
Abstract
The widespread nature of nucleocytoplasmic trafficking defects and protein accumulation suggests distinct yet overlapping mechanisms in a variety of neurodegenerative diseases. Detailed understanding of the cellular pathways involved in nucleocytoplasmic transport and its dysregulation are essential for elucidating neurodegenerative pathogenesis and pinpointing potential areas for therapeutic intervention. The transport of cargos from the nucleus to the cytoplasm is generally regulated by the structure and function of the nuclear pore as well as the karyopherin α/β, importin, exportin, and mRNA export mechanisms. The disruption of these crucial transport mechanisms has been extensively described in the context of neurodegenerative diseases. One common theme in neurodegeneration is the cytoplasmic aggregation of proteins, including nuclear RNA binding proteins, repeat expansion associated gene products, and tau. These cytoplasmic aggregations are partly a consequence of failed nucleocytoplasmic transport machinery, but can also further disrupt transport, creating cyclical feed-forward mechanisms that exacerbate neurodegeneration. Here we describe the canonical mechanisms that regulate nucleocytoplasmic trafficking as well as how these mechanisms falter in neurodegenerative diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative diseases describe a highly heterogeneous set of disorders where progressive and irreversible neuronal death leads to detrimental symptoms dependent upon the subset of neurons that are impacted. While patients will exhibit various degrees of heterogeneity in regard to the specific brain regions involved in their disease as well as the severity of symptom onset and progression, for most patient populations, the progression of these diseases will slowly cost patients their independence and ultimately their lives. While promising clinical trials appear to be on the horizon, very few existing therapeutics have disease modifying effects. A substantial hurdle to developing effective therapeutics is the lack of causative and targetable cellular pathways that directly lead to neurodegenerative phenotypes. Therefore, comprehensive understanding of the pathogenesis of neurodegenerative diseases is critical for finding successful therapies. Here we review the possible mechanisms leading to neurodegeneration in the context of altered nucleocytoplasmic transport.
The movement of proteins, RNA and other cargos between the nucleus and the cytoplasm broadly encompass the cellular mechanisms controlled by nucleocytoplasmic transport machinery. There is substantial evidence to suggest an association of nucleocytoplasmic trafficking deficits with neurodegeneration [1,2,3,4,5]. Furthermore, an overarching theme that has emerged in recent years is that the failure of cargo transport across the nuclear envelope is not unique to a single neurodegenerative disease, nor one genetic insult [1,2,3,4,5]. However, more work remains to be completed to determine, precisely, how nucleocytoplasmic trafficking deficits may cause neurodegeneration. To properly survey the disrupted landscape of nucleocytoplasmic trafficking in neurodegeneration, it is essential to understand how a healthy cell manages nucleocytoplasmic transport. Thus, we first review the principles and components of canonical nucleocytoplasmic transport, and then follow with how these highly regulated transport mechanisms are proposed to be disrupted in several neurodegenerative diseases.
Canonical Nucleocytoplasmic Transport
Structure and Architecture of the Nuclear Pore
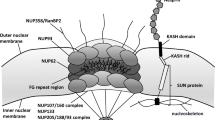
The nuclear membrane acts as a critical divide between the nuclear and cytoplasmic compartments of all eukaryotic cells [6, 7]. Its structural architecture provides a semi-permeable separation between the nucleus and the cytoplasm, which allows cytoplasmic access to genetic material, creating opportunities for complex gene regulation responses to stimuli outside of the nucleus. Unknown structures identified on the nuclear envelope using electron microscopy in the 1950s were first hypothesized to be regularly interspaced fusions of the inner and outer nuclear membrane and were later termed “nuclear pores” [8, 9]. These highly conserved structures are present in all eukaryotes, and are likely the result of early evolutionary events [10]. At the center of each nuclear pore is the massive, ~ 125,000 kDa nuclear pore complex (NPC) [8, 10,11,12]. The NPC displays eight-fold symmetry around a central channel, and additionally exhibits symmetry on an axis perpendicular to the central channel; where there exists a symmetrical nuclear and cytoplasmic ring on either side of the inner ring complex [12,13,14]. Additionally, asymmetric components of the NPC, the nuclear basket and cytoplasmic filaments, extend processes out from the NPC [14].
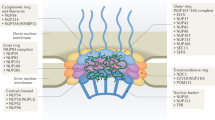
Each NPC consists of ~ 500–1000 proteins, which—due to the symmetry of the NPC—are comprised of multiple copies of only about 30 unique proteins known as nucleoporins (Nups) [14,15,16]. These ~ 30 unique Nups can be categorized into 5 groups, based on their structural role in the NPC: the coat nucleoporins, inner ring nucleoporins, transmembrane nucleoporins, cytoplasmic filament nucleoporins, and the nuclear basket nucleoporins (Fig. 1). Although the structure of nucleoporins and NPCs are highly conserved, they display poor genetic conservation, suggesting that broad structures not conserved by genetic sequence still perform similar functions across species [17, 18]. Figure 1 provides the human nomenclature, structural component, and the potential role in disease for each Nup [14, 19,20,21,22,23,24].

The structure and architecture of the nuclear pore complex. The structure of the human nuclear pore complex is comprised of 5 major groups. Coat nucleoporins (a), inner ring nucleoporins (b), transmembrane nucleoporins (c), cytoplasmic filaments (d), and the nuclear basket (e). Right panel: structural grouping of the human nucleoporins and the associations with ALS, AD, HD, and other non-neurological diseases
Coat Nucleoporins
This subset of Nups dictates the structure of the nuclear and cytoplasmic rings (Fig. 1a). In humans this “Y” shaped structure is composed of: Sec13, Seh1, Nup96, Nup75, Nup107, Nup160, Nup133, Nup37, Nup43, and ELYS [25, 26]. 16 coat nucleoporin complexes make up each of the nuclear and cytoplasmic rings by intricate interlocking of the upper arm with the center stalk of the “Y” region [12, 27]. The topographical structure of these complexes has been somewhat conserved, but species divergence does exist [25, 27].
Inner Ring Nucleoporins
Forming the central channel of the NPC, the inner ring structure contains Nup205, Nup188, Nup93, Nup155, Nup53, Nup54, Nup58, Nup62, Nup98 (Fig. 1b) [15, 28]. These Nups crystal structures have revealed that flexible linkers mediate the interaction of this complex, allowing for the flexibility of the inner ring [29]. The flexibility of these Nups and their ability to interact with cargos play crucial roles in proper trafficking through the nuclear pore [12].
Transmembrane Nucleoporins
The pore membrane proteins (POMs) act as anchors to secure the NPC in position by utilizing a transmembrane domain to interact with the nuclear envelope (Fig. 1c). The human nuclear pore contains NDC1, POM210, and POM121 [12, 15, 30]. These components are some of the most poorly characterized features of the NPC with regard to their structure and function [12, 30]. While the sequence conservation of POMs is particularly poor, it is likely that the function of transmembrane nucleoporins is conserved [12, 30]. POMs contain large often unstructured regions that have been hypothesized to interact either with the nuclear envelope or with the soluble components of the NPC [30]. While little is known about the function and regulation of human transmembrane nucleoporins, deletion of POMs in fungi produces no overt survival phenotype and they have been shown to be nonessential for cellular function [31, 32]. However, some studies indicate that these proteins play an important role in NPC assembly and nuclear membrane structure. Indeed, evidence suggests POM121 and NDC1 are important during NPC biogenesis and nuclear membrane homeostasis [33, 34].
Cytoplasmic Filaments and the Nuclear Basket
These two components of the nuclear pore are considered asymmetric because they have rotational symmetry like the components above, but not symmetry across the nuclear envelope. The cytoplasmic filaments are long projections that reach into the cytoplasm from the NPC (Fig. 1d) [35]. They consist of the following nucleoporins: Rae1, Nu42, Nup88, Nup214, DDX19, Gle1, and RanBP2 (also commonly referred to as Nup358) [12]. A principal role of these Nups is the spatial restriction of DDX19 and RanBP2, which are utilized for bulk mRNA and protein export, respectively, across the NPC to the cytoplasm [12, 36,37,38]. On the opposite side of the nuclear membrane, the human nuclear basket nucleoporins (Nup153, Nup50, and Tpr) also play roles in the organization of transport machinery across the nuclear pore by facilitating the recognition and binding of nuclear import and export factors in the nucleus (Fig. 1e) [12, 36].
Passive Diffusion Through the Nuclear Pore
The NPC regulates trafficking through many mechanisms. The first degree of regulation of transport through the nuclear pore is the diffusion barrier created by the intrinsically disordered phenylalanine—glycine (FG)-rich repeats [10, 12]. FG-repeats are common structural components of different nucleoporins spanning different structural regions and play multiple roles in regulating trafficking through the NPC. These FG-repeats form an intrinsically disordered domain in the central channel of the NPC that allows for the passive diffusion of small molecules (< 40 kDa) but creates an energetically inefficient method of transport for larger molecules [39]. It is interesting to note that the majority of the FG-repeat Nups are not essential, except for Nup98 which constitutes the largest contribution to the diffusion barrier [40, 41].
Facilitated Transport Through the Nuclear Pore
The Ran Gradient
The direction of—and energy for—cargo transport through the NPC is regulated by the gradient of Ras-related nuclear protein (Ran) [42]. This small GTPase adopts different conformations in its GTP or GDP bound state, modulating its affinity for transport factors [42]. The concentration of RanGTP, with higher levels in the nucleus, and RanGDP, with higher levels in the cytoplasm, is a tightly regulated, cyclical process resulting in an actively maintained Ran gradient in which the concentration of RanGTP to RanGDP mediates nucleocytoplasmic transport [42]. Cytoplasmic levels of RanGDP are maintained by the strictly cytoplasmic localization of Ran GTPase-activating protein (RanGAP) and Ran binding proteins (RanBPs) [43,44,45]. RanGAP together with RanBPs contribute to higher cytoplasmic levels of RanGDP by facilitating the hydrolysis of RanGTP that enters the cytoplasm (Fig. 2e, l, and q) [43,44,45]. Cytoplasmic RanGDP is shuttled into the nucleus by a dedicated transporter, nuclear transport factor 2 (NTF2) (Fig. 2f, m) [46]. Once in the nucleus, Ran’s chromatin-associated guanine nucleotide exchange factor (RCC1) promotes the exchange of GDP for GTP on Ran, increasing the concentration of nuclear RanGTP and completing the cyclical mechanism for the maintenance of the Ran gradient, essential for nuclear import and export (Fig. 2g, n) [47, 48].

Canonical nucleocytoplasmic trafficking mechanisms. Top left: Karyopherin α/β mediated import. KPNA binding NLS containing cargo with KPNB binding (a). The tripartite complex is translocated through the NPC (b) and dissociated by RanGTP binding to KPNB1 (c). KPNA recycling to the cytoplasm by XPO2 bound to RanGTP, while KPNB1 recycling is facilitated by its direct binding to RanGTP (d). On the cytoplasmic side of the NPC, the hydrolysis of RanGTP via RanGAP and RanBP dissociates KPNB1 and KPNA export complexes, producing RanGDP and releasing the karyopherins in the cytoplasm (e). RanGDP is shuttled to the nucleus by NTF2 (f), where RCC1 exchanges Ran’s GDP for GTP (g). Top right: Transportin. Cargo with a PY-NLS requires TNPO binding (h) and is transported through the NPC (i). Cargo is released after RanGTP binds TNPO (j). TNPO is recycled to the nucleus bound to RanGTP (k). TNPO and RanGTP dissociation is facilitated by RanGAP and RanBP (l), and the Ran gradient is maintained with NFT2 and RCC1 (m, n). Bottom left: Exportin. In the presence of RanGTP, exportins bind tRNA, miRNA, rRNA or NES containing protein as cargo (o). The resulting RanGTP and cargo bound exportin (p) is translocated through the NPC and dissociates after hydrolysis of RanGTP (q). Bottom right: Bulk mRNA transport. RNA export to the cytoplasm begins during transcription as the nascent strand emerges from RNA Pol II where the TREX complex and ALYREF bind the RNA between the EJC and the 5′ end of the pre-mRNA (r). This promotes the recruitment and conformational change of NXF1-NXT1, essential to the hand-off of RNA (r). After the dissociation of RNA Pol II and the binding of PABP to the 3′ polyA tail, the mRNP proceeds to the NPC (s). Releasing TREX and ALYREF, the mRNP is drawn through the NPC where NXF1 may interact with TREX2 as it enters the channel (t). On the cytoplasmic side of the NPC, DDX19, powered by ATP hydrolysis and stimulated by GLE1 bound to Ins6P, promotes the dissociation of the mRNP into its mRNA and export factor components (u). NXF1-NXT1 is recycled to the nucleus by either the TNPO or the KPNA/KPNB1 import pathways (v)
Karyopherin α/β Transport
Large protein complexes destined for the nucleus that struggle to passively diffuse across the FG-repeat domain of the NPC typically contain a nuclear localization signal (NLS) or a nuclear export signal (NES) [49,50,51]. These specific nuclear transport peptide sequences facilitate the binding of nuclear transport factors which promote transport through the NPC. In what is considered the “classical” transport pathway, karyopherin α (KPNA) binds directly to cargo containing a classical NLS [52]. Seven KPNA isoforms are thought to exist in humans and while there is some interesting specificity of the different isoforms, KPNA2 is considered to be the primary import protein for cargo with a classic NLS [53]. KPNA interacts directly with karyopherin β (KPNB1) through KPNA’s potent NLS domain, creating a tripartite cargo-KPNA-KPNB1 nuclear import complex (Fig. 2a) [16, 53]. Additionally, KPNB1 can also bind directly to some NLS sequences without the KPNA adaptor protein [16]. Once a cargo is bound to a nuclear import complex, the entire complex is guided through the NPC by KPNB1 due to its interactions with the intrinsically disordered FG-repeat domain of the central channel Nups (Fig. 2b) [54, 55]. After successful passage across the nuclear pore and into the nucleus, the import complex dissociates when RanGTP binds KPNB1 (Fig. 2c). KPNB1 recycling and return to the cytoplasm requires binding of RanGTP, while KPNA recycling requires RanGTP dependent nuclear export factor, exportin-2 (XPO2) (Fig. 2d) [56]. Immediately following translocation through the intrinsically disordered FG-repeats, the coordination of RanGAP with RanBPs promotes the innate GTPase activity of Ran, leading to hydrolysis of RanGTP, and disassembly of the karyopherin export complex, thereby releasing the export cargo (Fig. 2e) [57, 58].
Transportin Transport
A different nuclear transport mechanism was discovered by the identification of a new type of NLS, the PY-NLS, which is not recognized by the classical karyopherin α/β machinery, but instead by another group of nuclear transport receptors called transportins (TNPO) [59,60,61,62]. Consisting of Transportin1, 2A, and 2B, this transport family functions in a similar manner to the classical transport machinery described above, in which transportin binds to its cargo (Fig. 2h), mediates the interaction with FG-repeat Nups to allow for translocation through the NPC (Fig. 2i), dissociates from its cargo in a RanGTP dependent manner (Fig. 2j), is exported back to the cytoplasm in complex with RanGTP (Fig. 2k) and freed to bind new cargo following hydrolysis of RanGTP by RanGAP (Fig. 2l) [63, 64]. The difference in cargo recognition appears to be largely mediated by transportins’ recognition of cargo with the PY-NLS, unique from the classical NLS [65]. However, experimental evidence suggests that there are transportin dependent cargos that do not contain a PY-NLS, suggesting multiple recognition motifs for transportin mediated nuclear import [63, 66]. Additionally, post-translational modifications near the PY-NLS can alter transportin mediated transport [63].
Exportin Transport
Export of proteins and certain types of RNA from the nucleus, is mediated by seven exportin proteins in humans [67]. Exportins load their cargo in a RanGTP dependent manner, whereby the RanGTP binding increases exportins affinity for its cargo (Fig. 2o) [42]. Translocation of the exportin-cargo-RanGTP complex (Fig. 2p) through the NPC is mediated by exportin’s interactions with the FG-repeat Nups of the central channel. Once in the cytoplasm dissolution of the export complex and unloading of the cargo occurs through the hydrolysis of RanGTP via RanGAP, thus decreasing exportin’s affinity for its cargo (Fig. 2q) [57, 58]. Exportin 1 (XPO1) is the most prevalent and heavily utilized export receptor, which recognizes protein cargo with an NES [42]. Interestingly, XPO1 can also mediate rRNA, snRNA, and some mRNA export by binding to adaptor RNA binding proteins [42]. Different exportin isoforms have different targets. For example, XPOt is responsible for nuclear export of tRNA, and XPO5 is responsible for miRNA and rRNA export (Fig. 2o) [68, 69].
mRNA Transport
mRNA transport through the nuclear pore occurs through a different mechanism from the Ran mediated nucleocytoplasmic transport mechanism described above [69, 70]. Nuclear export of mRNA is a dynamic process where messenger ribonucleoprotein (mRNP) undergo various remodeling events from biogenesis to maturation and export [69]. The transcription and export (TREX) complex is a key player that initially associates with nascent RNA from transcribing RNA Pol II (Fig. 2r) [71,72,73]. During mRNA processing, recruitment of a TREX subunit, the RNA export factor ALYREF, is essential for nuclear export after RNA maturation (Fig. 2r) [74]. TREX and ALYREF contain binding sites that mediate the binding of the nuclear RNA export factor 1 (NXF1)—nuclear transport factor 2 like export factor 1 (NXT1) heterodimer [75, 76]. The NXF1-NXT1 interaction with ALYREF triggers the transfer of RNA from ALYREF to NXF1 (Fig. 2r) [77]. This hand-off of RNA is essential because NXF1 has the ability to drive the translocation through the FG-repeats of the nuclear pore [77]. These binding events signal the maturation of pre-mRNA into an NXF1-NXT1-mRNP complex (Fig. 2s), which then translocates to the nuclear basket, a process that may be conducted by TREX2 whose scaffolding component has been shown to directly interact with NXF1 (Fig. 2t) [78,79,80]. Once at the nuclear basket, recognition, docking, and subsequent translocation of the mRNP proceeds through interactions between FG-repeat Nups and NXF1 (Fig. 2u) [81]. As an mRNP passes through the NPC and enters the cytoplasm, its cargo mRNA and receptor complexes are dissociated via the ATP-dependent RNA helicase activity of DDX19 (human ortholog of Dbp5), in association with Nup214 stimulated by GLE1, which is bound to its co-factor inositol hexaphosphate (IP6) (Fig. 2u) [82]. The remodeling concludes when the cap-binding protein, eukaryotic translation initiation factor 4E (eIF4E), replaces the cap binding complex (CBC) added during the pre-mRNA stage, preparing the transcript for translation [83]. NXF1-NXT1 can be recycled to the nucleus by either the TNPO or the KPNA/KPNB1 pathway (Fig. 2v). Nuclear export of mRNA appears to be a relatively inefficient process. Studies have indicated that only about 30% of mRNPs that interact with the NPC are successfully passed through the diffusion barrier [84, 85]. While still unclear, it is postulated that the export receptor’s capacity for hydrophobic FG-repeat interactions is tempered by the size and composition of the mRNP, a balance that determines the solubility of the cargo-receptor complex in the hydrogel meshwork and, thus, its propensity to translocate through the nuclear pore [39, 86].
A Delicate Relationship Between Neurodegenerative Diseases and Nucleocytoplasmic Transport
The cytoplasmic aggregation of nuclear RNA binding proteins (RBPs), repeat expansion associated gene products, tau, and other proteins is often a common hallmark of neurodegenerative diseases. Emerging evidence indicates that the dysfunction of the dynamic and structural components of nucleocytoplasmic transport may be intricately tied to pathological protein aggregation [1, 87,88,89,90]. However, the exact mechanisms underlying this relationship and its implications on neurodegeneration are not fully understood. Disrupted RNA metabolism, as a downstream effect of aberrant RBP localization and aggregation, has become a widely accepted and commonly observed disease phenotype of ALS/FTD, although there has been limited evidence to support the idea that it directly leads to neuronal degeneration [1, 87, 91,92,93]. Similarly, although evidence suggests that aggregates of tau and repeat expanded gene products in Alzheimer’s Disease (AD) and Huntington’s Disease (HD), respectively, may directly impede nucleocytoplasmic transport and damage NPC machinery, it is not clear whether their presence is a driving force or a downstream by-product of disease progression. Here we will review proposed nucleocytoplasmic trafficking abnormalities across different neurodegenerative diseases, their relationship to aberrant protein aggregation, and how this complex dynamic may contribute to neuronal cell death.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that leads to the progressive loss of motor function [94]. Due to overlapping symptoms and genetics ALS is now commonly accepted as a spectrum disorder with frontotemporal dementia (FTD), where patients experience cognitive impairment due to the degeneration of the frontal and temporal lobes leading to social and behavioral changes [95,96,97]. The exact mechanisms leading to ALS/FTD remain unclear but nucleocytoplasmic trafficking abnormalities appear to play a central role in disease pathogenesis.
Chromosome 9 Open Reading Frame 72 (C9orf72)
The etiology of ALS/FTD is diverse and growing with over 40 genetic abnormalities currently associated with ALS [98, 99]. One of the most well characterized genetic abnormalities associated with the ALS/FTD spectrum to date is the chromosome 9 open reading frame 72 (C9orf72) repeat expansion [95,96,97]. First identified in 2011 the GGGGCC (G4C2) hexanucleotide repeat expansion in the first intron of the C9orf72 gene is responsible for ~ 50% of all familial ALS and ~ 20% of sporadic ALS cases [95,96,97]. Healthy individuals have less than 23 repeats of the intronic G4C2 sequence of C9orf72, while patients with C9orf72-mediated ALS/FTD can exhibit anywhere from 30 to several hundred repeats [95,96,97]. Three commonly accepted but not mutually exclusive hypotheses exist to explain the neurodegenerative phenotype introduced by C9orf72 repeat expansion: 1) C9orf72 haploinsufficiency, 2) accumulation of G4C2 sense and C4G2 antisense RNA-foci, and 3) the production of dipeptide repeats (DPRs) via repeat-associated non-AUG (RAN) translation (Fig. 3b) [100,101,102,103,104,105,106,107,108,109,110,111,112]. The direct mechanism for how the C9orf72 repeat expansion causes ALS/FTD pathogenesis remains elusive but is currently being extensively investigated.

Nucleocytoplasmic trafficking deficits caused by the C9orf72 repeat expansion. The sense and antisense transcription of the (G4C2) expansion of the C9orf72 gene between exon 1a and 1b leads the formation of sense and antisense RNA foci found to sequester Pur ɑ, RanGAP1, ADAR3, SRs, hnRNPs, and other proteins not shown (a). Expanded C9orf72 RNA results in cytoplasmic/perinuclear RNA foci, which colocalize with RanGAP1 and Nup205, and produce DPRs via RAN translation (b). DPRs aggregate in the cytoplasm and nucleus, where polyGA aggregates may cause the mislocalization of POM121 and RanGAP1 (c). PolyPR clogs the NPC due to strong interactions with the FG-repeats of the central channel nucleoporins, Nup54 and Nup98, whereby it may promote the conversion of the FG hydrogel sieve to a more solid, fibrillar state (d). Impaired import and subsequent accumulation of soluble RBPs (e) coupled with factors like stress, aging, and DPR interaction (f) leads to the formation of insoluble protein aggregates that further disrupt nucleocytoplasmic transport (g), resulting in a feed forward mechanism of toxicity. In addition, the depletion of RBPs from the nucleus has a detrimental effect on RNA metabolism (h). PolyA RNA retention in the nucleus (i) is another common feature of C9orf72 ALS/FTD models and may be a result of disrupted RNA metabolism and NPC defects
Early genetic screens in S. cerevisiae and D. melanogaster identified crucial components of the nucleocytoplasmic transport machinery as modifiers of C9orf72 disease models [113,114,115]. Specifically, in regards to the NPC, the coat nucleoporins, Nup107 and Nup160, the nuclear basket component, Nup50, as well as inner ring components, Nup155 and Nup98, were found to be suppressors of C9orf72 phenotype (Fig. 1) [113, 114]. In contrast, coat nucleoporin, Seh1, inner ring components, Nup62 and Nup93, nuclear basket components, Nup50, Nup153 and Tpr, the cytoplasmic filament component, GLE1, and the transmembrane nucleoporin, NDC1, were found to be enhancers of the observed C9orf72 phenotype (Fig. 1) [113,114,115]. Additionally, components central to nucleocytoplasmic transport, such as protein transport factors, Ran, RanGAP, RCC1, transportins, exportins, and karyopherin α and β, and RNA export factors, NXF1, GLE1, and ALYREF, were shown to be modifiers of C9orf72-phenotypes [113, 115].
In agreement with the idea that nucleocytoplasmic transport is altered in C9orf72-mediated ALS/FTD, RanGAP1, was shown to directly bind to the C9orf72 repeat expansion (Fig. 3a), and was later shown to form intranuclear inclusions [89, 116]. These findings suggest that RanGAP1′s ability to regulate the Ran gradient is altered in C9orf72-mediated ALS/FTD [89, 116]. Not surprisingly, disruptions of the Ran gradient have been observed in the presence of the C9orf72 repeat expansion, a phenotype that is rescued with overexpression of RanGAP1 [89]. Further evidence of disrupted nucleocytoplasmic transport components has been shown in mice overexpressing the GA dipeptide repeat, which form cytoplasmic and intranuclear aggregates that colocalize with RanGAP1 and POM121 (Fig. 3c) [117].
In addition to RNA-foci interaction with RanGAP1, evidence also suggests that C9orf72 RNA-foci sequester RNA-binding proteins, including ALYREF, Adenosine Deaminase Acting on RNA (ADAR3), heterogeneous nuclear ribonucleoproteins (hnRNPs), Pur-alpha, and serine arginine (SR) proteins (Fig. 3a) [116, 118, 119]. Additionally, the disruption of proper RBP function may be exacerbated by DPRs. GR and PR overexpression models in HEK293T cells identified over a hundred interactors of the arginine containing DPRs, including disease causing RBPs with low-complexity domains (LCD) [120, 121]. DPR interaction with LCD containing proteins reduces the capacity of membrane-less organelles for liquid–liquid phase separation (LLPS), a characteristic essential for the dynamic nature of RBPs, stress granules, nucleoli, and the diffusion barrier of the nuclear pore complex [120, 121]. The LLPS of proteins, a normally reversible process whereby proteins shift from a soluble or “de-mixed” state to liquid droplet state, can be a seeding point for the deposition of protein aggregates—commonly seen in ALS/FTD pathology—when aging, stress, and DPR interaction are in play (Fig. 3f) [122, 123]. Through similar mechanisms, PR repeat proteins have been shown to promote the fibrillization of the FG-repeats on Nup98 and Nup54, thereby clogging the NPC and reducing nuclear transport of RNA and proteins (Fig. 3d, i) [120, 124]. However, a recent study from Vanneste and colleagues provides evidence that these arginine containing DPRs do not directly interfere with nucleocytoplasmic transport and highlights a need for additional studies clarifying the role DPRs may play in the pathogenesis of C9orf72-mediated ALS/FTD [125].
Taken altogether, it is clear that nucleocytoplasmic transport can be disrupted in C9orf72 mediated ALS/FTD, through mechanisms leading to the sequestration of transport machinery and aberrant cytoplasmic RBP accumulation and aggregation (Fig. 3e–g). Aside from direct impairment of nuclear import and export, the nuclear depletion of RBPs causes a widespread disruption of RNA metabolism (Fig. 3h) that will be described in the context of TDP-43 and FUS below.
Transactive Response DNA Binding Protein (TARDBP)
Considering the diversity in the etiology and patient presentation within the ALS/FTD disease spectrum, it is surprising that there may be shared mechanisms leading to neurodegeneration in many patient subgroups [126,127,128]. Indeed, cytoplasmic accumulation and nuclear depletion of the RNA binding protein, encoded by the TARDBP gene, TAR-DNA binding protein-43 (TDP-43) is found in ~ 95% of all ALS cases and ~ 45% of FTD cases (FTD-TDP), irrespective of etiology [126, 127, 129, 130]. Notably, TDP-43 proteinopathies are not limited to ALS and FTD with TDP-43 pathology. In fact, TDP-43 positive inclusions are also a characteristic of certain cases of HD, AD, Parkinson’s disease, chronic traumatic encephalopathy, and certain inclusion body myopathies [126, 130,131,132,133]. Despite extensive research, it is unclear whether cytoplasmic TDP-43 accumulation leads to a nuclear loss of function or a cytoplasmic toxic gain of function, or a combination of both [93, 129, 134, 135]. Whether wild-type TDP-43 inclusions are drivers of disease progression, or simply a by-product of a degenerating neuron, remains to be seen. Considering that aberrant TDP-43 inclusions are a shared occurrence in multiple diseases we will discuss TDP-43′s functional role in cellular homeostasis and its potential contribution to neurodegenerative disease pathogenesis in more detail below.
TDP-43 is an essential RNA binding protein involved in many steps of RNA metabolism, including: transcription, splicing, maturation, stability, transport, translation, and micro and long non-coding RNA processing [134]. It has further been proposed to be involved in the formation and maintenance of stress granules, and the regulation of stress granule nucleating protein expression [136]. TDP-43 binds to RNA with high specificity via its two RNA recognition motifs (RRMs). Containing both an NLS and an NES, TDP-43 plays roles in both the nucleus and the cytoplasm and, therefore, its trafficking across the nuclear membrane is essential for its proper function. Nuclear import of TDP-43 is facilitated by the classical KPNA/KPNB1 pathway (Fig. 2a–e) [137]. Interestingly, expression of the nuclear export factor XPO2, which is required for KPNA recycling (Fig. 2d), is reduced in the brains of FTD-TDP patients, potentially contributing to the cytoplasmic accumulation of TDP-43 in these patients [137]. The nuclear export of TDP-43 has been predicted to be recognized as an NES-containing cargo by XPO1. Indeed TDP-43 contains two NES sequences that have been predicted to interact with XPO1 [138]. However, recent studies have suggested these two NES sequences are neither necessary nor sufficient for nuclear export, indicating they may be nonfunctional and that TDP-43 may not be actively transported out of the nucleus [138]. In support of this hypothesis, pharmacological and genetic disruption of facilitated nuclear export pathways (Fig. 2o–q), suggest that TDP-43 is not exported by any of the major export pathways (XPO1, XPO5 or with mRNA through bulk RNA export) [138, 139]. Instead, export of TDP-43 is thought to be mediated through passive diffusion [138].
Considering the prevalence of TDP-43 mislocalization it is essential to understand how the aberrant cytoplasmic accumulations (Fig. 4l) form and how these accumulations are involved in nucleocytoplasmic trafficking defects. The aggregation of TDP-43 is most commonly attributed to its LCD, housed within the C-terminus of the protein. The LCD is home to the majority of ALS associated TARDBP mutations [140]. In addition to being unusually disordered and aggregation prone, the C-terminus of TDP-43, in fragmented form, is a highly toxic and prevalent component of the cytoplasmic inclusions seen in ALS pathology [141, 142]. It seems possible that the ability of TDP-43 to undergo LLPS, may create opportunities for TDP-43 to form insoluble aggregates (Fig. 4n) [123, 143, 144]. Indeed, under stress, with age, and in mutated forms, cytoplasmic TDP-43 droplets become more solid and gel like, facilitating aggregation (Fig. 4p) [123, 144,145,146]. The specific mechanism detailing how endogenous TDP-43 aggregations form in neurodegenerative diseases remains unknown. Artificial introduction of TDP-43 inclusions recruit pathogenic species that mimic the TDP-43 inclusions observed in patients [147]. The low complexity domain on TDP-43 drives the formation of these neurotoxic TDP-43 inclusions and promotes further recruitment of nuclear TDP-43 to such inclusions [147]. Interestingly, aberrant TDP-43 liquid–liquid phase separation can be rescued by enhancing TDP-43 interaction with RNA [147], suggesting that the aggregation of TDP-43 in the cytoplasm may be the result of impaired mRNA export from the nucleus [147]. Mann et. al suggest the possibility that increased nuclear retention of RNA renders cytoplasmic TDP-43 more aggregation prone due to reduced cytoplasmic RNA [147].

Nucleocytoplasmic trafficking deficits in neurodegenerative diseases. Top Left: Alzheimer’s disease. The phosphorylation state of tau protein determines its capacity for LLPS, where hyperphosphorylation promotes a more inseparable phase (a). Dysregulation of tau phosphorylation from stress, aging, and/or mutations cause it to aggregate into NFTs (b). Phospho-tau positive NFTs interact with the nuclear envelope, where they disrupt the NPC, and co-aggregate with Nup98 and Nup62 (c). NFT’s physical occlusion of the NPC (d). Impaired Ran gradient, mislocalized or aggregated Nups, RBPs and transport factors act as additional evidence of nucleocytoplasmic trafficking disruptions (e). Top Right: Huntington’s disease. mHtt harbors over 36 glutamine repeats (f). The polyQ tract within mHtt promotes its LLPS (g) which can lead to its pathological aggregation through aging and cellular stress (h). Toxic perinuclear aggregates of mHtt disrupt the NPC, evidenced by their sequestration of Nups, Ran-related proteins, and other transport factors (i). Nuclear mHtt aggregates are also common in Huntington’s patients although their contribution to disease progression may be less severe (j). Similar to tau in AD, mHtt can lead to an impaired Ran gradient and the mislocalization and/or aggregation of proteins (k). Bottom: ALS/FTD. A common feature of ALS/FTD is the impaired import cytoplasmic accumulation of TDP-43 (l) and FUS (m). Both of these proteins can undergo LLPS distinct from stress granules, mediated by their LCDs (n, o). With aging, stress, and/or mutations in these LCDs, TDP-43 or FUS can shift to an aggregated state (p, q). TDP-43 aggregates can include at least 14 Nups as well as several nuclear transport factors (r). TDP-43 aggregates lead to further disruption of the NPC and induce mislocalization of POM121 and POM210 to perinuclear puncta (t). The binding interactions of FUS aggregates are less elucidated, although FTD-FUS co-aggregates with other FET proteins and TNPO1 (s, blue font). AD, HD, and ALS/FTD display a disrupted Ran gradient, and mislocalized RBPs and transport components (w). Nuclear loss of TDP-43 or FUS disrupts RNA metabolism (u, v). Nuclear accumulation of polyA-RNA (x) and distortions in the nuclear envelope (y)
TDP-43 aggregation may contribute to neurodegeneration by disrupting the nucleocytoplasmic transport machinery [92]. Overexpression of TDP-43 C-terminal fragments either co-aggregate with, or cause the mislocalization of coat Nups (Nup107 and Nup160), inner ring Nups (Nup35, Nup58, Nup62, Nup93, Nup98, Nup205), transmembrane Nups (POM121 and POM210), cytoplasmic filament Nups (Gle1, Nup88, Nup214, RanBP2) nuclear basket Nups (Nup153 and Nup155), and nuclear transport components (XPO5, NXF1) (Fig. 4r). This indicates that cytoplasmic aggregation of TDP-43 is highly disruptive to the NPC (Fig. 4t) [92]. While, the overexpressed system employed in these experiments may exaggerate the degree of NPC interaction, these studies strongly suggest that TDP-43 cytoplasmic inclusions have the capacity to disrupt the NPC and its components (Fig. 4t) [92]. Notably, despite the substantial dysregulation of the NPC and nucleocytoplasmic transport, the Ran gradient appears normal in these experiments [92].
In addition to the gain-of-toxic function due to TDP-43 aggregates, it is also possible that the nuclear depletion and loss of function of TDP-43 is a toxic contributor resulting from TDP-43 mislocalization to the cytoplasm [127]. These two mechanisms describing cellular stress from TDP-43 proteinopathy are likely not mutually exclusive. As stated above, TDP-43 is a master regulator of splicing and it has been shown to act as a splicing repressor for cryptic exons [148,149,150]. Proper TDP-43 function has been shown to prevent the inclusion of specific exons, termed cryptic exons, by negatively regulating splicing at these sites [150]. Loss of function of TDP-43 due to its depletion leads to the inclusion of these cryptic exons which could allow for improper handling throughout an RNAs translation and maturation often leading to the decay of the host transcript (Fig. 4u) [148,149,150]. Indeed, downregulated genes from RNA sequencing datasets of samples with depleted TDP-43 are enriched for cryptic exons [148]. The inclusion of cryptic exons has been observed in C9orf72-mediated ALS/FTD [150]. Interestingly, cryptic exons are highly variable in different cell types, a finding that may provide clues to explain selective vulnerability in neurodegeneration [149]. Considering that TDP-43 interacts with thousands of transcripts, substantial work remains to be completed to understand how the inclusion of cryptic exons is the result of TDP-43 mislocalization and what role they play in the pathogenesis of neurodegenerative diseases. One such possibility is TDP-43′s altered regulation of stathmin-2 in ALS [151, 152]. Depletion of TDP-43, mimicking loss of nuclear TDP-43, leads to the pre-polyadenylation of stathmin-2 and decreased protein expression [151, 152]. Interestingly, stathmin-2 is thought to play a role in neurite outgrowth and microtubule dynamics [151, 152]. The loss of stathmin-2 following depletion of TDP-43, causes failure of neuronal regeneration after axotomy, a phenotype that is rescued by stathmin-2 transduction and pharmacological inhibition of c-Jun N-terminal kinase [151, 152].
Fused in Sarcoma (FUS)
Another widely studied, dysregulated RBP implicated in neurodegenerative disease is the FUS protein [135, 153,154,155,156,157]. Historically, FUS pathology was considered to be a rare observation, only attributed to the abnormal aggregation of mutant FUS in ~ 5% of ALS cases (ALS-FUS), and the abnormal aggregation of wild-type FUS in ~ 10% of FTD patients (FTD-FUS) [153, 154, 158, 159]. While initial studies were focused primarily on pathological FUS inclusions, more recent work provides evidence that the spinal motor neurons of sporadic ALS patients display a diffuse cytoplasmic mislocalization of FUS, with no coinciding aggregation [157]. In agreement with these results, mice with mutation-induced cytoplasmic mislocalization of FUS without detectable cytoplasmic aggregation, exhibit age dependent progressive motor and cognitive deficits [160]. These recent studies suggest that the cytoplasmic inclusions of FUS observed in postmortem tissue may not be the only toxic contribution provided by FUS dysregulation [160]. Considering FUS may have a wider contribution to ALS/FTD pathogenesis than once predicted, it is essential to understand the mechanisms by which FUS dysregulation occur, how these differ between ALS-FUS and FTD-FUS patients, and how they are distinct from the mechanisms driving TDP-43 pathology.
The relationship between TDP-43 and FUS is interesting considering their structural and functional similarities: both have RNA recognition motifs, an LCD, and an NES and NLS; both are predominantly nuclear, yet play roles in mRNA stability and translation in the cytoplasm; and both are involved in transcription and splicing [161,162,163]. Indeed, the LCD of FUS is thought to mediate its capacity for LLPS, disruptions of which may seed FUS aggregation similar to the manner by which TDP-43 is thought to aggregate (Fig. 4o, q) [164]. Also, the nuclear export of FUS appears to be similar to TDP-43, given FUS has 2 predicted XPO1-mediated NES sequences that do not mediate its nuclear export [138]. Instead FUS export is regulated by passive diffusion through the nuclear pore and not by facilitated transport pathway [138, 165]. However, they have key differences that might explain why TDP-43 and FUS inclusions are mutually exclusive [153, 154, 158, 159]. For example, they utilize different transport pathways. The nuclear import of TDP-43 is facilitated by the KPNA/KPNB1 pathway (Fig. 2a–e), while FUS nuclear import is conducted by Transportin-1 (TNPO1) via the TNPO pathway (Fig. 2h–l) [63, 65, 162, 166]. FUS mutations in ALS-FUS patients, have been shown to disrupt the binding of the TNPO1 receptor, which would lead to FUS’s cytoplasmic accumulation in a manner independent from drivers of impaired TDP-43 nuclear import (Fig. 4m) [162, 166,167,168,169,170]. Interestingly, neuronal FUS has been shown to be dysregulated in conditions of stress due to failures of TNPO1 [165]. Intriguingly, astrocytes do not display FUS mislocalization under the same conditions, suggesting FUS may have cell-type specific contributions to disease [165]. Additionally, different manifestations of post-translational modifications of FUS, may play a role in its different presentations in ALS and FTD [171,172,173,174,175,176,177,178,179]. Specifically, the methylation of FUS residues adjacent to the PY-NLS can alter FUS’s interaction with TNPO1 [171,172,173,174,175,176,177,178,179]. Dimethylation, characteristic of ALS-FUS, reduces this interaction, and hypomethylation, characteristic of FTD-FUS, increases it [171,172,173,174,175,176,177,178,179]. In support of these findings, FTD-FUS inclusions are positive for TNPO1 and other members of the FET protein family, EWS and TAF15, while ALS-FUS inclusions are not (Fig. 4s) [180]. These differences are intricately tied to the unique presentations of each disease and parsing out why these differences occur is important to characterize disease progression.
Similar to TDP-43, one consequence of FUS aggregation and mislocalization may also be a loss of function due to a nuclear depletion. While TDP-43 and FUS have similar roles in RNA processing and regulation of alternative splicing, FUS is thought to regulate a largely distinct set of transcripts in comparison to TDP-43 [181]. Depletion of FUS leads to broad misregulation of RNAs, including the dysregulation of expression for over 600 mRNAs as well as the dysregulation of over 350 splicing patterns (Fig. 4v) [181]. Interestingly, while both TDP-43 and FUS are important regulators of RNA metabolism, only 86 RNAs are regulated by both proteins, suggesting that the disruption to RNA metabolism due to FUS dysfunction impacts a different group of RNA compared to TDP-43 dysfunction [181]. These studies suggest that neurodegeneration resulting from FUS dysfunction may be distinct from neurodegeneration caused by TDP-43 dysfunction [181].
hnRNP A1 and hnRNP A2B1
Both TDP-43 and FUS are members of a large subclass of RNA-binding proteins known as hnRNPs, which have been shown to have diverse transcriptional control through their roles in RNA maturation and neuronal RNA transport granules. Additionally, these proteins have been associated with stress granule formation, which is critical to the cellular stress response [182,183,184]. Like TDP-43 and FUS, rare dominant mutations in the LCD of hnRNP A1 and hnRNP A2B1 have been associated with ALS [182,183,184]. Interestingly, these mutations are most frequently associated with multisystem proteinopathy, a rare inherited disease in which patients experience the degeneration of muscle, bone and/or the central nervous system [182,183,184]. The mutations in hnRNP A1 and hnRNP A2B1 have been shown to cause their mislocalization from the nucleus to the cytoplasm and to pathologically alter stress granule dynamics by enhancing their tendency to fibrillize and thereby disrupting their capacity to undergo LLPS [182,183,184]. These cytoplasmic inclusions have also been shown to colocalize with cytoplasmic TDP-43 aggregates and have been associated with TDP-43 nuclear depletion [182,183,184].
ADAR2
Our lab has shown that the RNA editing enzyme Adenosine Deaminase Acting on double-stranded RNA 2 (ADAR2) forms cytoplasmic inclusions in C9orf72-mediated ALS/FTD and in Alzheimer’s Disease (Fig. 4e, w) [185]. ADAR2 utilizes KPNA1 and KPNA3 to traffic into the nucleus [186, 187]. While it is unclear how ADAR2 accumulations form in ALS and AD it is possible that dysregulation of KPNA1 or 3 would lead to abnormal cytoplasmic inclusions of ADAR2. In addition to its cytoplasmic accumulation we provided evidence to suggest aberrant function of A to I editing in C9orf72-mediated ALS/FTD with enrichment of A to I editing aberrations in the EIF2 pathway, which may lead to global translational inhibition [185]. ADAR2 is a nuclear enzyme and its presence in the cytoplasm alters the function of the enzyme, suggesting that the altered cellular localization in disease represents a mechanism for its dysregulation [185]. There has been a revolution in the understanding of the function of A to I editing in both normal cellular function and in disease [188]. Dysregulation of ADAR2 and its normal cellular function would lead to altered regulation of RNA transcripts that would have yet unknown cellular consequences. While many RNA editing abnormalities have been associated with neurodegeneration [188], due to the massive number of uncharacterized A to I editing sites further experiments are needed to establish the specific role of altered RNA A to I editing events in the pathogenesis of neurodegenerative diseases.
Superoxide Dismutase 1 (SOD1)
Mutations in the SOD1 gene were the first genetic association with ALS [189]. Patients with SOD1 mutations do not have TDP-43 or FUS pathology [190,191,192,193]. However, large SOD1 accumulations exist in patients with these SOD1 mutations [190,191,192,193]. The dichotomy that appears to exist between neurons with SOD1 accumulations compared to TDP-43 and FUS accumulations is interesting and suggests different mechanisms leading to neurodegeneration [190,191,192,193]. Further evidence to support the unique nature of SOD1 suggests that SOD1 exhibits a distinct transcriptome compared to that of C9orf72 patients [194]. It is possible that some components of the nuclear pore and nucleocytoplasmic transport are disrupted as a result of the SOD1 G93A mutation [195]. However, conflicting reports suggest that more experiments are required to understand nucleocytoplasmic transport deficits in SOD1 mediated ALS [92].
Sporadic ALS
In contrast to familial ALS, there have been few studies which describe nucleocytoplasmic transport deficits in sporadic ALS. Limited number of studies report impaired nucleoporins (Nup62) and KPNB1 in cells with TDP-43 aggregates [196, 197]. Considering the challenges associated with studying and modeling sporadic forms of ALS it is not surprising that few studies have begun to describe nucleocytoplasmic trafficking deficits. However, as stated above, TDP-43 aggregations have the capacity to disrupt the nucleocytoplasmic trafficking machinery and it is possible there will be future disruptions identified in sporadic ALS.
Huntington’s Disease
The degeneration of striatal medium spiny projection neurons as well as cortical pyramidal neurons due to the presence of an expanded CAG repeat in the Huntingtin (HTT) gene is known as Huntington’s disease [198]. Individuals with more than 39 CAG repeats in the HTT gene develop progressive motor, cognitive and psychiatric symptoms leading to a steady decline in physical health and quality of life culminating in death 10—30 years after disease onset [199]. Translation of HTT with an abnormal number of CAG repeats, leads to huntingtin protein with an expanded poly-glutamine (polyQ) tract (mHtt) (Fig. 4f). Due to this polyQ tract and proline-rich region, mHtt undergoes LLPS (Fig. 4g), potentially leading to cytoplasmic, perinuclear, and nuclear aggregates through factors such as aging and other stressors (Fig. 4h–j) [200]. There is substantial evidence that suggests these aggregates disrupt the NPC and nucleocytoplasmic trafficking in Huntington's disease.
Immune-electron microscopy performed on cells overexpressing mHtt show distortion of the nuclear membrane (Fig. 4y) and cytoplasmic inclusions of NPC proteins in cells with perinuclear inclusions of mHtt (Fig. 4k) [201]. Additionally, early analysis of protein interactions suggested that polyQ accumulations interact with nuclear pore components, specifically FG-repeat Nups found in the cytoplasmic filaments (DDX19, RanBP2, and Nup214), the nuclear basket (Nup153), and the inner ring nucleoporin Nup62 [202].
More recent studies elaborated on these previous publications, firmly associating nucleocytoplasmic trafficking abnormalities with HD [90, 203]. First Gasset-Rosa et al. confirmed that mutant huntingtin accumulations lead to abnormal nuclear envelope morphology [203]. Using a mHtt mouse model, an increasingly severe nuclear envelope pathology in the homozygous compared to heterozygous animals suggested a dose-dependent phenotype [203]. These findings were confirmed in human induced pluripotent neural progenitor cells and in human postmortem tissue further associating mutant huntingtin and nuclear membrane abnormalities with disease state [203]. In addition to nuclear membrane pathology, HD models and patients displays overt nucleocytoplasmic trafficking deficits [203]. mRNA export is dysregulated, as evidenced by increased mRNA levels in the nuclei of cortical regions in a mouse models and Huntington’s disease human postmortem tissue (Fig. 4x) [203]. Concurrently, work done by Grima et al. showed a disruption of the Ran gradient in motor neurons differentiated from hiPSCs from HD patients [90]. Evidence for disruption of the NPC comes from the discovery in human post mortem tissue showing that patients with Huntington’s disease exhibit a mislocalization or aggregation of the Ran GTPase activating protein, RanGAP1, in the frontal cortex, striatum, cerebellum, and hiPSC neurons (Fig. 4k) [90]. Additionally, they found that Nup62, a component of the central channel of the inner ring of the NPC, was mislocalized only in the striatum and displayed a diffuse cytoplasmic pathology in human tissue and hiPSC neuronal models (Fig. 4k) [90]. The Ran gradient deficit can be rescued both by inhibition of nuclear export via Exportin-1 inhibition with KPT-350 treatment; and interestingly, inhibition of O-GlcNAcylation, a common post translational modification for nucleoporins [90].
Alzheimer’s Disease
AD is the leading cause of dementia worldwide [204]. The age related progressive cognitive decline of episodic memory is the most common clinical feature of Alzheimer’s disease [204]. Early studies utilizing electron microscopy identified a close association between neurofibrillary tangles (NFTs) and the NPC in neurons of post mortem tissue from patients with Alzheimer’s disease (Fig. 4d) [205]. Furthermore, recent studies using iPSC-differentiated neurons have shown that tau accumulation disrupts nuclear lamina and nucleocytoplasmic transport [206]. Mechanisms for tau aggregation have parallels to RBP and mHtt aggregation—mutation and the phosphorylation states of tau have been implicated in its aberrant LLPS (Fig. 4a, b) [207, 208]. Given the prevalence of tau-pathology in AD and the emerging link between nucleocytoplasmic trafficking deficits and neurodegenerative diseases, elucidating the impact of tau inclusions on the NPC is critical to understanding disease pathogenesis.
Conflicting reports exist on the nature of Nup62s localization in Alzheimer’s disease postmortem tissue [88, 209]. Early studies found that Nup62 staining on abnormal nuclei in neurons with NFTs was localized primarily to the nuclear membrane and identified no aberrant localization [209]. More recent studies have suggested that similar to Huntington’s disease, Nup62 is mislocalized in neurons from AD patients (Fig. 4e) [88]. Due to difficulties in Nup62 labeling the authors suggest it is difficult to determine if cytoplasmic aggregation exists due to lipofuscin autofluorescence, but instead suggest diffuse cytosolic mislocalization and depletion at the nuclear membrane [88]. Nup414 labeling, which includes Nup62, further suggests the dysregulation of FG-repeat Nups [88]. Indeed, Nup98, the largest contributor to the FG-repeat diffusion barrier, interacts directly with phospho-tau and is mislocalized in AD hippocampal neurons [88]. Not surprisingly, the direct interaction of tau with the NPC leads to disruption of the NPC diffusion barrier and the Ran gradient (Fig. 4c) [88]. However, Nup54, POM121, Nup88, Nup153, Nup133, and Nup214 were shown to not have apparent differences in expression or distribution in neurons positive for tau tangles, which suggests normal function of the transmembrane, cytoplasmic filaments, nuclear basket, and coat nucleoporins, as well as some of the inner ring nucleoporins, indicating there is not a complete breakdown of the NPC [88, 209]. Nonetheless, similar to nuclear envelope aberrations found in other neurodegenerative diseases, neurons from patients with Alzheimer’s disease exhibit aberrantly shaped nuclei (Fig. 4y) [209].
Neurons with and without NFTs also contain cytoplasmic accumulations of the RanGDP transporter, NTF2 (Fig. 4e) [209]. Failure of the nuclear import of RanGDP would lead to the disruption of the Ran gradient in AD and disruption of Ran mediated nucleocytoplasmic transport [210]. This supports the disruption of the Ran gradient in AD CA1 hippocampal tissue, with phospho-tau inclusions [88]. Considering that the interactions of the karyopherin family with NTF2 are thought to expedite transport kinetics, it would not be surprising that NTF2 accumulations accompany additional nuclear import deficits [211]. Interestingly, induced cytoplasmic artificial beta sheets that form amyloid-like aggregates, inhibit nuclear export of mRNA, likely due to their forced mislocalization and co-aggregation of KPNA2 and KPNA4, RanBP1, and THOC2 suggesting that the presence of beta-sheets in NFTs found in AD may further disrupt nuclear trafficking adding additional sources of cellular stress (Fig. 4e) [212, 213].
Therapeutically Targeting Nucleocytoplasmic Transport Defects
The prevalence of nucleocytoplasmic trafficking throughout neurodegenerative diseases creates an enticing opportunity for therapeutic intervention. To this point the nuclear export inhibitors KPT-350, KPT-335, and KPT-276 have been shown to have neuroprotective effects [89, 90, 92, 139]. However, the mechanism of this neuroprotection remains unclear [139]. Considering the recent evidence suggesting that TDP-43 and FUS are not targets of exportin mediated transport, it is unknown whether KPT compounds are directly modulating TDP-43 or FUS nucleocytoplasmic transport [138, 139, 165]. Biogen has recently initiated a Phase1 safety trial with an XPO1 inhibitor in ALS patients to be completed by the end of the fiscal year 2020 [214]. Interestingly though, low non-toxic concentrations of KPT compounds are not able to inhibit nuclear export, emphasizing even more that the detailed mechanisms for how KPT compounds might confer neuroprotection in neurodegenerative disease have yet to be identified [139].
Considering the widespread presence of protein aggregation in these diseases, removing these aberrant protein aggregates may alleviate the associated neurotoxic phenotypes [146, 215]. Interestingly, nuclear import receptors can disassemble aberrant insoluble protein aggregates that have been associated with neurodegenerative diseases [216]. Specifically, KNPA and KNPB have been shown to be able to disaggregate TDP-43 aggregates and TNPO1 was able to disaggregate FUS aggregates as well as other aggregates of proteins with a PY-NLS [216]. These findings suggest that failure of nuclear import can be rescued by supporting the possibly overwhelmed endogenous nuclear import receptors (Ex. KNPA, KNPB, and TNPO1) [216]. If disaggregation of aberrant fibrils using nuclear import receptors can also restore normal transport of RNA binding proteins to the nucleus, it may be possible to rescue toxic cytoplasmic aggregates, alleviate nuclear depletion of RBPs and thereby protect against neuronal loss in neurodegenerative diseases [215, 216].
Conclusion
The widespread prevalence of impaired elements of nucleocytoplasmic trafficking machinery throughout instances of neurodegeneration suggest the existence of a common mechanism of dysfunction. The potential of overlapping causative disease mechanisms creates exciting opportunities for multifaceted therapeutic interventions. While an enormous body of work remains to fully understand the mechanisms behind possible therapeutic interventions in nucleocytoplasmic transport, early evidence suggests multiple exciting opportunities. These trafficking aberrations have been well established and their role in neurodegenerative diseases has been increasingly strengthened. However, the initiating factor that leads directly to detrimental nucleocytoplasmic trafficking deficits remains unclear. Nevertheless, it seems likely that regardless of how trafficking deficits begin, the consequences act in a feed-forward mechanism that will further disrupt nucleocytoplasmic trafficking resulting in more severe consequences. Once nucleocytoplasmic trafficking defects have taken place, how this cellular disruption causes neurodegeneration is still largely unknown. It is likely that nuclear depletion and increased cytoplasmic concentration leading to aberrant aggregations will contribute to cellular stress through feed-forward disruptions of RNA metabolism and nucleocytoplasmic transport. The identification of toxic species that result from trafficking deficits may result in additional opportunities for therapeutic interventions.
References
Hutten S, Dormann D (2019) Nucleocytoplasmic transport defects in neurodegeneration—cause or consequence? Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2019.05.020
Ferreira PA (2019) The coming-of-age of nucleocytoplasmic transport in motor neuron disease and neurodegeneration. Cell Mol Life Sci 76(12):2247–2273. https://doi.org/10.1007/s00018-019-03029-0
Boeynaems S, Bogaert E, Van Damme P, Van Den Bosch L (2016) Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol 132(2):159–173. https://doi.org/10.1007/s00401-016-1586-5
Kim HJ, Taylor JP (2017) Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 96(2):285–297. https://doi.org/10.1016/j.neuron.2017.07.029
Haeusler AR, Donnelly CJ, Rothstein JD (2016) The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci 17(6):383–395. https://doi.org/10.1038/nrn.2016.38
Hetzer MW (2010) The role of the nuclear pore complex in aging of post-mitotic cells. Aging (Albany NY) 2(2):74–75. https://doi.org/10.18632/aging.100125
Ungricht R, Kutay U (2017) Mechanisms and functions of nuclear envelope remodelling. Nat Rev Mol Cell Biol 18(4):229–245. https://doi.org/10.1038/nrm.2016.153
Watson ML (1955) The nuclear envelope; its structure and relation to cytoplasmic membranes. J Biophys Biochem Cytol 1(3):257–270. https://doi.org/10.1083/jcb.1.3.257
Callan HG, Tomlin SG (1950) Experimental studies on amphibian oocyte nuclei. I. Investigation of the structure of the nuclear membrane by means of the electron microscope. Proc R Soc Lond B 137(888):367–378
Wente SR, Rout MP (2010) The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol 2(10):a000562. https://doi.org/10.1101/cshperspect.a000562
Franke WW (1966) Isolated nuclear membranes. J Cell Biol 31(3):619–623. https://doi.org/10.1083/jcb.31.3.619
Lin DH, Hoelz A (2019) The structure of the nuclear pore complex (an update). Annu Rev Biochem 88:725–783. https://doi.org/10.1146/annurev-biochem-062917-011901
Gall JG (1967) Octagonal nuclear pores. J Cell Biol 32(2):391–399. https://doi.org/10.1083/jcb.32.2.391
Hoelz A, Debler EW, Blobel G (2011) The structure of the nuclear pore complex. Annu Rev Biochem 80:613–643. https://doi.org/10.1146/annurev-biochem-060109-151030
Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT, Matunis MJ (2002) Proteomic analysis of the mammalian nuclear pore complex. J Cell Biol 158(5):915–927. https://doi.org/10.1083/jcb.200206106
Floch AG, Palancade B, Doye V (2014) Fifty years of nuclear pores and nucleocytoplasmic transport studies: multiple tools revealing complex rules. Methods Cell Biol 122:1–40. https://doi.org/10.1016/B978-0-12-417160-2.00001-1
DeGrasse JA, DuBois KN, Devos D, Siegel TN, Sali A, Field MC, Rout MP, Chait BT (2009) Evidence for a shared nuclear pore complex architecture that is conserved from the last common eukaryotic ancestor. Mol Cell Proteomics 8(9):2119–2130. https://doi.org/10.1074/mcp.M900038-MCP200
Neumann N, Lundin D, Poole AM (2010) Comparative genomic evidence for a complete nuclear pore complex in the last eukaryotic common ancestor. PLoS ONE 5(10):e13241. https://doi.org/10.1371/journal.pone.0013241
Maimon T, Elad N, Dahan I, Medalia O (2012) The human nuclear pore complex as revealed by cryo-electron tomography. Structure 20(6):998–1006. https://doi.org/10.1016/j.str.2012.03.025
Pumroy RA, Nardozzi JD, Hart DJ, Root MJ, Cingolani G (2012) Nucleoporin Nup50 stabilizes closed conformation of armadillo repeat 10 in importin alpha5. J Biol Chem 287(3):2022–2031. https://doi.org/10.1074/jbc.M111.315838
Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Sali A, Rout MP (2007) The molecular architecture of the nuclear pore complex. Nature 450(7170):695–701. https://doi.org/10.1038/nature06405
Nofrini V, Di Giacomo D, Mecucci C (2016) Nucleoporin genes in human diseases. Eur J Hum Genet 24(10):1388–1395. https://doi.org/10.1038/ejhg.2016.25
Griffis ER, Craige B, Dimaano C, Ullman KS, Powers MA (2004) Distinct functional domains within nucleoporins Nup153 and Nup98 mediate transcription-dependent mobility. Mol Biol Cell 15(4):1991–2002. https://doi.org/10.1091/mbc.e03-10-0743
Chemudupati M, Osmani AH, Osmani SA (2016) A mitotic nuclear envelope tether for Gle1 also impacts nuclear and nucleolar architecture. Mol Biol Cell. https://doi.org/10.1091/mbc.E16-07-0544
Bui KH, von Appen A, DiGuilio AL, Ori A, Sparks L, Mackmull MT, Bock T, Hagen W, Andres-Pons A, Glavy JS, Beck M (2013) Integrated structural analysis of the human nuclear pore complex scaffold. Cell 155(6):1233–1243. https://doi.org/10.1016/j.cell.2013.10.055
Lutzmann M, Kunze R, Buerer A, Aebi U, Hurt E (2002) Modular self-assembly of a Y-shaped multiprotein complex from seven nucleoporins. EMBO J 21(3):387–397. https://doi.org/10.1093/emboj/21.3.387
Kelley K, Knockenhauer KE, Kabachinski G, Schwartz TU (2015) Atomic structure of the Y complex of the nuclear pore. Nat Struct Mol Biol 22(5):425–431. https://doi.org/10.1038/nsmb.2998
Kosinski J, Mosalaganti S, von Appen A, Teimer R, DiGuilio AL, Wan W, Bui KH, Hagen WJ, Briggs JA, Glavy JS, Hurt E, Beck M (2016) Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science 352(6283):363–365. https://doi.org/10.1126/science.aaf0643
Fischer J, Teimer R, Amlacher S, Kunze R, Hurt E (2015) Linker Nups connect the nuclear pore complex inner ring with the outer ring and transport channel. Nat Struct Mol Biol 22(10):774–781. https://doi.org/10.1038/nsmb.3084
Upla P, Kim SJ, Sampathkumar P, Dutta K, Cahill SM, Chemmama IE, Williams R, Bonanno JB, Rice WJ, Stokes DL, Cowburn D, Almo SC, Sali A, Rout MP, Fernandez-Martinez J (2017) Molecular architecture of the major membrane ring component of the nuclear pore complex. Structure 25(3):434–445. https://doi.org/10.1016/j.str.2017.01.006
Osmani AH, Davies J, Liu HL, Nile A, Osmani SA (2006) Systematic deletion and mitotic localization of the nuclear pore complex proteins of Aspergillus nidulans. Mol Biol Cell 17(12):4946–4961. https://doi.org/10.1091/mbc.e06-07-0657
Liu HL, De Souza CP, Osmani AH, Osmani SA (2009) The three fungal transmembrane nuclear pore complex proteins of Aspergillus nidulans are dispensable in the presence of an intact An-Nup84-120 complex. Mol Biol Cell 20(2):616–630. https://doi.org/10.1091/mbc.E08-06-0628
Funakoshi T, Clever M, Watanabe A, Imamoto N (2011) Localization of Pom121 to the inner nuclear membrane is required for an early step of interphase nuclear pore complex assembly. Mol Biol Cell 22(7):1058–1069. https://doi.org/10.1091/mbc.E10-07-0641
Mansfeld J, Guttinger S, Hawryluk-Gara LA, Pante N, Mall M, Galy V, Haselmann U, Muhlhausser P, Wozniak RW, Mattaj IW, Kutay U, Antonin W (2006) The conserved transmembrane nucleoporin NDC1 is required for nuclear pore complex assembly in vertebrate cells. Mol Cell 22(1):93–103. https://doi.org/10.1016/j.molcel.2006.02.015
Kiseleva E, Allen TD, Rutherford S, Bucci M, Wente SR, Goldberg MW (2004) Yeast nuclear pore complexes have a cytoplasmic ring and internal filaments. J Struct Biol 145(3):272–288. https://doi.org/10.1016/j.jsb.2003.11.010
Fernandez-Martinez J, Kim SJ, Shi Y, Upla P, Pellarin R, Gagnon M, Chemmama IE, Wang J, Nudelman I, Zhang W, Williams R, Rice WJ, Stokes DL, Zenklusen D, Chait BT, Sali A, Rout MP (2016) Structure and function of the nuclear pore complex cytoplasmic mrna export platform. Cell 167(5):1215–1228. https://doi.org/10.1016/j.cell.2016.10.028
Alcazar-Roman AR, Tran EJ, Guo S, Wente SR (2006) Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat Cell Biol 8(7):711–716. https://doi.org/10.1038/ncb1427
Weirich CS, Erzberger JP, Flick JS, Berger JM, Thorner J, Weis K (2006) Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat Cell Biol 8(7):668–676. https://doi.org/10.1038/ncb1424
Ribbeck K, Gorlich D (2002) The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J 21(11):2664–2671. https://doi.org/10.1093/emboj/21.11.2664
Strawn LA, Shen T, Shulga N, Goldfarb DS, Wente SR (2004) Minimal nuclear pore complexes define FG repeat domains essential for transport. Nat Cell Biol 6(3):197–206. https://doi.org/10.1038/ncb1097
Hulsmann BB, Labokha AA, Gorlich D (2012) The permeability of reconstituted nuclear pores provides direct evidence for the selective phase model. Cell 150(4):738–751. https://doi.org/10.1016/j.cell.2012.07.019
Fried H, Kutay U (2003) Nucleocytoplasmic transport: taking an inventory. Cell Mol Life Sci 60(8):1659–1688. https://doi.org/10.1007/s00018-003-3070-3
Stewart M (2007) Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol 8(3):195–208. https://doi.org/10.1038/nrm2114
Mahajan R, Delphin C, Guan T, Gerace L, Melchior F (1997) A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 88(1):97–107. https://doi.org/10.1016/s0092-8674(00)81862-0
Matunis MJ, Wu J, Blobel G (1998) SUMO-1 modification and its role in targeting the Ran GTPase-activating protein, RanGAP1, to the nuclear pore complex. J Cell Biol 140(3):499–509. https://doi.org/10.1083/jcb.140.3.499
Ribbeck K, Lipowsky G, Kent HM, Stewart M, Gorlich D (1998) NTF2 mediates nuclear import of Ran. EMBO J 17(22):6587–6598. https://doi.org/10.1093/emboj/17.22.6587
Bischoff FR, Ponstingl H (1991) Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature 354(6348):80–82. https://doi.org/10.1038/354080a0
Bischoff FR, Ponstingl H (1991) Mitotic regulator protein RCC1 is complexed with a nuclear ras-related polypeptide. Proc Natl Acad Sci USA 88(23):10830–10834. https://doi.org/10.1073/pnas.88.23.10830
Kalderon D, Roberts BL, Richardson WD, Smith AE (1984) A short amino acid sequence able to specify nuclear location. Cell 39(3 Pt 2):499–509. https://doi.org/10.1016/0092-8674(84)90457-4
Chook YM (1813) Suel KE (2011) Nuclear import by karyopherin-betas: recognition and inhibition. Biochim Biophys Acta 9:1593–1606. https://doi.org/10.1016/j.bbamcr.2010.10.014
Wen W, Meinkoth JL, Tsien RY, Taylor SS (1995) Identification of a signal for rapid export of proteins from the nucleus. Cell 82(3):463–473. https://doi.org/10.1016/0092-8674(95)90435-2
Lange A, McLane LM, Mills RE, Devine SE, Corbett AH (2010) Expanding the definition of the classical bipartite nuclear localization signal. Traffic 11(3):311–323. https://doi.org/10.1111/j.1600-0854.2009.01028.x
Miyamoto Y, Yamada K, Yoneda Y (2016) Importin alpha: a key molecule in nuclear transport and non-transport functions. J Biochem 160(2):69–75. https://doi.org/10.1093/jb/mvw036
Bayliss R, Littlewood T, Stewart M (2000) Structural basis for the interaction between FxFG nucleoporin repeats and importin-beta in nuclear trafficking. Cell 102(1):99–108. https://doi.org/10.1016/s0092-8674(00)00014-3
Bayliss R, Kent HM, Corbett AH, Stewart M (2000) Crystallization and initial X-ray diffraction characterization of complexes of FxFG nucleoporin repeats with nuclear transport factors. J Struct Biol 131(3):240–247. https://doi.org/10.1006/jsbi.2000.4297
Kutay U, Bischoff FR, Kostka S, Kraft R, Gorlich D (1997) Export of importin alpha from the nucleus is mediated by a specific nuclear transport factor. Cell 90(6):1061–1071. https://doi.org/10.1016/s0092-8674(00)80372-4
Bischoff FR, Klebe C, Kretschmer J, Wittinghofer A, Ponstingl H (1994) RanGAP1 induces GTPase activity of nuclear Ras-related Ran. Proc Natl Acad Sci USA 91(7):2587–2591. https://doi.org/10.1073/pnas.91.7.2587
Gorlich D, Seewald MJ, Ribbeck K (2003) Characterization of Ran-driven cargo transport and the RanGTPase system by kinetic measurements and computer simulation. EMBO J 22(5):1088–1100. https://doi.org/10.1093/emboj/cdg113
Nakielny S, Siomi MC, Siomi H, Michael WM, Pollard V, Dreyfuss G (1996) Transportin: nuclear transport receptor of a novel nuclear protein import pathway. Exp Cell Res 229(2):261–266. https://doi.org/10.1006/excr.1996.0369
Victoria W, Pollard WMM, Nakielny S, Siomi MC, Wang F, Dreyfuss G (1996) A novel receptor-mediated nuclear protein import pathway. Cell 86:985–994
Robert A, Fridell RT, Leigh Thorne R, Benson E, Cullen BR (1997) Nuclear import of hnRNP A1 is mediated by a novel cellular cofactor related to karyopherin-β. J Cell Sci 110:1325–1331
Bonifaci N, Moroianu J, Radu A, Blobel G (1997) Karyopherin beta2 mediates nuclear import of a mRNA binding protein. Proc Natl Acad Sci USA 94(10):5055–5060. https://doi.org/10.1073/pnas.94.10.5055
Twyffels L, Gueydan C, Kruys V (2014) Transportin-1 and Transportin-2: protein nuclear import and beyond. FEBS Lett 588(10):1857–1868. https://doi.org/10.1016/j.febslet.2014.04.023
Terry LJ, Shows EB, Wente SR (2007) Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science 318(5855):1412–1416. https://doi.org/10.1126/science.1142204
Lee BJ, Cansizoglu AE, Suel KE, Louis TH, Zhang Z, Chook YM (2006) Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell 126(3):543–558. https://doi.org/10.1016/j.cell.2006.05.049
Jakel S, Gorlich D (1998) Importin beta, transportin, RanBP5 and RanBP7 mediate nuclear import of ribosomal proteins in mammalian cells. EMBO J 17(15):4491–4502. https://doi.org/10.1093/emboj/17.15.4491
Okada N, Ishigami Y, Suzuki T, Kaneko A, Yasui K, Fukutomi R, Isemura M (2008) Importins and exportins in cellular differentiation. J Cell Mol Med 12(5B):1863–1871. https://doi.org/10.1111/j.1582-4934.2008.00437.x
Wild T, Horvath P, Wyler E, Widmann B, Badertscher L, Zemp I, Kozak K, Csucs G, Lund E, Kutay U (2010) A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol 8(10):e1000522. https://doi.org/10.1371/journal.pbio.1000522
Okamura M, Inose H, Masuda S (2015) RNA export through the NPC in Eukaryotes. Genes (Basel) 6(1):124–149. https://doi.org/10.3390/genes6010124
Boehringer A, Bowser R (2018) RNA nucleocytoplasmic transport defects in neurodegenerative diseases. Adv Neurobiol 20:85–101. https://doi.org/10.1007/978-3-319-89689-2_4
Heath CG, Viphakone N, Wilson SA (2016) The role of TREX in gene expression and disease. Biochem J 473(19):2911–2935. https://doi.org/10.1042/BCJ20160010
Bjork P, Wieslander L (2015) The balbiani ring story: synthesis, assembly, processing, and transport of specific messenger RNA-protein complexes. Annu Rev Biochem 84:65–92. https://doi.org/10.1146/annurev-biochem-060614-034150
Meinel DM, Burkert-Kautzsch C, Kieser A, O'Duibhir E, Siebert M, Mayer A, Cramer P, Soding J, Holstege FC, Strasser K (2013) Recruitment of TREX to the transcription machinery by its direct binding to the phospho-CTD of RNA polymerase II. PLoS Genet 9(11):e1003914. https://doi.org/10.1371/journal.pgen.1003914
Rodrigues JP, Rode M, Gatfield D, Blencowe BJ, Carmo-Fonseca M, Izaurralde E (2001) REF proteins mediate the export of spliced and unspliced mRNAs from the nucleus. Proc Natl Acad Sci USA 98(3):1030–1035. https://doi.org/10.1073/pnas.031586198
Kohler A, Hurt E (2007) Exporting RNA from the nucleus to the cytoplasm. Nat Rev Mol Cell Biol 8(10):761–773. https://doi.org/10.1038/nrm2255
Herold A, Suyama M, Rodrigues JP, Braun IC, Kutay U, Carmo-Fonseca M, Bork P, Izaurralde E (2000) TAP (NXF1) belongs to a multigene family of putative RNA export factors with a conserved modular architecture. Mol Cell Biol 20(23):8996–9008. https://doi.org/10.1128/mcb.20.23.8996-9008.2000
Hung ML, Hautbergue GM, Snijders AP, Dickman MJ, Wilson SA (2010) Arginine methylation of REF/ALY promotes efficient handover of mRNA to TAP/NXF1. Nucleic Acids Res 38(10):3351–3361. https://doi.org/10.1093/nar/gkq033
Jani D, Lutz S, Hurt E, Laskey RA, Stewart M, Wickramasinghe VO (2012) Functional and structural characterization of the mammalian TREX-2 complex that links transcription with nuclear messenger RNA export. Nucleic Acids Res 40(10):4562–4573. https://doi.org/10.1093/nar/gks059
Wickramasinghe VO, McMurtrie PI, Mills AD, Takei Y, Penrhyn-Lowe S, Amagase Y, Main S, Marr J, Stewart M, Laskey RA (2010) mRNA export from mammalian cell nuclei is dependent on GANP. Curr Biol 20(1):25–31. https://doi.org/10.1016/j.cub.2009.10.078
Wickramasinghe VO, Stewart M, Laskey RA (2010) GANP enhances the efficiency of mRNA nuclear export in mammalian cells. Nucleus 1(5):393–396. https://doi.org/10.4161/nucl.1.5.12351
Fribourg S, Braun IC, Izaurralde E, Conti E (2001) Structural basis for the recognition of a nucleoporin FG repeat by the NTF2-like domain of the TAP/p15 mRNA nuclear export factor. Mol Cell 8(3):645–656. https://doi.org/10.1016/s1097-2765(01)00348-3
Delaleau M, Borden KL (2015) Multiple export mechanisms for mRNAs cells 4(3):452–473. https://doi.org/10.3390/cells4030452
Jackson RJ, Hellen CU, Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11(2):113–127. https://doi.org/10.1038/nrm2838
Ma WK, Cloutier SC, Tran EJ (2013) The DEAD-box protein Dbp2 functions with the RNA-binding protein Yra1 to promote mRNP assembly. J Mol Biol 425(20):3824–3838. https://doi.org/10.1016/j.jmb.2013.05.016
Siebrasse JP, Kaminski T, Kubitscheck U (2012) Nuclear export of single native mRNA molecules observed by light sheet fluorescence microscopy. Proc Natl Acad Sci USA 109(24):9426–9431. https://doi.org/10.1073/pnas.1201781109
Rout MP, Aitchison JD, Magnasco MO, Chait BT (2003) Virtual gating and nuclear transport: the hole picture. Trends Cell Biol 13(12):622–628
Jovičić A, Paul Iii JW, Gitler AD (2016) Nuclear transport dysfunction: a common theme in amyotrophic lateral sclerosis and frontotemporal dementia. J Neurochem 138(S1):134–144. https://doi.org/10.1111/jnc.13642
Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ, Dujardin S, Amaral AS, Grima JC, Bennett RE, Tepper K, DeTure M, Vanderburg CR, Corjuc BT, DeVos SL, Gonzalez JA, Chew J, Vidensky S, Gage FH, Mertens J, Troncoso J, Mandelkow E, Salvatella X, Lim RYH, Petrucelli L, Wegmann S, Rothstein JD, Hyman BT (2018) Tau protein disrupts nucleocytoplasmic transport in Alzheimer's disease. Neuron 99(5):925–940. https://doi.org/10.1016/j.neuron.2018.07.039
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525(7567):56–61. https://doi.org/10.1038/nature14973
Grima JC, Daigle JG, Arbez N, Cunningham KC, Zhang K, Ochaba J, Geater C, Morozko E, Stocksdale J, Glatzer JC, Pham JT, Ahmed I, Peng Q, Wadhwa H, Pletnikova O, Troncoso JC, Duan W, Snyder SH, Ranum LPW, Thompson LM, Lloyd TE, Ross CA, Rothstein JD (2017) Mutant huntingtin disrupts the nuclear pore complex. Neuron 94(1):93–107. https://doi.org/10.1016/j.neuron.2017.03.023
Gitler AD, Tsuiji H (2016) There has been an awakening: emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res 1647:19–29. https://doi.org/10.1016/j.brainres.2016.04.004
Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG, Chen YH, Duong DM, Seyfried NT, Powers MA, Kukar T, Hales CM, Gearing M, Cairns NJ, Boylan KB, Dickson DW, Rademakers R, Zhang YJ, Petrucelli L, Sattler R, Zarnescu DC, Glass JD, Rossoll W (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci 21(2):228–239. https://doi.org/10.1038/s41593-017-0047-3
Ling S-C, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79(3):416–438. https://doi.org/10.1016/j.neuron.2013.07.033
Vucic S, Rothstein JD, Kiernan MC (2014) Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci 37(8):433–442. https://doi.org/10.1016/j.tins.2014.05.006
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Baumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin JJ, De Deyn PP, Cruts M, Van Broeckhoven C (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11(1):54–65. https://doi.org/10.1016/S1474-4422(11)70261-7
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(2):245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Consortium I, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72(2):257–268. https://doi.org/10.1016/j.neuron.2011.09.010
Taylor JP, Brown RH Jr, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539(7628):197–206. https://doi.org/10.1038/nature20413
Mathis S, Goizet C, Soulages A, Vallat J-M, Masson GL (2019) Genetics of amyotrophic lateral sclerosis: a review. J Neurol Sci 399:217–226. https://doi.org/10.1016/j.jns.2019.02.030
Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E (2013) Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol 74(2):180–187. https://doi.org/10.1002/ana.23946
Therrien M, Rouleau GA, Dion PA, Parker JA (2013) Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C elegans. PLoS ONE 8(12):e83450. https://doi.org/10.1371/journal.pone.0083450
Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, Keith J, Zinman L, Rogaeva E, Robertson J (2015) Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol 78(4):568–583. https://doi.org/10.1002/ana.24469
Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, Pregent L, Daughrity L, Baker MC, Rademakers R, Boylan K, Patel TC, Dickson DW, Petrucelli L (2013) Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol 126(6):895–905. https://doi.org/10.1007/s00401-013-1199-1
Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ (2014) Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 35(7):1779.e1775–1779.e1713. https://doi.org/10.1016/j.neurobiolaging.2014.01.016
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J, Gong G, Zambrowicz B, Valenzuela D, Yancopoulos G, Murphy A, Thurston G, Lai KM (2016) C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep 6:23204. https://doi.org/10.1038/srep23204
Burberry A, Suzuki N, Wang J-Y, Moccia R, Mordes DA, Stewart MH, Suzuki-Uematsu S, Ghosh S, Singh A, Merkle FT, Koszka K, Li Q-Z, Zon L, Rossi DJ, Trowbridge JJ, Notarangelo LD, Eggan K (2016) Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 8(347):347ra93
Shi Y, Lin S, Staats KA, Li Y, Chang W-H, Hung S-T, Hendricks E, Linares GR, Wang Y, Son EY, Wen X, Kisler K, Wilkinson B, Menendez L, Sugawara T, Woolwine P, Huang M, Cowan MJ, Ge B, Koutsodendris N, Sandor KP, Komberg J, Vangoor VR, Senthilkumar K, Hennes V, Seah C, Nelson AR, Cheng T-Y, Lee S-JJ, August PR, Chen JA, Wisniewski N, Hanson-Smith V, Belgard TG, Zhang A, Coba M, Grunseich C, Ward ME, van den Berg LH, Pasterkamp RJ, Trotti D, Zlokovic BV, Ichida JK (2018) Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 24:313. https://doi.org/10.1038/nm.4490
Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80(2):415–428. https://doi.org/10.1016/j.neuron.2013.10.015
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li H-R, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling S-C, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu X-D, Bennett CF, Cleveland DW, Ravits J (2013) Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci 110(47):E4530
Lee Y-B, Chen H-J, Peres João N, Gomez-Deza J, Attig J, Štalekar M, Troakes C, Nishimura Agnes L, Scotter Emma L, Vance C, Adachi Y, Sardone V, Miller Jack W, Smith Bradley N, Gallo J-M, Ule J, Hirth F, Rogelj B, Houart C, Shaw Christopher E (2013) Hexanucleotide repeats in ALS/FTD form length-dependent RNA Foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5(5):1178–1186. https://doi.org/10.1016/j.celrep.2013.10.049
Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, DeGroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao F-B (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol 126(3):385–399. https://doi.org/10.1007/s00401-013-1149-y
McCauley ME, Baloh RH (2019) Inflammation in ALS/FTD pathogenesis. Acta Neuropathol 137(5):715–730. https://doi.org/10.1007/s00401-018-1933-9
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee K-H, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao F-B, Taylor JP (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525(7567):129–133. https://doi.org/10.1038/nature14974
Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, Verstrepen KJ, Callaerts P, Rousseau F, Schymkowitz J, Cruts M, Van Broeckhoven C, Van Damme P, Gitler AD, Robberecht W, Van Den Bosch L (2016) Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. https://doi.org/10.1038/srep20877
Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18(9):1226–1229. https://doi.org/10.1038/nn.4085
Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80(2):415–428. https://doi.org/10.1016/j.neuron.2013.10.015
Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M, Lin WL, Garrett A, Stankowski JN, Daughrity L, Tong J, Perkerson EA, Yue M, Chew J, Castanedes-Casey M, Kurti A, Wang ZS, Liesinger AM, Baker JD, Jiang J, Lagier-Tourenne C, Edbauer D, Cleveland DW, Rademakers R, Boylan KB, Bu G, Link CD, Dickey CA, Rothstein JD, Dickson DW, Fryer JD, Petrucelli L (2016) C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci 19(5):668–677. https://doi.org/10.1038/nn.4272
Cooper-Knock J, Bury JJ, Heath PR, Wyles M, Higginbottom A, Gelsthorpe C, Highley JR, Hautbergue G, Rattray M, Kirby J, Shaw PJ (2015) C9ORF72 GGGGCC expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosis. PLoS ONE 10(5):e0127376. https://doi.org/10.1371/journal.pone.0127376
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, Jin P (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci 110(19):7778–7783. https://doi.org/10.1073/pnas.1219643110
Lin Y, Mori E, Kato M, Xiang S, Wu L, Kwon I, McKnight SL (2016) Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 167(3):789–802. https://doi.org/10.1016/j.cell.2016.10.003
Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, Maxwell BA, Kim NC, Temirov J, Moore J, Kolaitis RM, Shaw TI, Bai B, Peng J, Kriwacki RW, Taylor JP (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167(3):774–788. https://doi.org/10.1016/j.cell.2016.10.002
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162(5):1066–1077. https://doi.org/10.1016/j.cell.2015.07.047
Boeynaems S, Alberti S, Fawzi NL, Mittag T, Polymenidou M, Rousseau F, Schymkowitz J, Shorter J, Wolozin B, Van Den Bosch L, Tompa P, Fuxreiter M (2018) Protein phase separation: a new phase in cell biology. Trends Cell Biol 28(6):420–435. https://doi.org/10.1016/j.tcb.2018.02.004
Shi KY, Mori E, Nizami ZF, Lin Y, Kato M, Xiang S, Wu LC, Ding M, Yu Y, Gall JG, McKnight SL (2017) Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc Natl Acad Sci USA 114(7):E1111–E1117. https://doi.org/10.1073/pnas.1620293114
Vanneste J, Vercruysse T, Boeynaems S, Sicart A, Van Damme P, Daelemans D, Van Den Bosch L (2019) C9orf72-generated poly-GR and poly-PR do not directly interfere with nucleocytoplasmic transport. Sci Rep 9(1):15728. https://doi.org/10.1038/s41598-019-52035-6
Neumann M (2009) Molecular neuropathology of TDP-43 proteinopathies. Int J Mol Sci. https://doi.org/10.3390/ijms10010232
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130. https://doi.org/10.1126/science.1134108
Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK (2019) Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci 12:25
Weskamp K, Barmada SJ (2018) TDP43 and RNA instability in amyotrophic lateral sclerosis. Brain Res 1693:67–74. https://doi.org/10.1016/j.brainres.2018.01.015
Kawakami I, Arai T, Hasegawa M (2019) The basis of clinicopathological heterogeneity in TDP-43 proteinopathy. Acta Neuropathol. https://doi.org/10.1007/s00401-019-02077-x
Heyburn L, Sajja VSSS, Long JB (2019) The role of TDP-43 in military-relevant TBI and chronic neurodegeneration. Front Neurol 10:680
Smith C, Malek N, Grosset K, Cullen B, Gentleman S, Grosset DG (2019) Neuropathology of dementia in patients with Parkinson’s disease: a systematic review of autopsy studies. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2019-321111
Matej R, Tesar A, Rusina R (2019) Alzheimer's disease and other neurodegenerative dementias in comorbidity: a clinical and neuropathological overview. Clin Biochem. https://doi.org/10.1016/j.clinbiochem.2019.08.005
Birsa N, Bentham MP, Fratta P (2019) Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2019.05.023
Lagier-Tourenne C, Polymenidou M, Cleveland DW (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19(R1):R46–64. https://doi.org/10.1093/hmg/ddq137
McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande Velde C (2011) TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet 20(7):1400–1410. https://doi.org/10.1093/hmg/ddr021
Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, Gallo JM, Hortobagyi T, Shaw CE, Rogelj B (2010) Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 133(Pt 6):1763–1771. https://doi.org/10.1093/brain/awq111
Ederle H, Funk C, Abou-Ajram C, Hutten S, Funk EBE, Kehlenbach RH, Bailer SM, Dormann D (2018) Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci Rep 8(1):7084. https://doi.org/10.1038/s41598-018-25007-5
Archbold HC, Jackson KL, Arora A, Weskamp K, Tank EM, Li X, Miguez R, Dayton RD, Tamir S, Klein RL, Barmada SJ (2018) TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci Rep 8(1):4606. https://doi.org/10.1038/s41598-018-22858-w
Buratti E (2015) Chapter one—functional significance of TDP-43 mutations in disease. In: Friedmann T, Dunlap JC, Goodwin SF (eds) Advances in genetics, vol 91. Academic Press, New York, pp 1–53. https://doi.org/10.1016/bs.adgen.2015.07.001.
Santamaria N, Alhothali M, Alfonso MH, Breydo L, Uversky VN (2016) Intrinsic disorder in proteins involved in amyotrophic lateral sclerosis. Cell Mol Life Sci 74(7):1297–1318. https://doi.org/10.1007/s00018-016-2416-6
Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, Gass J, Rangachari V, Buratti E, Baralle F, Golde TE, Dickson DW, Petrucelli L (2009) Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA 106(18):7607–7612. https://doi.org/10.1073/pnas.0900688106
Conicella AE, Zerze GH, Mittal J, Fawzi NL (2016) ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure (Lond, Engl) 24(9):1537–1549. https://doi.org/10.1016/j.str.2016.07.007
Gomes E, Shorter J (2019) The molecular language of membraneless organelles. J Biol Chem 294(18):7115–7127
Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, Shorter J, Da Cruz S, Cleveland DW (2019) Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102(2):339–357. https://doi.org/10.1016/j.neuron.2019.02.038
Lin Y, Protter David SW, Rosen Michael K, Parker R (2015) Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol Cell 60(2):208–219. https://doi.org/10.1016/j.molcel.2015.08.018
Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG, Copley KE, Hurtle B, Portz B, Pyles NJ, Guo L, Calder CB, Wills ZP, Pandey UB, Kofler JK, Brodsky JL, Thathiah A, Shorter J, Donnelly CJ (2019) RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102(2):321–338. https://doi.org/10.1016/j.neuron.2019.01.048
Humphrey J, Emmett W, Fratta P, Isaacs AM, Plagnol V (2017) Quantitative analysis of cryptic splicing associated with TDP-43 depletion. BMC Med Genomics. https://doi.org/10.1186/s12920-017-0274-1
Jeong YH, Ling JP, Lin SZ, Donde AN, Braunstein KE, Majounie E, Traynor BJ, LaClair KD, Lloyd TE, Wong PC (2017) Tdp-43 cryptic exons are highly variable between cell types. Mol Neurodegener 12(1):13. https://doi.org/10.1186/s13024-016-0144-x
Ling JP, Pletnikova O, Troncoso JC, Wong PC (2015) TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349(6248):650–655. https://doi.org/10.1126/science.aab0983
Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, Ohkubo T, Rodriguez M, Lin N, Wu D, Bennett CF, Rigo F, Da Cruz S, Ravits J, Lagier-Tourenne C, Cleveland DW (2019) Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci 22(2):180–190. https://doi.org/10.1038/s41593-018-0293-z
Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K (2019) ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci 22(2):167–179. https://doi.org/10.1038/s41593-018-0300-4
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323(5918):1205–1208. https://doi.org/10.1126/science.1166066
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211. https://doi.org/10.1126/science.1165942
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132(Pt 11):2922–2931. https://doi.org/10.1093/brain/awp214
Fiesel FC, Kahle PJ (2011) TDP-43 and FUS/TLS: cellular functions and implications for neurodegeneration. FEBS J 278(19):3550–3568. https://doi.org/10.1111/j.1742-4658.2011.08258.x
Tyzack GE, Luisier R, Taha DM, Neeves J, Modic M, Mitchell JS, Meyer I, Greensmith L, Newcombe J, Ule J, Luscombe NM, Patani R (2019) Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain 142(9):2572–2580. https://doi.org/10.1093/brain/awz217
Rademakers R, Stewart H, Dejesus-Hernandez M, Krieger C, Graff-Radford N, Fabros M, Briemberg H, Cashman N, Eisen A, Mackenzie IR (2010) Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve 42(2):170–176. https://doi.org/10.1002/mus.21665
Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9(10):995–1007. https://doi.org/10.1016/S1474-4422(10)70195-2
Lopez-Erauskin J, Tadokoro T, Baughn MW, Myers B, McAlonis-Downes M, Chillon-Marinas C, Asiaban JN, Artates J, Bui AT, Vetto AP, Lee SK, Le AV, Sun Y, Jambeau M, Boubaker J, Swing D, Qiu J, Hicks GG, Ouyang Z, Fu XD, Tessarollo L, Ling SC, Parone PA, Shaw CE, Marsala M, Lagier-Tourenne C, Cleveland DW, Da Cruz S (2018) ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 100(4):816–830. https://doi.org/10.1016/j.neuron.2018.09.044
Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 121(Pt 22):3778–3785. https://doi.org/10.1242/jcs.038950
Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, Neumann M, Haass C (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29(16):2841–2857. https://doi.org/10.1038/emboj.2010.143
Buratti E, Baralle FE (2010) The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol 7(4):420–429. https://doi.org/10.4161/rna.7.4.12205
Kang J, Lim L, Lu Y, Song J (2019) A unified mechanism for LLPS of ALS/FTLD-causing FUS as well as its modulation by ATP and oligonucleic acids. PLoS Biol 17(6):e3000327. https://doi.org/10.1371/journal.pbio.3000327
Hock EM, Maniecka Z, Hruska-Plochan M, Reber S, Laferriere F, Sahadevan MKS, Ederle H, Gittings L, Pelkmans L, Dupuis L, Lashley T, Ruepp MD, Dormann D, Polymenidou M (2018) Hypertonic stress causes cytoplasmic translocation of neuronal, but not astrocytic, FUS due to impaired transportin function. Cell Rep 24(4):987–1000. https://doi.org/10.1016/j.celrep.2018.06.094
Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, Ruepp MD, Simons M, Niessing D, Madl T, Dormann D (2018) Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 173(3):706–719. https://doi.org/10.1016/j.cell.2018.03.004
Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ Jr, Sapp P, McKenna-Yasek D, Brown RH Jr, Hayward LJ (2010) Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet 19(21):4160–4175. https://doi.org/10.1093/hmg/ddq335
Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, Zhu H (2011) Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging 32(12):2323–2340. https://doi.org/10.1016/j.neurobiolaging.2010.06.010
Zhang ZC, Chook YM (2012) Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the Fused in Sarcoma protein (FUS). Proc Natl Acad Sci USA 109(30):12017–12021. https://doi.org/10.1073/pnas.1207247109
Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, Urwin H, Manser C, Miller CC, Hortobagyi T, Dragunow M, Rogelj B, Shaw CE (2013) ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet 22(13):2676–2688. https://doi.org/10.1093/hmg/ddt117
Suarez-Calvet M, Neumann M, Arzberger T, Abou-Ajram C, Funk E, Hartmann H, Edbauer D, Kremmer E, Gobl C, Resch M, Bourgeois B, Madl T, Reber S, Jutzi D, Ruepp MD, Mackenzie IR, Ansorge O, Dormann D, Haass C (2016) Monomethylated and unmethylated FUS exhibit increased binding to transportin and distinguish FTLD-FUS from ALS-FUS. Acta Neuropathol 131(4):587–604. https://doi.org/10.1007/s00401-016-1544-2
Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IR, Neumann M, Haass C (2012) Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J 31(22):4258–4275. https://doi.org/10.1038/emboj.2012.261
Rappsilber J, Friesen WJ, Paushkin S, Dreyfuss G, Mann M (2003) Detection of arginine dimethylated peptides by parallel precursor ion scanning mass spectrometry in positive ion mode. Anal Chem 75(13):3107–3114. https://doi.org/10.1021/ac026283q
Hung CJ, Lee YJ, Chen DH, Li C (2009) Proteomic analysis of methylarginine-containing proteins in HeLa cells by two-dimensional gel electrophoresis and immunoblotting with a methylarginine-specific antibody. Protein J 28(3–4):139–147. https://doi.org/10.1007/s10930-009-9174-3
Jobert L, Argentini M, Tora L (2009) PRMT1 mediated methylation of TAF15 is required for its positive gene regulatory function. Exp Cell Res 315(7):1273–1286. https://doi.org/10.1016/j.yexcr.2008.12.008
Bentmann E, Neumann M, Tahirovic S, Rodde R, Dormann D, Haass C (2012) Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem 287(27):23079–23094. https://doi.org/10.1074/jbc.M111.328757
Tradewell ML, Yu Z, Tibshirani M, Boulanger MC, Durham HD, Richard S (2012) Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum Mol Genet 21(1):136–149. https://doi.org/10.1093/hmg/ddr448
Yamaguchi A, Kitajo K (2012) The effect of PRMT1-mediated arginine methylation on the subcellular localization, stress granules, and detergent-insoluble aggregates of FUS/TLS. PLoS ONE 7(11):e49267. https://doi.org/10.1371/journal.pone.0049267
Scaramuzzino C, Monaghan J, Milioto C, Lanson NA Jr, Maltare A, Aggarwal T, Casci I, Fackelmayer FO, Pennuto M, Pandey UB (2013) Protein arginine methyltransferase 1 and 8 interact with FUS to modify its sub-cellular distribution and toxicity in vitro and in vivo. PLoS ONE 8(4):e61576. https://doi.org/10.1371/journal.pone.0061576
Gittings LM, Foti SC, Benson BC, Gami-Patel P, Isaacs AM, Lashley T (2019) Heterogeneous nuclear ribonucleoproteins R and Q accumulate in pathological inclusions in FTLD-FUS. Acta Neuropathol Commun 7(1):18. https://doi.org/10.1186/s40478-019-0673-y
Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling S-C, Liang TY, Mazur C, Wancewicz E, Kim AS, Watt A, Freier S, Hicks GG, Donohue JP, Shiue L, Bennett CF, Ravits J, Cleveland DW, Yeo GW (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15(11):1488–1497. https://doi.org/10.1038/nn.3230
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495(7442):467–473. https://doi.org/10.1038/nature11922
Zhao M, Kim JR, van Bruggen R, Park J (2018) RNA-binding proteins in amyotrophic lateral sclerosis. Mol Cells 41(9):818–829. https://doi.org/10.14348/molcells.2018.0243
Purice MD, Taylor JP (2018) Linking hnRNP function to ALS and FTD pathology. Front Neurosci 12:326. https://doi.org/10.3389/fnins.2018.00326
Moore S, Alsop E, Lorenzini I, Starr A, Rabichow BE, Mendez E, Levy JL, Burciu C, Reiman R, Chew J, Belzil VV, Robertson J, Staats KA, Ichida JK, Petrucelli L, Van Keuren-Jensen K, Sattler R (2019) ADAR2 mislocalization and widespread RNA editing aberrations in C9orf72-mediated ALS/FTD. Acta Neuropathol 138(1):49–65. https://doi.org/10.1007/s00401-019-01999-w
Maas S, Gommans WM (2009) Identification of a selective nuclear import signal in adenosine deaminases acting on RNA. Nucleic Acids Res 37(17):5822–5829. https://doi.org/10.1093/nar/gkp599
Maas S, Gommans WM (2009) Novel exon of mammalian ADAR2 extends open reading frame. PLoS ONE 4(1):e4225. https://doi.org/10.1371/journal.pone.0004225
Lorenzini I, Moore S, Sattler R (2018) RNA editing deficiency in neurodegeneration. Adv Neurobiol 20:63–83. https://doi.org/10.1007/978-3-319-89689-2_3
Rosen DR (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364(6435):362. https://doi.org/10.1038/364362c0
Mackenzie IRA, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VMY, Trojanowski JQ (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61(5):427–434. https://doi.org/10.1002/ana.21147
Maekawa S, Leigh PN, King A, Jones E, Steele JC, Bodi I, Shaw CE, Hortobagyi T, Al-Sarraj S (2009) TDP-43 is consistently co-localized with ubiquitinated inclusions in sporadic and Guam amyotrophic lateral sclerosis but not in familial amyotrophic lateral sclerosis with and without SOD1 mutations. Neuropathology 29(6):672–683. https://doi.org/10.1111/j.1440-1789.2009.01029.x
Robertson J, Sanelli T, Xiao S, Yang W, Horne P, Hammond R, Pioro EP, Strong MJ (2007) Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci Lett 420(2):128–132. https://doi.org/10.1016/j.neulet.2007.03.066
Tan C-F, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A, Nishizawa M, Kakita A, Takahashi H (2007) TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 113(5):535–542. https://doi.org/10.1007/s00401-007-0206-9
Wong CO, Venkatachalam K (2019) Motor neurons from ALS patients with mutations in C9ORF72 and SOD1 exhibit distinct transcriptional landscapes. Hum Mol Genet 28(16):2799–2810. https://doi.org/10.1093/hmg/ddz104
Shang J, Yamashita T, Nakano Y, Morihara R, Li X, Feng T, Liu X, Huang Y, Fukui Y, Hishikawa N, Ohta Y, Abe K (2017) Aberrant distributions of nuclear pore complex proteins in ALS mice and ALS patients. Neuroscience 350:158–168. https://doi.org/10.1016/j.neuroscience.2017.03.024
Kinoshita MY, Ito H, Hirano A, Fujita K, Wate R, Nakamura M, Kaneko S, Nakano S, Kusaka H (2009) Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 68(11):1184–1192
Aizawa H, Yamashita T, Kato H, Kimura T, Kwak S (2019) Impaired nucleoporins are present in sporadic amyotrophic lateral sclerosis motor neurons that exhibit mislocalization of the 43-kDa TAR DNA-binding protein. J Clin Neurol 15(1):62–67. https://doi.org/10.3988/jcn.2019.15.1.62
Finkbeiner S (2011) Huntington's disease. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a007476
McColgan P, Tabrizi SJ (2018) Huntington's disease: a clinical review. Eur J Neurol 25(1):24–34. https://doi.org/10.1111/ene.13413
Peskett TR, Rau F, O'Driscoll J, Patani R, Lowe AR, Saibil HR (2018) A liquid to solid phase transition underlying pathological huntingtin Exon1 aggregation. Mol Cell 70(4):588–601. https://doi.org/10.1016/j.molcel.2018.04.007
Liu KY, Shyu YC, Barbaro BA, Lin YT, Chern Y, Thompson LM, James Shen CK, Marsh JL (2015) Disruption of the nuclear membrane by perinuclear inclusions of mutant huntingtin causes cell-cycle re-entry and striatal cell death in mouse and cell models of Huntington's disease. Hum Mol Genet 24(6):1602–1616. https://doi.org/10.1093/hmg/ddu574
Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH (2001) Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol 153(2):283–294. https://doi.org/10.1083/jcb.153.2.283
Gasset-Rosa F, Chillon-Marinas C, Goginashvili A, Atwal RS, Artates JW, Tabet R, Wheeler VC, Bang AG, Cleveland DW, Lagier-Tourenne C (2017) Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron 94(1):48–57.e44. https://doi.org/10.1016/j.neuron.2017.03.027
Lane CA, Hardy J, Schott JM (2018) Alzheimer's disease. Eur J Neurol 25(1):59–70. https://doi.org/10.1111/ene.13439
Metuzals J, Robitaille Y, Houghton S, Gauthier S, Leblanc R (1988) Paired helical filaments and the cytoplasmic-nuclear interface in Alzheimer's disease. J Neurocytol 17(6):827–833. https://doi.org/10.1007/bf01216709
Paonessa F, Evans LD, Solanki R, Larrieu D, Wray S, Hardy J, Jackson SP, Livesey FJ (2019) Microtubules deform the nuclear membrane and disrupt nucleocytoplasmic transport in tau-mediated frontotemporal dementia. Cell Rep 26(3):582–593. https://doi.org/10.1016/j.celrep.2018.12.085
Wegmann S, Eftekharzadeh B, Tepper K, Zoltowska KM, Bennett RE, Dujardin S, Laskowski PR, MacKenzie D, Kamath T, Commins C, Vanderburg C, Roe AD, Fan Z, Molliex AM, Hernandez-Vega A, Muller D, Hyman AA, Mandelkow E, Taylor JP, Hyman BT (2018) Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. https://doi.org/10.15252/embj.201798049
Ambadipudi S, Biernat J, Riedel D, Mandelkow E, Zweckstetter M (2017) Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat Commun 8(1):275. https://doi.org/10.1038/s41467-017-00480-0
Sheffield LG, Miskiewicz HB, Tannenbaum LB, Mirra SS (2006) Nuclear pore complex proteins in Alzheimer disease. J Neuropathol Exp Neurol 65(1):45–54. https://doi.org/10.1097/01.jnen.0000195939.40410.08
Mastroeni D, Chouliaras L, Grover A, Liang WS, Hauns K, Rogers J, Coleman PD (2013) Reduced RAN expression and disrupted transport between cytoplasm and nucleus; a key event in Alzheimer's disease pathophysiology. PLoS ONE 8(1):e53349. https://doi.org/10.1371/journal.pone.0053349
Wagner RS, Kapinos LE, Marshall NJ, Stewart M, Lim RYH (2015) Promiscuous binding of Karyopherinbeta1 modulates FG nucleoporin barrier function and expedites NTF2 transport kinetics. Biophys J 108(4):918–927. https://doi.org/10.1016/j.bpj.2014.12.041
Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer-Hartl M, Hartl FU, Vabulas RM (2011) Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144(1):67–78. https://doi.org/10.1016/j.cell.2010.11.050
Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, Hipp MS (2016) Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351(6269):173–176. https://doi.org/10.1126/science.aad2033
Biogen (2019) A study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of BIIB100 administered orally to adults with amyotrophic lateral sclerosis. https://www.clinicaltrials.gov/ct2/show/NCT03945279?term=biib100&rank=1
Guo L, Fare CM, Shorter J (2019) Therapeutic dissolution of aberrant phases by nuclear-import receptors. Trends Cell Biol 29(4):308–322. https://doi.org/10.1016/j.tcb.2018.12.004
Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N, Zhang ZC, Coughlin M, Sweeny EA, DeSantis ME, Jackrel ME, Rodell CB, Burdick JA, King OD, Gitler AD, Lagier-Tourenne C, Pandey UB, Chook YM, Taylor JP, Shorter J (2018) Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173(3):677–692.e620. https://doi.org/10.1016/j.cell.2018.03.002
Acknowledgements
We would like to thank Dr. Lauren Gittings and Mo Roberts as well as the entire Sattler laboratory for helpful comments and insightful discussion leading to production of this review. Specifically, we would like to thank Doug Clough, a member of the Sattler lab currently fighting ALS both in the lab and at home, for constant inspiration. This work was supported by the Muscular Dystrophy Association (RS); the ALS Association (RS); the Robert Packard Center for ALS Research (RS); and the Barrow Neurological Foundation (RS). We would finally like to thank Cindy Giljames and Chinami Michaels of Neuroscience Publications at Barrow Neurological Institute for assistance with illustration preparation. Figures 2, 3, and 4 in this review were created with BioRender.com.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Special Issue: In Honor of Professor Michael Robinson.
Rights and permissions
About this article

Cite this article
Moore, S., Rabichow, B.E. & Sattler, R. The Hitchhiker’s Guide to Nucleocytoplasmic Trafficking in Neurodegeneration. Neurochem Res 45, 1306–1327 (2020). https://doi.org/10.1007/s11064-020-02989-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-020-02989-1