 23(6):063002
Jiang B, Zheng J, Qiu S, Wu M, Zhang Q, Yan Z, Xue Q (2014) Chem Ing J 236:348–368
Yan J, Du C, Li X, Cheron B, Ni M, Cen K (2006) Plasma Chem Plasma Process 26(1):31–41
Grabowski L, Veldhuzen E, Pemen A, Rutgers W (2006) Plasma Chem Plasma Process 26(1):3–17
Lukes P, Locke B (2005) J Phys D Appl Phys 38(22):4074–4081
Liu D, Bruggeman P, Iza F, Rong M, Kong M (2010) Plasma Sources Sci Technol 19(2):025018
Mirakami T, Niemi K, Gans T, O’Connel D, Graham W (2013) Plasma Sources Sci Technol 22(1):01503
Van Gaens W, Bogaerts A (2013) J Phys D Appl Phys 46(27):275201
Bobkova E, Smirnov S, Zalipaeva Y, Rybkin V (2014) Plasma Chem Plasma Process 34(4):721–743
Maksimov A, Rybkin V (1982) J Appl Spectrosc 37(5):1233–1236
Smirnov S, Rybkin V, Ivanov A, Titov V (2007) High Temp 45(3):291–297
Shutov D, Smirnov S, Bobkova E, Rybkin V (2015) Plasma Chem Plasma Process 35(1):107–132
Titov V, Rybkin V, Maximov A, Choi H-S (2005) Plasma Chem Plasma Process 25(5):502–518
Gordiets B, Ferreira C, Guerra V, Loureiro J, Nahorny J, Pagnon D, Touzeau M, Vialle M (1995) IEEE Trans Plasma Sci 23(23):750–768
Kajita S, Ushiroda S, Kondo Y (1990) J Phys D Appl Phys 67(9):4015–4023
Rybkin V, Titov V, Kholodkov V (2008) Izv Vyssh Uchebn Zaved Khim Khim Tekhnol 51(3):2–10 (in Russian)
Rybkin V, Titov V, Kholodkov I (2009) Izv Vyss Ucheb Zaved Khim Khim Tekhnol 52(12):3–10 (in Russian)
Laher R, Gilmore F (1990) J Phys Chem Ref Data 19(1):277–304
Diamy A, Legrand J, Smirnov S, Rybkin V (2005) Contrib Plasma Phys 45(1):5–21
Titov V, Rybkin V, Smirnov S, Kulentsan A, Choi H (2006) Plasma Chem Plasma Process 26(6):543–555
Schwartz R, Slawsky Z, Herzfeld K (1952) J Chem Phys 20(10):1591–1599
Capitelli M (ed) (1986) Nonequilibrium vibrational kinetics. Springer, Berlin
Kiefer J (1972) J Chem Phys 57(5):1938–1956
Mnatsakanyan A, Naidis G (1985) High Temp (Teplofyzika Vysokikh Temperatur) 23(4):640–646 (in Russian)
Salnikov VA, Starik AM (1995) High Temp (Teplofyzika Vysokikh Temperatur) 33(1):121–133 (in Russian)
Wyson I (1994) J Chem Phys 101(4):2800–2810
Frost M, Islam M, Smith I (1994) Can J Chem 72(3):606–611
Huber K, Herzberg G (1979) Molecular spectra and molecular structure. IV. Constants of diatomic molecules. Litton Educational Publishing Inc., New York
Glushko VP (ed) (1978) Thermodynamic properties of individual substances. Handbook. Moscow, Nauka (in Russian)
Radtsig A, Smirnov B (1980) Handbook on atomic and molecular physics. Atomizdat, Moscow (in Russian)
Raizer Y (1987) Gas discharge physics. Springer, Berlin
Li L, Nikiforov A, Xiong Q, Lu X, Taghizadeh L, Leys C (2012) J Phys D Appl Phys 45(12):125201
Verreycken T, Schram DC, Leys C, Bruggeman P (2010) Plasma Sources Sci Technol 19(4):045004
Smirnov S, Shutov D, Bobkova S, Rybkin V (2015) Plasma Chem Plasma Process 35(4):639–657
Grigoriev I, Meiylikhov E (ed) (1991) Physical quantities. Handbook. Energoatomizdat, Moscow (in Russian)
Reid RC, Sherwood TK (1966) The properties of gases and liquids. Their estimation and correlation. McGraw-Hill Company, New York
Chernyiy G, Losev S (eds) (1995) Physical–chemical processes in a gas dynamics. V. 1. Dynamics of physical–chemical processes in gas and plasma. Moscow State University, Moscow (in Russian)
Moore C (1976) Selected tables of atomic spectra, atomic energy levels and multiplet tables—OI. Nat. Bur. Stand. US
Zang S, van Gessel A, van Grootel S, Bruggeman P (2014) Plasma Sources Sci Technol 23(2):025012
Nikiforov A, Li L, Xiong Q, Leys C, Lu XP (2011) Eur Phys J Appl Phys 56(2):24009–24013
Aleksandrov N (1988) Adv Phys Sci (Uspekhi Fizicheskikh Nauk) 154(2):177–206 (in Russian)
Eliasson B, Kogelschatz U (1986) Basic data for modeling of electrical discharges in gases: oxygen. Brown Boveri Research Report. KLR86-11C
Fehsenfeld F, Albritton D, Burt J, Schiff H (1969) Can J Chem 47(10):1793–1795
Phelps A (1969) Can J Chem 47(10):1783–1793
Kenner R, Ogryzlo E (1980) Int J Chem Kinet 12(7):502–508
Fehsenfeld F, Ferguson E, Schmeltekopf A (1966) J Chem Phys 45(5):1844–1845
Slanger T, Black G (1979) J Chem Phys 70(7):3434–3443
Dvoryankin A, Ibragimov L, Kulagin Yu, Shelepin L (1987) Mechanisms of electron relaxation in atomic–molecular media. Plasma chemistry. Energoatomizdat, Moscow (in Russian)
Zinn J, Sutherland C, Stone S, Dunkan L (1982) J Atmos Terr Phys 44(12):1143–1171
Young R, Black G (1967) J Chem Phys 47(7):2311–2318
Smirnov B (1982) Excited atoms. Energoatomizdat, Moscow (in Russian)
Slanger T, Black G (1981) J Chem Phys 75(5):2247–2251
Slanger T, Black G (1976) J Chem Phys 64(9):3763–3766
Atkinson R, Welge K (1972) J Chem Phys 57(9):3689–3693
Kossyi I, Kostinsky A, Matveyev A, Silakov V (1992) Plasma Sources Sci Technol 1(3):207–220
Aleksandrov N (1978) Tech Phys (Zhurn. Tekh. Fiziki) 48(7):1428–1431 (in Russian)
Smith K, Thomson R (1978) Computer modeling of gas lasers. Plenum Press, New York
Krivonosova O, Losev S, Nalivaiyko V (1987) Recommended data on rate constants of chemical reactions between molecules consisting of N and O atoms. Plasma chemistry. Energoatomizdat, Moscow (in Russian)
Jannuzzi M, Jeffries J, Kaufman F (1982) Chem Phys Lett 87(6):570–574
Piper L, Caledonia G, Kennelaly J (1981) J Chem Phys 75(6):2847–2853
Silakov B (1990) Mechanism of supporting the long-lived plasma in molecular nitrogen at high pressure. Preprint MIFI 010-09 12 (in Russian)
Slovetskiy D (1980) Mechanisms of chemical reactions in non-equilibrium plasma. Moscow, Nauka (in Russian)
Piper L (1982) J Chem Phys 77(5):2373–2377
Young R, Black G, Slanger T (1970) J Chem Phys 51(1):116–121
Piper L (1987) J Chem Phys 87(3):1625–1629
Yaron M, Von Engel A, Vidaud P (1976) Chem Phys Lett 37(1):159–161
Didyukov A, Kulagin Yu, Shelepin L, Yarygina V (1989) Quantum Electron 16(5):892–904 (in Russian)
O’Brien R, Myers G (1970) J Chem Phys 53(10):3832–3835
Yau A, Shepherd G (1979) Planet Space Sci 27(4):481–490
Husain D, Mitra S, Young A (1974) J Chem Soc Faradey Trans Part II 70(10):1721–1731
Delcroix J, Ferreira C, Ricard F (1973) Proc of the XI intern conf phenomena in ionized gases: invited paper Prague 301
Baulch D, Cox R, Crutzen P (1982) J Phys Chem Ref Data 11(2):327–496
Dmitrieva I, Zenevich V (1984) Russ J Phys Chem B Focus Phys (Zhurnal Khimicheskoi Fiziki) 3(8):1075–1080 (in Russian)
Burch D, Gryvnak A (1979) Appl Optics 8(7):1493–1499
Ivanov V, Klopovskii K, Lopaev D, Rakhimov A, Rakhimova T (2000) Plasma Phys Rep 26(1):980–990
Bastien F, Haug R, Lecuiller M (1975) J Chim Phys 72(1):105–112
Pace L (1978) IEEE J Quant Electron 14(4):263–274
Smirnov B (1978) Negative ions. Atomizdat, Moscow (in Russian)
Baulch D, Cobos C, Cox R, Esser C, Franec P, Just Th, Kerr J, Pilling M, Troe J, Walker R, Warnatz J (1992) J Phys Chem Ref Data 21(3):411–429
Peyrous R, Rignolet P, Held B (1989) J Appl Phys D Appl Phys 22(11):1658–1667
Chirokov A (2003) Self-organization of microdischarges in DBD plasma. Master Thesis, Dreaxel University
Tochikubo F, Ushida S, Watanabe T (2004) Jpn J Appl Phys 43(1):315–320
Tsang W, Herron J (1991) J Phys Chem Ref Data 20(4):609–663
Hipples H, Krasteva N, Nasterlack S, Striebel F (1999) J Chem Phys A 110(14):6781–6788
Smedt F, Bui X, Nguyen T, Peeters J, Vereecken L (2005) J Phys Chem 109(1):2401–2409
Pontiga F, Soria C, Casrellanos A, Skalny J (2002) Ozone Sci Eng 24(6):447–462
Young R, Black G, Slanger T (1969) J Chem Phys 50(1):303–308
Gauthier M, Snelling D (1975) J Photochem 4(1–2):27–50
DeMore W, Sander S, Golden D, Hampson R, Kurylo M, Howard C, Ravishankara A, Kolb C, Molina M (1997) Chemical kinetics and photochemical data for use in stratospheric modeling. Evaluation number 12 (JPL Publication 97-4)
Su M, Kumaran S, Lim K, Michael J, Wagner A, Hardling L, Fang D (2002) J Phys Chem 106(36):8261–8270
Taylor G, Setser D (1971) J Am Chem Soc 93(19):4930–4932
Clark W, Setser D (1980) J Phys Chem 84(18):2228–2233
Arnold J, Comes F (1980) J Chem Phys 47(1):125–130
Smith C, Molina L, Lamb J, Molina M (1984) Int J Chem Kinet 16(1):41–45
Herron J (1999) J Phys Chem Ref Data 28(5):1453–1484
Clyne M, Stedman D (1967) J Phys Chem 71(9):3071–3073
Javoy S, Naudet V, Abid S, Paillard C (2003) Exp Thermal Fluid Sci 27(4):371–377
Warnatz J (2000) Rate coefficients in the C/H/O system. In gas-phase combustion chemistry. Springer, New York
Filseth S, Stuhl F, Welge K (1970) J Chem Phys 52(1):239–243
Nakayma T, Takahashi K, Matsumi Y (2004) Chem Phys Lett 398(1–3):163–167
Heimerl J, Coffee T (1979) Combust Flame 35:117–123
Shuman N, Miller T, Viggiano A (2012) J Chem Phys 136(12):124307
Doroshenko V, Kudryavtsev N, Smetanin V (1992) High Energy Chem 26(2):227–231
Moseley J, Aberth W, Peterson J (1972) J Geophys Res 77(1):255–259
Smith D (1983) Adams N Physics of ion-ion and electron-ions collisions. Plenum Press, New York
Shimamura I (1988) Sci Pap IPCR 82:1–51
Kogelschatz U (2003) Plasma Chem Plasma Process 23(1):1–46
Acknowledgments
This study was supported by the RFBR Grant, Project Number 14-02-01113 A.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Smirnov, S.A., Shutov, D.A., Bobkova, E.S. et al. Chemical Composition, Physical Properties and Populating Mechanism of Some O(I) States for a DC Discharge in Oxygen with Water Cathode. Plasma Chem Plasma Process 36, 415–436 (2016). https://doi.org/10.1007/s11090-015-9669-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-015-9669-y