
Abstract
The identification of potential therapeutic agents for the treatment of epilepsy requires the use of seizure models. Except for some early treatments, including bromides and phenobarbital, the antiseizure activity of all clinically used drugs was, for the most part, defined by acute seizure models in rodents using the maximal electroshock and subcutaneous pentylenetetrazole seizure tests and the electrically kindled rat. Unfortunately, the clinical evidence to date would suggest that none of these models, albeit useful, are likely to identify those therapeutics that will effectively manage patients with drug resistant seizures. Over the last 30 years, a number of animal models have been developed that display varying degrees of pharmacoresistance, such as the phenytoin- or lamotrigine-resistant kindled rat, the 6-Hz mouse model of partial seizures, the intrahippocampal kainate model in mice, or rats in which spontaneous recurrent seizures develops after inducing status epilepticus by chemical or electrical stimulation. As such, these models can be used to study mechanisms of drug resistance and may provide a unique opportunity for identifying a truly novel antiseizure drug (ASD), but thus far clinical evidence for this hope is lacking. Although animal models of drug resistant seizures are now included in ASD discovery approaches such as the ETSP (epilepsy therapy screening program), it is important to note that no single model has been validated for use to identify potential compounds for as yet drug resistant seizures, but rather a battery of such models should be employed, thus enhancing the sensitivity to discover novel, highly effective ASDs. The present review describes the previous and current approaches used in the search for new ASDs and offers some insight into future directions incorporating new and emerging animal models of therapy resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
For the last 80 years, animal models have been the foundation on which many new therapies have been identified for the treatment of epilepsy (Fig. 1). The numerous novel antiseizure drugs (ASDs; previously termed antiepileptic or anticonvulsant drugs), which have been discovered by testing of large numbers of investigational compounds in animal models over the last 40 years (Fig. 2), have undoubtedly expanded the therapeutic options. This has been particularly important for those in need for a change in medical regimen [1], clearly supporting the value of animal models in the early identification of promising new drugs for the patient with epilepsy. The value of animal models in ASD development also demonstrates that animal models resemble human seizures in their response to ASDs, which is a logical prerequisite for any drug development program, but is often dismissed in the clinical arena. Indeed, animal models with a similarly high predictive value do not exist for other central nervous system (CNS) disorders, such as bipolar disorders or migraine [2].

Milestones in the development of animal models for antiseizure drug (ASD) discovery and development. All models, except the electroshock threshold method in cats described by Putnam and Merritt [3], are still used in development of new epilepsy therapies. Furthermore, various other models, not shown in the figure, are important for different purposes in epilepsy research [4]. The figure has been modified from Löscher et al. [2]. ASP Anticonvulsant screening project, EST electroshock threshold, ETSP Epileptic therapy screening program, GAERS Genetic absence epilepsy rat from Strasbourg, MES maximal electroshock seizure, NIH National Institutes of Health, NINDS National Institutes of Neurological Disorders and Stroke, PTZ pentylenetetrazole, SE status epilepticus, SRS spontaneous recurrent seizures

Introduction of AEDs to the market from 1853 to 2016 (adapted from Löscher and Schmidt [5]). Licensing varied from country to country. Here, the year of first licensing or the first mention of clinical use in a country of Europe, the US or Japan is shown. As described in the text, ASDs have assigned into three generations. Some derivatives of listed ASDs (e.g., fosphenytoin) or ASDs used solely for treatment of SE are not included. Furthermore, 2nd generation drugs such as phethenylate, methbarbital, benzchlorpropamide or aminoglutethimide, which were marketed as ASDs but withdrawn later, are not shown. The start of drug screening in an animal model (in ~1937 by Merritt and Putnam) as well as the start the Anticonvulsant screening project (ASP) in 1975 are also indicated in the figure
Unfortunately, despite this success, approximately 30% of patients with epilepsy fail to achieve full seizure control or suffer intolerable adverse events [5]. Furthermore, many of the new drugs are not really much improved in terms of adverse events and side effects, but they often tend to have less efficacy than some of the classical, older drugs, such as carbamazepine in head-to-head comparisons [5]. As such, no one would disagree with the idea that there is a clear need for more effective and better tolerated therapies for the treatment of epilepsy. Several interesting concepts how to achieve this goal have been presented in recent years [2, 4, 6–11]. However, whether any of these concepts will be successful cannot be readily determined.
In this review, I will shortly describe and discuss the past, present, and future role of animal models for the discovery of ASDs. There are several excellent published reviews on the history of animal models and ASD development [12–22], which I partly used as sources for the first section of this review. I strongly believe that sophisticated animal models and differentiated drug testing in such models will eventually allow discovering drugs in the future that are also effective in patients (at least subpopulations of patients) with as yet drug resistant epilepsy. H. Steve White and his group has played a strong role in improving and extending the animal model armamentarium for drug testing in the past and will certainly do so in the future [1, 16, 23–37]. However, at the end only a concerted effort of animal modelers and clinicians will lead to better therapies for the epilepsies. The history of antiseizure drug discovery impressively illustrates how important such a concerted effort is.
The History of Discovery of Antiseizure Drugs
The history of the drug discovery for treatment of epilepsy is an intriguing and tangled story [18, 19]. The following account cannot be comprehensive and is in many ways a personal view. The early years of ASD discovery were characterized by observation and serendipity, rather than a rational, targeted approach to drug development. Control of seizures was seen as the primary aim of therapy, with much less focus on safety and tolerability. However, the thalidomide (Contergan) tragedy of the early 1960s brought safety to the fore, resulting in an era of much tighter regulatory control that still persists today [20].
From Serendipitous Clinical Drug Discovery to First Animal Models of Seizures for Drug Testing
Epileptic disorders have been treated for thousands of years with a variety of botanicals and herbs [18]. Potassium bromide was the first single compound, which was used for the treatment of epilepsy following the serendipitous (chance) discovery of its activity in this area by Sir Charles Locock in 1857 [18]. During the mid-1800s, a number of inorganic salts of bromide were reported to produce good sedative effect and were accepted into medical practice. Sir Charles Locock, in 1857, aware that potassium bromide had been noted to cause impotence, used it to treat catamenial seizures. He reported its success to the Royal Medical and Chirurgical Society, introducing drug therapy for the epilepsies. This development led other physicians to administer bromides to their patients whose seizures were uncontrolled by other therapies. Bromide’s ability to reduce the seizure frequency in many of these patients led to the establishment of its efficacy and its subsequent widespread replacement of nondrug therapy [13].
Because bromide salts were the only drug available for the treatment of epilepsy at the time, these were used regularly for the next 50 years, despite their limited efficacy and serious adverse effects, including severe skin eruptions and psychoses. Other medicinal therapies such as borax, employed in this period when bromide salts failed, are now largely forgotten [18].
Phenobarbital was the second single compound discovered serendipitously for the treatment of epilepsy [38]. The history of the barbiturates started with the synthesis of barbituric acid (malonylurea) by the German chemist and Nobel Prize winner Adolf von Baeyer in 1864. Although barbituric acid itself does not exert any effects on the CNS, probably because it is not lipophilic enough to penetrate sufficiently through the blood–brain barrier, it formed the backbone for all subsequently developed CNS-active barbiturates. In 1903, two German chemists working at Bayer, Emil Fischer and Joseph von Mering, discovered that the more lipophilic barbituric acid derivative barbital (5,5-diethylbarbituric acid) was very effective in inducing sleep in dogs. The same group synthesized phenobarbital (5-ethyl-5-phenylbarbituric acid) in 1911, and both barbital (Veronal®) and phenobarbital (Luminal®) were soon marketed by Bayer as hypnotic drugs for patients with insomnia. In 1912, the German psychiatrist Alfred Hauptmann discovered the anticonvulsant activity of phenobarbital [39]. Alfred Hauptmann, then a young clinical assistant in Freiburg, gave phenobarbital to his epilepsy patients as a tranquilizer and observed that their epileptic seizures were also suppressed. Subsequent clinical experience substantiated the drug’s antiseizure efficacy. It proved to be more effective than the bromide salts and patients taking it did not suffer their overt toxicity. Consequently, phenobarbital supplanted the bromides as the major ASD [38].
Based on the large medical success of these first barbiturates, more than 2500 barbiturates were synthesized in following decades, some 50 of which were eventually employed clinically as sedatives and hypnotics. One of these, mephobarbital, demonstrated good antiseizure activity when given to man [40, 41] and was marketed in the United States in 1935. Controlled trials comparing its antiepileptic efficacy with phenobarbital’s indicated that in some patients mephobarbital was more effective and produced less sedation than phenobarbital [42, 43].
In the absence of validated animal models in which to test antiseizure activity, the discovery of the antiseizure effect of the bromides and, later, phenobarbital and mephobarbital was the result of keen observation on the part of clinicians who used them to treat epilepsy [13]. The demonstration of their widespread efficacy occurred after they were marketed. The paucity of effective drugs for epilepsy (Fig. 2) and the widespread need favored investment in ASDs by the pharmaceutical industry. For testing large numbers of drugs for antiseizure activity in an industry setting, predictive animal models were needed.
In 1882, the Italian physiologist Pietro Albertoni reported the induction of experimental seizures in dogs by direct electrical stimulation of the motor cortex [44]. He was among the first to use such a procedure to test chemicals for antiseizure activity. Although Albertoni successfully investigated the properties of the bromides, atropine and a cinchona alkaloid, the laborious preparation of his canine seizure model for identifying potential ASDs prevented its use for drug screening.
The convulsant properties of a number of naturally occurring chemicals were explored beginning in the late 1880s [13]. Pentylenetetrazole (PTZ; also termed Metrazol®, Cardiazol®, or pentetrazole) was synthesized in 1924 and used in several laboratories to test drugs for antiseizure activity soon after the first report of its convulsant action [45]. Although it supplanted most other chemical convulsants, its use for screening antiseizure chemicals was not yet standardized and was considered unreliable [3].
Reports of a new electroshock technique for producing experimental convulsions in the intact animal [46] and of a similar technique for testing chemicals for antiseizure activity [3, 47] opened the door for experimental evaluation of chemicals prior to their first administration to humans [13].
The Discovery of Novel, More Effective Antiseizure Drugs by Animal Models: How It Started
In the 1930s, Tracy Putnam and Houston Merritt, working at the Neurological Unit of the Boston City Hospital, codiscovered the antiseizure properties of phenytoin, by using an electroshock seizure model in cats [3]. Their triumph is among the first translational partnerships between academia and pharmaceutical companies in which a chemical “library” that had been screened for efficacy in an animal model was used. Putnam and Merritt collaborated in trials that led to the verification of the antiseizure properties of phenytoin in humans. This singular triumph places Putnam and Merritt at the forefront in the history of therapeutic development and clinical neuroscience [17].
Although Merritt and Putnam were not the first to use electroshock for seizure induction in animals, they devised a simple and reliable method, the so-called electroshock threshold (EST) method in cats, to assess drugs for antiseizure effects. In this model, tonic seizures were induced in cats by applying rectangular current to the animal’s head through mouth and scalp electrodes attached to a 45-V battery. Merritt and Putnam used the model to systematically screen more than 700 compounds between 1936 and 1945. Among the first compounds tested was phenytoin [3], which had been first synthesized in 1908 by the German chemist Heinrich Biltz, who sold his discovery to Parke-Davis, which did not find an immediate use for it. In the experiments of Merritt and Putnam, phenytoin was among the most potent compounds and more effective than either bromide or phenobarbital in protecting animals from electrically induced convulsions. Because toxicology studies showed phenytoin to be well tolerated by laboratory animals, it was subjected to clinical trials. Merritt and Putnam [48] reported phenytoin’s clinical efficacy in 1938, the same year in which it was marketed. The time that elapsed between the first report of phenytoin’s antiseizure effect in the cat and its availability on the market slightly exceeded 1 year. The large number of epileptic patients for whom barbiturates or bromides were ineffective, their dramatic improvement when phenytoin was added to barbiturate therapy, and the absence of a pronounced sedative effect were factors in this rapid marketing [13].
Merritt and Putnam’ s demonstration of the antiseizure efficacy of phenytoin was a keystone event (Fig. 1), not only because it provided a successful therapy for many patients with uncontrolled epilepsy but also because of its effect on the process of drug study and development [13]. The electroshock test was later modified for mice and rats, and the maximal electroshock seizure (MES) test created by Toman et al. [49] (Fig. 1) is still the most commonly used first screen in the search for new ASDs, being quite effective in identifying drugs that block generalized tonic–clonic seizures in humans [1].
Following the discovery by Merritt and Putnam that electroshock seizures in animals can be used to identify clinically effective ASDs, the 2nd milestone discovery in the development of animal models was reported by Everett and Richards in 1944 [50] (Fig. 1). They used the PTZ seizure model in mice to demonstrate the antiseizure effect of trimethadione, which was subsequently demonstrated to block absence seizures in humans and introduced for this indication in 1946 (Fig. 2). Everett and Richards [50] also showed that phenytoin was ineffective in the PTZ model, which is in line with its lack of efficacy against absence seizures in patients. Therefore, two simple animal models, the MES and PTZ tests, could be used to differentiate ASDs with different clinical effects, which subsequently formed the basis for Swinyard [51] and Swinyard et al. [52] to propose the MES and subcutaneous (s.c.) PTZ seizure tests in mice and rats as standard procedures for predicting clinical antiseizure activity of investigational drugs. Some years after the discovery of trimethadione, the PTZ test was crucial to the successful identification of the succinimides [53], including phensuximide, methsuximide, and ethosuximide (Fig. 2), which rapidly replaced oxazolidinediones such as trimethadione because of their superior tolerability. Furthermore, numerous other drugs were evaluated by the MES and PTZ seizure tests in rodents, resulting in several clinically effective ASDs that were marketed in the 1950s and 60 s (Fig. 2).
Historically, ASDs can be classified into three generations (Fig. 2). The first generation, entering the market from 1857 to 1958, includes potassium bromide, phenobarbital, and a variety of drugs that were derived mainly by modification of the barbiturate structure, including mephobarbital, phenytoin, primidone, trimethadione, and ethosuximide [5, 13]. The second generation ASDs, including carbamazepine, valproate, and the benzodiazepines, which were introduced between 1960 and 1975 (Fig. 2), differed chemically from the barbiturates and exhibited superior tolerability compared to barbiturate-based structures [5].
By the early 1970s, there were few new ASDs approaching clinical trial. Consequently, the Epilepsy Branch of the National Institute of Neurological Disorders and Stroke (NINDS) of the U.S. National Institutes of Health (NIH), which was formed in 1966, began to turn its attention toward ways in which it could spur development in the preclinical ASD identification phases [13], leading to the implementation of the Anticonvulsant Screening Project (ASP) in 1975. The ASP was founded by Kiffin Penry (director of the Epilepsy Branch) with Ewart Swinyard (Department of Pharmacology, University of Utah, where the ASP was based) and Harvey Kupferberg (head of the preclinical section of the Epilepsy Branch) with the aid of Roger Porter (Fig. 1) [54]. Harvey Kupferberg at the Epilepsy Branch of the NIH successfully liaised for many years with the ASP [55].
The Establishment of the Anticonvulsant Screening Project (ASP)
When the ASP, the first preclinical segment of the NINDS Antiepileptic Drug Development (ADD) Program was established in 1975 (Fig. 1), the main goal of the program was to increase the involvement of both industry and academia throughout the world in the discovery and development of new ASDs by inviting them to submit chemicals for testing [56]. From its start, the drug testing in animal models was performed at a contract facility based at the University of Utah on a blinded and confidential basis and at no cost to the ASP participants. Several of the pioneers of modelling seizures in animals and using such models for drug testing, including Ewart Swinyard, Louis Goodman, and Dixon Woodbury, all faculty members in the Department of Pharmacology and Toxicology in Salt Lake City, were responsible for the NINDS-sponsored ASP in Utah. Later (1986) Steve White, who had been trained by Dixon Woodbury, joined the ASP as a junior faculty member in Pharmacology and Toxicology and assumed the role of Senior Scientist until 2001 when he followed Harold Wolf as the PI of the Utah Screening Contract until his move to the University of Washington in early 2016 to become chair of the Department of Pharmacy of the University of Washington in Seattle. After he left, Karen Wilcox (University of Utah) became responsible for the program in Salt Lake City. During his almost 30 year tenure with the Utah Program, White played a pivotal role in the introduction of several of the newer models currently in use for the early identification and characterization of investigational ASDs.
After its start in 1975, the ASP rapidly enjoyed a renewed interest of pharmaceutical industry in epilepsy and the drugs used to treat it, and it has played an important role in the development of various new ASDs (Fig. 2). For the pharmaceutical companies, the ASP offered an opportunity to evaluate new antiseizure compounds in rodent seizure models against a large number and variety of compounds in a standardized, consistent manner. Until now, more than 32,000 compounds have been tested. Since its establishment in 1975, the program has made important contributions to the development of several FDA-approved drugs for epilepsy, including felbamate, topiramate, lacosamide, and retigabine [57].
Initially, the ASP was primarily based on drug testing in three models, the MES test, the s.c. PTZ seizure test, and the rotarod neurotoxicity test [56] (Fig. 3), but later additional models were added, such as the 6-Hz test of ASD-resistant partial seizures and the lamotrigine-resistant kindled rat model of partial seizures [27]. Furthermore, in recent years the aims of the ASP were broadened to include also the search for disease-modifying and antiepileptogenic treatments.

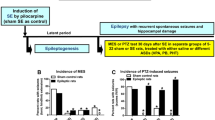
Schematic illustration of drug screening by the Anticonvulsant Screening Project (ASP) shortly after its start in 1975. The figure has been redrawn and modified from its original version shown by Krall et al. [13]. MES maximal electroshock seizure, PTZ pentylenetetrazole
The New Epilepsy Therapy Screening Program (ETSP)
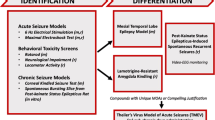
In 2015, it was decided to change the name of the ASP to ETSP to reflect the emphasis on identifying differentiated agents to address the unmet medical needs of epilepsy [57]. Furthermore, an External Consultant Board (ECB) of scientists from academia and industry was formed to provide regular feedback to the ETSP. One of the major aims of the ETSP is to identify drugs with efficacy against pharmacoresistant seizures. As shown in Fig. 4, a battery of rodent and in vitro tests is used for this aim. As described in detail by Kehne et al. [57] in this issue of Neurochemical Research, testing is divided into an initial “Identification” phase, followed by a “Differentiation” phase (Fig. 4). Evaluation of compounds begins with assessment in two acute seizure models in normal mice, the MES test and the 6-Hz test for partial seizures, which is performed at a current intensity of 44 mA, at which many clinically established ASDs do not suppress the seizures [4, 37]. The inclusion of this “high-hurdle”, acute seizure assay at the initial stage of the Identification phase is intended to raise the threshold for advancing compounds forward, thereby increasing the probability that agents with improved efficacy relative to existing agents will be detected [57]. After a compound has been tested in the MES and 6-Hz 44 mA models in mice, it is usually also tested in the MES model in rats. Furthermore, the rat 6 Hz model, which is relatively new [58], is available on an “as needed” basis. For example, if a compound is not active in the MES model, it can be used to generate rat dosing information for the lamotrigine-resistant amygdala kindled rat model of the Differentiation phase (see below).

Pharmacoresistance epilepsy work flow for the Epilepsy Therapy Screening Program (ETSP). Models in mice are indicated by “m” and those in rats by “r”. The figure has been provided by John Kehne and slightly modified for consistency with the text of this review. For details see text and Kehne et al. [57]. m mice, MOA mechanism of action, r rats
To avoid the possibility that compounds with novel mechanisms of action that are not effective in the MES or 6 Hz 44 mA tests, but may have potential as improved antiseizure agents, are missed, there are additional assay options available in the Identification phase to evaluate the activity of such compounds (Fig. 4). These assays include the corneal kindled mouse and an in vitro assay in which spontaneous electrical bursting occurs in a hippocampal/entorhinal cortex-containing brain slice removed from rats made chronically-epileptic by prior exposure to kainate. Finally, in addition to antiseizure activity, in vivo assessments of neurotoxicity are obtained using performance on the rotarod in mice, and locomotor activity and neurological scores in the rat, in addition to overall behavioral observations (Fig. 4). The quantification of ED50s in these tests allows an estimation of therapeutic index of test compounds.
Recently, the Identification phase of the new ETSP preclinical testing platform for pharmacoresistant epilepsy has been evaluated by testing a number of mechanistically diverse, commercially available ASDs as well as several experimental compounds [59]. These studies suggested that the Identification phase of this testing platform will identify numerous compounds to be advanced to further differentiation studies in etiologically-relevant models of epilepsy.
The Differentiation phase of the ETSP flow chart is currently comprised of three assays, one in mice and two in rats (Fig. 4): the intrahippocampal kainate model of mesial temporal lobe epilepsy (TLE) in mice, the lamotrigine-resistant amygdala kindled rat, and a chronic epilepsy rat model, in which epilepsy develops after induction of status epilepticus (SE) by kainate [57]. Furthermore, as shown in Fig. 4, for compounds with unique mechanisms of action or compelling justification, testing in Theiler’s virus-treated mice (a model of viral encephalitis-induced epilepsy) may be added. These models will be discussed in more detail in the next section of this review.
Furthermore, the ETSP has initiated activity for identifying antiepileptogenic compounds using several chronic epilepsy models, including the kainate post-SE model of TLE in rats and Theiler’s model of viral encephalitis-induced TLE in C57BL/6 mice [57]. The latter model has been developed by Steve White in cooperation with Robert Fujinami and Karen Wilcox [30] and represents the first infection-induced rodent model of epilepsy. This is an important contribution to the experimental armamentarium of drug discovery and development, because epilepsies are frequent consequences of brain infections in humans, particularly in developing countries [60].
Finally, an important milestone for the ETSP has been the release of a publicly-accessible database referred to by the acronym PANAChE (Public Access to Neuroactive and Anticonvulsant Chemical Evaluations; http://panache.ninds.nih.gov) [57]. PANAChE provides detailed information on tests, procedures, and flowcharts used by the ETSP, both currently and historically. Furthermore, it provides a searchable repository for non-confidential data on compounds tested by the program.
Models of Seizures or Epilepsy Beyond Simple Screening Tests in Rodents
Simple seizure tests, such as the MES and PTZ tests, have been instrumental to identify many of the ASDs that are clinically used today (Fig. 2) [1]. One important reason is that such simple seizure tests, particularly the MES test, allow mass-screening of novel compounds, which is also an advantage of the 6-Hz partial seizure test in mice, which was first described by Toman in 1951 [61] and later proposed as a model for pharmacoresistant partial seizures by Steve White’s group in 2001 [37] (Fig. 1) and subsequently added to the ASP/ETSP. However, it is questionable whether these three tests alone are capable of discovering new compounds that are effective in the about 30% of patients with drug resistant epilepsy. This was the reason to add additional models to the ETSP as shown in Fig. 4. Overall, choosing an optimal model for the search of more effective compounds is a complex endeavour in the quest to balance out the requirements of mass-screening versus the validity of models to discover drugs for previously drug-resistant epilepsies [5]. The chronic epilepsy models now used in the Differentiation phase of the ETSP (Fig. 4) certainly represent a valid approach to include the complex brain alterations that are associated with chronic difficult-to-treat epilepsy in drug evaluation; however, these models only reflect one type of epilepsy in adults, i.e., TLE.
The Amygdala Kindling Rat Model of Temporal Lobe Epilepsy
Whereas the MES, PTZ, and 6-Hz seizure models induce acute seizures in healthy, neurologically intact rodents, amygdala kindling is a chronic model in which the repeated application of electrical stimuli via a depth electrode in the basolateral amygdala of rats induces permanently enhanced seizure susceptibility and other enduring brain alterations that are similar to those occurring in human TLE [62, 63]. Both partial and secondarily generalized seizures in fully kindled rats are more difficult to suppress by most clinically established ASDs than seizures in the MES test, so that amygdala kindling was the first proposed animal model of drug resistant partial epilepsy [64]. Furthermore, the rat kindling model is one of the few models that adequately predicted the clinical utility of novel ASDs such as levetiracetam against partial seizures in patients with epilepsy [4]. It has been used extensively in studying mechanisms underlying epileptogenesis and thus represents a true milestone in the development of animal models (Fig. 1). However, it is often argued that this model, at best, may only reflect partial epileptogenesis, because kindled rats do not exhibit spontaneous recurrent seizures (SRS) at the fully kindled stage [65]. On the other hand, the possibility to induce partial and secondarily generalized seizures at any time in fully kindled rats largely simplifies drug testing in this model. Different modifications of the amygdala kindling model, such as the lamotrigine-resistant kindled rat [32] or the phenytoin-resistant kindled rat [66] have been developed in an attempt to obtain new models of drug resistant epilepsy that can be used in the search for more effective ASDs. Furthermore, such models are important to better understand the mechanisms underlying drug resistance [2].
The Corneal Kindling Model in Mice
To my knowledge, the first demonstration that animals can be noninvasively kindled by an electrical 60-Hz stimulus applied via corneal electrodes was by Sangdee et al. [67], using male CF1 mice. Matagne and Klitgaard [68] reported corneal kindling in male NMRI mice and characterized and validated this new model, including testing of several ASDs. Based on their data, they proposed that corneally kindled mice represent a very sensitive and valid noninvasive screening model that may improve the preclinical evaluation of efficacy and adverse effect potential of drug candidates being developed for the treatment of partial epilepsy [68].
In a subsequent study by Potschka and Löscher [69], the antiseizure efficacy of phenytoin in corneally kindled NMRI mice and amygdala kindled Wistar rats was compared. Large groups of kindled mice were used to examine whether phenytoin non-responders can be selected in the corneal kindling model as reported previously for amygdala kindled rats [66]. Furthermore, in view of the enhanced adverse effect potential of NMDA antagonists in amygdala kindled rats [70] and patients with TLE [71], it was evaluated whether corneally kindled mice also differ in this respect from non-kindled animals. Phenytoin proved highly potent and efficacious to block corneally kindled seizures. Only one non-responder could be selected out of 75 fully kindled mice repeatedly tested with phenytoin. At 6 days after the last kindled seizure, kindled mice were more sensitive than non-kindled mice to phencyclidine-like behavioral adverse effects of the competitive NMDA antagonist D-CPPene, but this altered sensitivity was not long-lasting, having almost disappeared 27 days after the last seizure, indicating that, in contrast to traditional kindling, brain alterations after corneal kindling are not permanent. Potschka and Löscher [69] concluded that although corneal kindling may have advantages for the identification of new drugs during initial screening of large numbers of compounds, it cannot replace traditional electrical kindling during later phases of drug development.
Rowley and White [29] further evaluated the utility of the corneal kindled mouse model as a tool for rapid screening of investigational ASDs. Results obtained with nine ASDs (valproic acid, lamotrigine, phenytoin, carbamazepine, levetiracetam, vigabatrin, topiramate, tiagabine, and ezogabine) with varying mechanisms of action and clinical spectrums, as well as six investigational compounds were evaluated in the corneal kindled mouse, using CF1 mice. ED50 values were compared to those obtained in the hippocampal kindled rat, the mouse MES model, the 6-Hz partial psychomotor seizure model, and the s.c. PTZ test.
With the exception of topiramate, all of the ASDs tested displayed a dose-dependent protection against secondarily generalized seizures in the corneal kindled mouse model. Importantly, the corneal kindled mouse model was the only model to demonstrate efficacy of all tested prototype ASDs at non-toxic doses. The authors concluded that the corneal kindled mouse model of partial and secondarily generalized epilepsy is a sensitive and valid screening model, and that early demonstration of efficacy of an investigational ASD in the corneal kindled mouse could provide sufficient proof-of-concept in a chronic seizure model to support more extensive studies in the labor-intensive electrical kindled rat [29]. As shown in Fig. 4, the corneal kindled mouse model is part of the Identification phase of the ETSP.
More recently, Leclercq et al. [72] reported that corneal kindling of NMRI mice with 6-Hz stimuli may be more advantageous than the previously described 50/60-Hz corneal kindling models due to its robustness and persistence of the fully kindled state. When assessing the protective efficacy of four mechanistically different ASDs (clonazepam, valproate, carbamazepine and levetiracetam) in 6 and 50-Hz fully kindled NMRI mice, all tested ASDs showed a relatively lower potency in the 6-Hz kindling model, and a limited efficacy against partial seizures was observed with carbamazepine and levetiracetam. The authors suggested that the observed low potency and limited efficacy of ASDs in 6-Hz fully kindled mice indicate that this model could be a useful tool in the discovery of novel ASDs targeting treatment-resistant epilepsy.
Post-Status Epilepticus Models of Temporal Lobe Epilepsy
Another important category of chronic models of epilepsy are models in which SRS develop after chemical or electrical induction of a sustained SE. One of these models uses kainate, a cyclic analog of L-glutamate and an agonist of the ionotropic kainate receptors [73]. Although it was first shown by Nadler et al. [74] that hippocampal pyramidal cells are highly sensitive to damage induced by kainate, the use of this drug as a model of TLE was originally proposed by Ben-Ari et al. [75], who reported that intra-amygdala injections of kainate in rodents induce behavioral seizures and produce neuropathological lesions in the hippocampus that are similar to those occurring in patients with TLE (Fig. 1). The initial SE produced by kainate was followed days later by the occurrence of spontaneous seizures [76].
More recently, the intrahippocampal kainate mouse model of mesial TLE has been characterized pharmacologically [77–79], and this model is now part of the Differentiation phase of the ETSP (Fig. 4). The advantage of the latter model is the occurrence of highly frequent electrographic seizures in the hippocampal kainate focus that allow drug testing on spontaneous seizures with only short periods of EEG recording [80]. These frequent electrographic seizures are resistant to several ASDs, so that the model has been proposed to be suited for discovering novel drugs with higher efficacy against difficult-to-treat focal seizures [79]. Interestingly, such frequent electrographic seizures are not observed in the rat intrahippocampal kainate model [81].
When kainate is injected systemically for SE induction, more wide-spread and bilateral neuronal damage is observed, particularly in limbic regions [73]. In contrast to the intrahippocampal kainate mouse model, only a few studies have tested the effects of ASDs on the SRS developing after systemic administration of kainate in rats [82–85]. Such studies are complicated and extremely laborious because of at least two problems. First, frequency of SRS in the kainate rat model is extremely variable, so that continuous (24/7) video-EEG monitoring over weeks is needed to correctly determine a drug’s efficacy to suppress the seizures. Second, most drugs are much more rapidly eliminated by rodents than by humans, so that studies necessitating chronic treatment have to resolve the problem of maintaining effective drug concentrations in the animals over the period of prolonged drug administration. Steve White’s group has developed a novel computer-automated pellet delivery system which allows for tight experimenter control of drug treatment in rodents using a drug-in-food protocol [33]. The problem of large inter-individual variation in SRS frequency can be partially resolved by selecting epileptic rats with high SRS frequency for drug testing. By using the latter approach, the ETSP, which includes the systemic kainate rat model in its Differentiation phase (Fig. 4) has started to characterize the pharmacological profile of SRS in this model, using a pragmatic vehicle/drug crossover design with only 5 days of treatment for each drug and repeated use of the same epileptic rats for several consecutive drug trials, thus allowing for intermediate drug screening [86]. First experiments with this approach showed that treatment with high doses of carbamazepine (90 mg/kg/day) and levetiracetam (300 mg/kg/day) led to a significant reduction in seizure frequency compared to vehicle, whereas lamotrigine (60 mg/kg/day) and VPA (600 mg/kg/day) did not [86]. It will be important to determine whether individual rats respond differently to ASDs so that, similar to other post-SE models of TLE [4, 87], drug responders and nonresponders can also be selected in the rat kainate model. If so, such nonresponders would be extremely interesting animals for testing of promising novel compounds.
An ex vivo approach for drug testing in the kainate rat model has been described by West et al. [88] and this approach has been included in the Identification phase of the ETSP (Fig. 4) [59]. In this approach, compounds are tested for their ability to eliminate the spontaneous electrographic bursting observed in the medial entorhinal cortex-hippocampus of brain slices obtained from rats that have experienced kainate-induced SE. To improve throughput, 8–10 brain slices are recorded simultaneously. Although phenytoin, carbamazepine, lamotrigine, lacosamide, ezogabine, clobazam, midazolam, phenobarbital, tiagabine, vigabatrin, and topiramate significantly attenuated spontaneous electrographic bursts in this model, these compounds (with the exception of carbamazepine) did so only at concentrations between 2 and 200 times the effective plasma concentrations reported in rats and/or humans [59, 88]. Furthermore, ethosuximide, gabapentin, levetiracetam, valproic acid, and felbamate failed to affect spontaneous bursts at any concentration tested. This model’s profile is therefore consistent with accepted definitions of pharmacoresistance and may be useful for the early identification of compounds effective against pharmacoresistant seizures [59, 88].
The most frequently used model of post-SE TLE is the pilocarpine model [73, 89]. This model, which uses the cholinergic muscarinic agonist pilocarpine, was first described by Turski and Cavalheiro [90], who showed that systemic intraperitoneal administration of pilocarpine in rodents was followed by a sequence of automatisms and motor limbic seizures evolving into SE. Analysis of the brain of these animals revealed widespread damage in the olfactory cortex, amygdala, thalamus, neocortex, hippocampus and substantia nigra [91]. As with kainate, pilocarpine-treated animals showed spontaneous seizures approximately 2 weeks after the initial SE, which established the model as a model of TLE [92], reproducing both the typical histopathological alterations and spontaneous chronic seizures seen in patients with TLE (Fig. 1). Esper Cavalheiro’s group was also the first to describe the pharmacology of the SRS in the pilocarpine model [93]. Later, Löscher’s group showed interindividual differences in the response of SRS in this model to levetiracetam [94] and phenobarbital [95], indicating that, similar to the phenytoin response in kindled rats [66] and patients with TLE, two individuals with seemingly similar spontaneous seizures may differ in their response to ASDs.
In addition to chemical SE induction, SE can be induced by sustained electrical stimulation of hippocampus or amygdala, resulting in development of SRS following a seizure-free latent period [96]. In one of these electrical SE models, in which SE was induced by electrical stimulation of the basolateral amygdala, Brandt et al. [97] demonstrated that individual epileptic rats differ strikingly in their response to phenobarbital. The majority of phenobarbital nonresponders also did not respond to phenytoin [98], so that this model fulfills the minimum criteria of a model of drug resistant epilepsy, i.e., persistent seizure activity that does not respond, or respond poorly, to monotherapy at tolerable doses with at least two current ASDs [99]. By using this model, we tried to identify the factors that determine whether an individual epileptic rat responds or does not response to ASD treatment [87]. Factors thus identified to be associated with drug resistance included high seizure density before onset of treatment, hippocampal damage (which does not occur in all rats of this model), alterations in brain drug target composition, behavioral abnormalities, and increased expression of the multidrug transporter P-glycoprotein (P-gp) in the epileptic focus [87]. All these factors have also been described in patients with drug resistant TLE [2], demonstrating that the rat model reflects clinically relevant alterations that could be targeted for obtaining more effective therapies. One interesting finding in this regard is that inhibition of P-gp in phenobarbital nonresponders restores the antiseizure effect of this ASD [100]. Thus, imaging of P-gp expression and functionality by positron emission tomography may be a biomarker for a mechanism of pharmacoresistance, thus allowing selective treatment of affected patients by either coadministration of a P-gp inhibitor or switching treatment to an ASD that is not transported by P-gp [101]. Although this possibility needs to be proven clinically, it demonstrates how advanced animal models can guide novel strategies for therapy [2].
Induction of Acute Seizures in Epileptic Rodents
Most of the post-SE models of TLE described above are not suited for drug screening, because drug testing on SRS necessitates laborious video-EEG seizure monitoring. More recently, it was proposed that, instead of monitoring SRS, chemical or electrical induction of acute seizures in epileptic rodents may be used as a surrogate for testing the efficacy of novel ASDs against refractory SRS [102]. Indeed, several ASDs were shown to lose their efficacy on acute seizures, when such seizures were induced by PTZ in epileptic rather than nonepileptic rats, whereas this was not observed when using the MES test [102]. Subsequent studies confirmed the loss of anti-seizure efficacy of valproate against PTZ-induced seizures in epileptic mice, but several other ASDs, including valproate, phenytoin, and phenobarbital, were more potent against PTZ in epileptic than nonepileptic mice [103]. This was also observed when using the 6-Hz model of partial seizures in epileptic mice, in which the potency of levetiracetam, in particular, was markedly increased compared to nonepileptic animals [104, 105]. Furthermore, when using the MES test in epileptic mice, the efficacy of a bumetanide/phenobarbital combination was significantly larger compared to this test in nonepileptic mice [106], indicating that, similar to the findings with levetiracetam in the 6-Hz test [104, 105], the targets for these drugs are altered in epileptic rodents [106]. Overall, these observations suggest that performing acute seizure tests in epileptic rodents provides valuable information on the pharmacological profile of ASDs, in particular those with mechanisms inherent to disease-induced brain alterations. However, it appears that further work is needed to define optimal approaches for acute seizure induction and generation of epileptic/drug refractory animals that would permit reliable screening of new ASDs with improved potential to provide seizure control in patients with pharmacoresistant epilepsy [107].
Animal Models of Epilepsy Developing After Traumatic Brain Injury and Stroke
One inherent problem of models in which normal, healthy adult rodents are used to induce SE and subsequent epilepsy is the fact that de novo SE is rare in adult humans and thus not a frequent cause of acquired epilepsy. The most common causes of acquired epilepsy in adult humans are traumatic brain injury (TBI), stroke, and CNS infections [108]. Limited clinical evidence suggests that the susceptibility of seizures to ASDs may differ as a function of the underlying cause of epilepsy [109–111]. If so, it would be important to include animal models of TBI, stroke, and brain infections in the Differentiation phase of drug development. However, as yet models of post-traumatic epilepsy or of post-stroke epilepsy are rarely used for pharmacological studies in the preclinical arena, because in most of these models, only 20–50% of the animals develop epilepsy after several months, so that drug experiments in such models are difficult [112, 113]. As a consequence, the pharmacology of SRS developing in models of TBI and stroke is largely unknown with some exceptions [114, 115]. Interestingly, in models of TBI and viral encephalitis, valproate reduced seizure burden, while carbamazepine was ineffective, indicating commonalities in pharmacology [35, 114]. Thus, models of infection-induced epilepsy should be included during differentiation of novel compounds.
Models of Infection-Induced Epilepsy: Theiler’s Virus Model of Encephalitis-Induced Epilepsy in Mice
Theiler’s model of viral encephalitis-induced seizures in C57BL/6 mice exhibits two types of seizures, frequent early (encephalitis-associated) seizures in the first week after infection and less frequent late seizures developing in the months after encephalitis [30, 116]. Wilcox et al. [117] have recently evaluated the effect of prototype ASDs on early seizures in this novel model. Of the compounds evaluated, clonazepam and ethosuximide were without effect in this model. Gabapentin, lacosamide, tiagabine, topiramate, valproate, and lamotrigine reduced cumulative seizure burden. The anti-inflammatory compound minocycline also reduced cumulative seizure burden [36]. Thus, these studies suggest that novel compounds can be evaluated in this assay for anti-inflammatory, anti-seizure, or mixed mechanisms of action [36, 59, 117]. In addition to evaluating the effect of drugs on acute seizures, the model can be used to study disease-modifying or antiepileptogenic drug effects [36].
Animal Models of Pediatric Epilepsies
Despite the fact that 70% of epilepsy begins in childhood, drug screening is typically done in young adult rodents. Although one may argue that age probably does not have a major impact on preclinical screening, it is a concern of pediatric neurologists. Indeed, the pathophysiology and hence pharmacology of epileptogenesis in the developing brain may differ markedly [118, 119]. Several epilepsy or seizure models in neonatal rats or mice, including models of febrile seizures and infantile spasms, have been described and are increasingly being used for developing new therapies [4, 34, 120–122]. In addition, genetic mouse models of pediatric epilepsies are available [34].
Genetic Animal Models of Epilepsy
The identification of spontaneous, and also induced, genetic epilepsies in mice has proven invaluable for modeling human epilepsies [11]. Furthermore, spontaneous genetic epilepsies have been found in several other species, including rats, non-human primates, and dogs [4, 123]. Such genetic models are providing essential insight into the role of a specific mutation in ictogenesis and epileptogenesis [4]. Furthermore, models such as the audiogenic seizure susceptible DBA/2 mouse or the Genetic Absence Epilepsy Rat from Strasbourg (GAERS) are widely used for testing drugs for antiseizure properties. While audiogenic seizures in DBA/2 mice do not discriminate between different categories of ASDs, the GAERS model in Wistar rats, first described by Christan Marescaux’s group in Strasbourg in 1982 [124] (Fig. 1), exhibits absence-like spike-wave discharges that are particularly sensitive to anti-absence drugs such as ethosuximide and valproate, but not to drugs that do not exhibit anti-absence effects in the clinic [125]. Thus, the GAERS model is better suited than the PTZ test to identify drugs with anti-absence efficacy. Shortly after the discovery of the GAERS rat, the WAG/Rij rat model of absence epilepsy was discovered; importantly, the WAG/Rij rat exhibits similar pharmacological characteristics to that of the GAERS rat [126].
Many of the genetic models are emerging as important tools for validating novel targets for the treatment and prevention of epilepsy [34]. One example here is the use of models of channelopathies to predict efficacy of ASDs in the increasing number of human epilepsies associated with channel dysfunction [34, 127]. Furthermore, genetic models are interesting tools to study antiepileptogenic or disease-modifying drug potential as exemplified by interesting data on ethosuximide, levetiracetam, and zonisamide. Thus, in the WAG/Rij rat model of absence epilepsy, early prophylactic treatment with ethosuximide, levetiracetam or zonisamide (but not carbamazepine) before the onset of SWDs in the EEG suppressed the development of such absence-like seizures [128–130]. This phenomenon was subsequently also observed in the GAERS model of absence epilepsy [131]. These findings suggest that models are available in which epileptogenesis can be controlled and that early treatment during development may provide a strategy for preventing genetic epilepsy in susceptible individuals. In addition to inducing epileptogenic mutations in mice, such mutations can be induced in zebrafish, a vertebrate genetic model organism with tremendous potential for modeling acute seizures and genetic epilepsies [11]. For instance, zebrafish with a mutation in the SCN1A homologue recapitulate spontaneous seizure activity and mimic the convulsive behavioral movements observed in Dravet syndrome [11]. Griffin et al. [132] recently reported that phenotypic screening of drug libraries in zebrafish scn1 mutants rapidly and successfully identifies new therapeutics. The latter study demonstrated that drugs acting on serotonin signalling pathways, e.g. trazodone and lorcaserin, block seizures in zebrafish SCN1 mutants, and this finding could be translated to medically intractable Dravet syndrome patients [132].
The Future Role of Animal Models for the Discovery of Antiseizure Drugs
As in other fields of biomedical research, there is not one “ideal” animal model or battery of animal models for ASD discovery, but rather the “fit-for-purpose” paradigm should be used when choosing animal models for a specific clinical condition, such as discovering novel drugs for as yet pharmacoresistant seizures [4]. An essential requirement for improving success in drug development is the availability of animal models with high predictive validity for a therapeutic drug response. Animal models when carefully selected, designed and conducted are important parts of any translational drug development strategy. The translational value of animal models can be further enhanced when combined with other translational tools such as quantitative systems pharmacology, biomarkers or experimental clinical trials [4, 133]. However, it should be kept in mind that an animal model is a simple representation of a complex system. Consequently, an animal model for a human disease is by no means attempting to reproduce the human disease with all its complexities but rather to model specific aspects of the disease, for instance drug resistant partial seizures. Whenever using animal models, it is thus of utmost importance to define a specific question and to ensure that the chosen model is fit-for-purpose [4, 133–135]. As recently outlined by Harward and McNamara [9], development of novel therapies for the epilepsies requires properly aligning the animal model with the clinical syndrome, necessitating continuous and effective interactions of skilled clinicians and basic scientists. Apart from animal models, the intuition and creativity of experienced scientists is essential to discover novel targets or correctly interpret unexpected findings that may ultimately lead to novel drugs that really make a difference. In this respect, new pharmacology and gene discovery strategies are likely to permit targeting sub-populations of patients with drug resistant epilepsy rather than searching for the magic bullet that is effective in all types of difficult-to-treat epilepsies. Advanced animal models such as those discussed in this review will play a decisive role in how the utilization of such novel strategies will contribute to the identification and development of drugs that lead to marked changes for treating the drug resistant patient population.
Abbreviations
- ADD:
-
Antiepileptic drug development
- ASD:
-
Antiseizure drugs
- ASP:
-
Anticonvulsant Screening Project
- CNS:
-
Central nervous system
- ECB:
-
External Consultant Board
- EST:
-
Electroshock threshold
- ETSP:
-
Epilepsy Therapy Screening Program
- GAERS:
-
Genetic Absence Epilepsy Rat from Strasbourg
- MES:
-
Maximal electroshock seizure
- NIH:
-
National Institutes of Health
- NINDS:
-
National Institutes of Neurological Disorders and Stroke
- NMDA:
-
N-methyl-d-aspartate
- PANAChE:
-
Public access to neuroactive and anticonvulsant chemical evaluations
- P-gp:
-
P-glycoprotein
- PTZ:
-
Pentylenetetrazole
- s.c.:
-
Subcutaneous
- SRS:
-
Spontaneous recurrent seizures
- TBI:
-
Traumatic brain injury
- TLE:
-
Temporal lobe epilepsy
References
Bialer M, White HS (2010) Key factors in the discovery and development of new antiepileptic drugs. Nat Rev Drug Discov 9:68–82
Löscher W, Klitgaard H, Twyman RE, Schmidt D.(2013) New avenues for antiepileptic drug discovery and development. Nat Rev Drug Discov 12:757–776
Putnam TJ, Merritt HH (1937) Experimental determination of the anticonvulsant properties of some phenyl derivatives. Science 85:525–526
Löscher W (2016) Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res 126:157–184
Löscher W, Schmidt D (2011) Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 52:657–678
French JA, White HS, Klitgaard H, Holmes GL, Privitera MD, Cole AJ, Quay E, Wiebe S, Schmidt D, Porter RJ, Arzimanoglou A, Trinka E, Perucca E (2013) Development of new treatment approaches for epilepsy: unmet needs and opportunities. Epilepsia 54(Suppl 4):3–12
O’Brien TJ, Ben Menachem E, Bertram EH III, Collins SD, Kokaia M, Lerche H, Klitgaard H, Staley KJ, Vaudano E, Walker MC, Simonato M (2013) Proposal for a “phase II” multicenter trial model for preclinical new antiepilepsy therapy development. Epilepsia 54(Suppl 4):70–74
Wilcox KS, Dixon-Salazar T, Sills GJ, Ben Menachem E, White HS, Porter RJ, Dichter MA, Moshe SL, Noebels JL, Privitera MD, Rogawski MA (2013) Issues related to development of new antiseizure treatments. Epilepsia 54(Suppl 4):24–34
Harward SC, McNamara JO (2014) Aligning animal models with clinical epilepsy: where to begin? Adv Exp Med Biol 813:243–251
Simonato M, Brooks-Kayal AR, Engel J Jr, Galanopoulou AS, Jensen FE, Moshe SL, O’Brien TJ, Pitkänen A, Wilcox KS, French JA (2014) The challenge and promise of anti-epileptic therapy development in animal models. Lancet Neurol 13:949–960
Grone BP, Baraban SC (2015) Animal models in epilepsy research: legacies and new directions. Nat Neurosci 18:339–343
Purpura DP, Penry JK, Tower D, Woodbury DM, Walter R (1972) Experimental models of epilepsy—A manual for the laboratory worker. Raven Press, New York
Krall RL, Penry JK, Kupferberg HJ, Swinyard EA (1978) Antiepileptic drug development: I. History and a program for progress. Epilepsia 19:393–408
Friedlander WJ (1986) Putnam, Merritt, and the discovery of Dilantin. Epilepsia 27(Suppl 3):S1–S20
Kupferberg H (2001) Animal models used in the screening of antiepileptic drugs. Epilepsia 42(Suppl 4):7–12
White HS (2003) Preclinical development of antiepileptic drugs: past, present, and future directions. Epilepsia 44(Suppl 7):2–8
Kaminski HJ (2009) The legacy of Tracy J. Putnam and H. Houston Merritt: modern neurology in the United States. New Engl J Med 360:941–942
Shorvon SD (2009) Drug treatment of epilepsy in the century of the ILAE: the first 50 years, 1909–1958. Epilepsia 50(Suppl 3):69–92
Shorvon SD (2009) Drug treatment of epilepsy in the century of the ILAE: the second 50 years, 1959–2009. Epilepsia 50(Suppl 3):93–130
Arzimanoglou A, Ben-Menachem E, Cramer J, Glauser T, Seeruthun R, Harrison M (2010) The evolution of antiepileptic drug development and regulation. Epileptic Disord 12:3–15
Brodie MJ (2010) Antiepileptic drug therapy the story so far. Seizure 19:650–655
Sidiropoulou K, Diamantis A, Magiorkinis E (2010) Hallmarks in 18th- and 19th-century epilepsy research. Epilepsy Behav 18:151–161
White HS, Johnson M, Wolf HH, Kupferberg HJ (1995) The early identification of anticonvulsant activity: role of the maximal electroshock and subcutaneous pentylenetetrazol seizure models. Ital J Neurol Sci 16:73–77
White HS (1997) Clinical significance of animal seizure models and mechanism of action studies of potential antiepileptic drugs. Epilepsia 38:S9–S17
White HS (1999) Comparative anticonvulsant and mechanistic profile of the established and newer antiepileptic drugs. Epilepsia 40:S2–S10
White HS (2002) Animal models of epileptogenesis. Neurology 59:S7–S14
White HS, Smith-Yockman M, Srivastava A, Wilcox KS (2006) Therapeutic assays for the identification and characterization of antiepileptic and antiepileptogenic drugs. In: Pitkänen A, Schwartzkroin PA, and Moshé SL (eds.), Models of seizures and epilepsy. Elsevier, Amsterdam, pp 539–549
White HS, Wolf HH, Woodhead JH, Kupferberg HJ (1998) The National Institutes of Health Anticonvulsant Drug Development Program: screening for efficacy. Adv Neurol 76:29–39
Rowley NM, White HS (2010) Comparative anticonvulsant efficacy in the corneal kindled mouse model of partial epilepsy: correlation with other seizure and epilepsy models. Epilepsy Res 92:163–169
Stewart KA, Wilcox KS, Fujinami RS, White HS (2010) Development of postinfection epilepsy after Theiler’s virus infection of C57BL/6 mice. J Neuropathol Exp Neurol 69:1210–1219
White HS (2012) Animal models for evaluating antiepileptogenesis. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, and Delgado-Escueta AV (eds.), Jasper’s Basic Mechanisms of the Epilepsies, 4th edition. Oxford University Press, New York, pp 1041–1054
Srivastava AK, White HS (2013) Carbamazepine, but not valproate, displays pharmacoresistance in lamotrigine-resistant amygdala kindled rats. Epilepsy Res 104:26–34
Thomson KE, White HS (2014) A novel open-source drug-delivery system that allows for first-of-kind simulation of nonadherence to pharmacological interventions in animal disease models. J Neurosci Methods 238:105–111
White HS, Löscher W (2014) Searching for the ideal antiepileptogenic agent in experimental models: single treatment versus combinatorial treatment strategies. Neurother 11:373–384
Barker-Haliski ML, Dahle EJ, Heck TD, Pruess TH, Vanegas F, Wilcox KS, White HS (2015) Evaluating an etiologically relevant platform for therapy development for temporal lobe epilepsy: effects of carbamazepine and valproic acid on acute seizures and chronic behavioral comorbidities in the Theiler’s murine encephalomyelitis virus mouse model. J Pharmacol Exp Ther 353:318–329
Barker-Haliski ML, Heck TD, Dahle EJ, Vanegas F, Pruess TH, Wilcox KS, White HS (2016) Acute treatment with minocycline, but not valproic acid, improves long-term behavioral outcomes in the Theiler’s virus model of temporal lobe epilepsy. Epilepsia 57:1958–1967
Barton ME, Klein BD, Wolf HH, White HS (2001) Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res 47:217–228
Löscher W, Rogawski MA (2012) How theories evolved concerning the mechanism of action of barbiturates. Epilepsia 53(Suppl 8):12–25
Hauptmann A (1912) Luminal bei Epilepsie. Münch Med Wochenschr 59:1907–1909
Blum E (1932) Die Bekämpfung epileptischer Anfälle und ihrer Folgeerscheinungen mit Prominal. Dtsch Med Wochenschr 58:696–698
Weese H (1932) Pharmakologie des Prominal. Dtsch Med Wochenschr 58:696
Page LGM (1936) Prominal in epilepsy. Br Med J 1:531
Millman CG (1939) Report on five years’ use of prominal as routine treatment for epileptics. J Ment Sci 85:971–975
Albertoni P (1882) Untersuchungen über die Wirkung einiger Arzneimittel auf die Erregbarkeit des Grosshirns nebst Beiträgen zur Therapie der Epilepsie. Arch Exp Pathol Pharmakol 15:248–288
Hildebrandt F (1926) Pentamethylentetrazol (Cardiazol). I. Mitteilung. Naunyn-Schmiedeberg’s Arch Exp Pathol Pharmacol 116:100–109
Spiegel EA (1937) Quantitative determination of the convulsive reactivity by electrical stimulation of the brain with the skull intact. J Lab Clin Med 22:1274–1276
Merritt HH, Putnam TJ, Schwab DM (1938) A new series of anticonvulsant drugs tested by experiments on animals. Arch Neurol Psychiatry 39:1003–1015
Merritt HH, Putnam TJ (1938) Sodium diphenyl hydantoinate in the treatment of convulsive disorders. JAMA 111:1068–1073
Toman JEP, Swinyard EA, Goodman LS (1946) Properties of maximal seizures and their alteration by anticonvulsant drugs and other agents. J Neurophysiol 9:231–239
Everett GM, Richards RK (1944) Comparative anticonvulsive action of 3,5,5-trimethyloxazolidine-2,4-dione (Tridione), Dilantin and phenobarbital. J Pharmacol Exp Ther 81:402–407
Swinyard EA (1949) Laboratory assay of clinically effective antiepileptic drugs. J Am Pharm Assoc 38:201–204
Swinyard EA, Brown WC, Goodman LS (1952) Comparative assay of antiepileptic drugs in mice and rats. J Pharmacol Exp Ther 106:319–330
Chen G, Portman R, Ensor CR, Bratton AC Jr (1951) The anticonvulsant activity of o-phenyl succinimides. J Pharmacol Exp Ther 103:54–61
Porter RJ, Kupferberg HJ (2017) The anticonvulsant screening program of the National Institute of Neurological Disorders and Stroke, NIH: history and contributions to clinical care in the 20th century and beyond. Neurochem Res (in press)
Löscher W, Schmidt D (2012) Seizing the moment for the future: the U.S. Anticonvulsant Screening Project. Epilepsia 53:1841–1842
Krall RL, Penry JK, White BG, Kupferberg HJ, Swinyard EA (1978) Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia 19:409–428
Kehne JH (2017) National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP). Neurochem Res (in press)
Metcalf C, West P, Rueda C, Thomson K, Lu Z, Smith M, Wilcox K (2016) Development and pharmacologic characterization of the rat 6 HZ model. American Epilepsy Society 70th Annual Meeting Abstracts Online Abst. 1.058.
Barker-Haliski ML, Johnson K, Billingsley P, Huff J, Handy LJ, Khaleel R, Lu Z, Mau MJ, Pruess TH, Rueda C, Saunders G, Underwood TK, Vanegas F, Smith MD, West PJ, Wilcox KS (2017) Validation of a preclinical testing platform for pharmacoresistant epilepsy. Neurochem Res (in press)
Vezzani A, Fujinami RS, White HS, Preux PM, Blümcke I, Sander JW, Löscher W (2016) Infections, inflammation and epilepsy. Acta Neuropathol 131:211–234
Toman JEP (1951) Experimental psychomotor seizures. Electroencephalogr Clin Neurophysiol 3:253
Goddard GV, McIntyre DC, Leech CK (1969) A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol 25:295–330
Sato M, Racine RJ, McIntyre DC (1990) Kindling: basic mechanisms and clinical validity. Electroenceph Clin Neurophysiol 76:459–472
Löscher W, Jäckel R, Czuczwar SJ (1986) Is amygdala kindling in rats a model for drug-resistant partial epilepsy? Exp Neurol 93:211–226
Löscher W, Brandt C (2010) Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol Rev 62:668–700
Löscher W, Rundfeldt C (1991) Kindling as a model of drug-resistant partial epilepsy: selection of phenytoin-resistant and nonresistant rats. J Pharmacol Exp Ther 258:483–489
Sangdee P, Turkanis SA, Karler R (1982) Kindling-like effect induced by repeated corneal electroshock in mice. Epilepsia 23:471–479
Matagne A, Klitgaard H (1998) Validation of corneally kindled mice: a sensitive screening model for partial epilepsy in man. Epilepsy Res 31:59–71
Potschka H, Löscher W (1999) Corneal kindling in mice: behavioral and pharmacological differences to conventional kindling. Epilepsy Res 37:109–120
Löscher W, Hönack D (1991) Responses to NMDA receptor antagonists altered by epileptogenesis. Trends Pharmacol Sci 12:52
Sveinbjornsdottir S, Sander JWAS, Upton D, Thompson PJ, Patsalos PN, Hirt D, Emre M, Lowe D, Duncan JS (1993) The excitatory amino acid antagonist D-CPP-ene (SDZ EAA-494) in patients with epilepsy. Epilepsy Res 16:165–174
Leclercq K, Matagne A, Kaminski RM (2014) Low potency and limited efficacy of antiepileptic drugs in the mouse 6 Hz corneal kindling model. Epilepsy Res 108:675–683
Levesque M, Avoli M, Bernard C (2016) Animal models of temporal lobe epilepsy following systemic chemoconvulsant administration. J Neurosci Methods 260:45–52
Nadler JV, Perry BW, Cotman CW (1978) Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature 271:676–677
Ben Ari Y, Lagowska J, Tremblay E, Le Gal LS (1979) A new model of focal status epilepticus: intra-amygdaloid application of kainic acid elicits repetitive secondarily generalized convulsive seizures. Brain Res 163:176–179
Cavalheiro EA, Riche DA, Le Gal LS (1982) Long-term effects of intrahippocampal kainic acid injection in rats: a method for inducing spontaneous recurrent seizures. Electroencephalogr Clin Neurophysiol 53:581–589
Riban V, Bouilleret V, Pham L, Fritschy JM, Marescaux C, Depaulis A (2002) Evolution of hippocampal epileptic activity during the development of hippocampal sclerosis in a mouse model of temporal lobe epilepsy. Neuroscience 112:101–111
Klein S, Bankstahl M, Löscher W (2015) Inter-individual variation in the effect of antiepileptic drugs in the intrahippocampal kainate model of mesial temporal lobe epilepsy in mice. Neuropharmacology 90:53–62
Duveau V, Pouyatos B, Bressand K, Bouyssieres C, Chabrol T, Roche Y, Depaulis A, Roucard C (2016) Differential effects of antiepileptic drugs on focal seizures in the intrahippocampal kainate mouse model of mesial temporal lobe epilepsy. CNS Neurosci Ther 22:497–506
Guillemain I, Kahane P, Depaulis A (2012) Animal models to study aetiopathology of epilepsy: what are the features to model? Epileptic Disord 14:217–225
Klee R, Brandt C, Töllner K, Löscher W (2017) Various modifications of the intrahippocampal kainate model of mesial temporal lobe epilepsy in rats fail to resolve the marked rat-to-mouse differences in type and frequency of spontaneous seizures in this model. Epilepsy Behav 68:129–140
Grabenstatter HL, Ferraro DJ, Williams PA, Chapman PL, Dudek FE (2005) Use of chronic epilepsy models in antiepileptic drug discovery: the effect of topiramate on spontaneous motor seizures in rats with kainate-induced epilepsy. Epilepsia 46:8–14
Grabenstatter HL, Clark S, Dudek FE (2007) Anticonvulsant effects of carbamazepine on spontaneous seizures in rats with kainate-induced epilepsy: comparison of intraperitoneal injections with drug-in-food protocols. Epilepsia 48:2287–2295
Grabenstatter HL, Dudek FE (2008) A new potential AED, carisbamate, substantially reduces spontaneous motor seizures in rats with kainate-induced epilepsy. Epilepsia 49:1787–1794
Ali A, Dua Y, Constance JE, Franklin MR, Dudek FE (2012) A once-per-day, drug-in-food protocol for prolonged administration of antiepileptic drugs in animal models. Epilepsia 53:199–206
Thomson K, West P, Newell T, Metcalf C, Wilcox K (2016) Rapid screening for antiseizure therapies utilizing repeated dosing in chronically epileptic rats. American Epilepsy Society 70th annual meeting abstracts online abst. 3.224.
Löscher W (2011) Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 20:359–368
West P, Saunders G, Billingsley P, Smith M, Metcalf C, White H, Wilcox K (2016) Spontaneous electrographic bursting in the medial entorhinal cortex of kainate-lesioned rats is refractory to multiple classes of anti-seizure drugs. American Epilepsy Society 70th annual meeting abstracts online abst. 3.061.
Curia G, Longo D, Biagini G, Jones RS, Avoli M (2008) The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods 172:143–157
Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L (1983) Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res 9:315–335
Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA (1989) Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: A novel model of intractable epilepsy. Synapse 3:154–171
Cavalheiro EA, Leite JP, Bortolotto ZA, Turski WA, Ikonomidou C, Turski L (1991) Long-term effects of pilocarpine in rats: structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia 32:778–782
Leite JP, Cavalheiro EA (1995) Effects of conventional antiepileptic drugs in a model of spontaneous recurrent seizures in rats. Epilepsy Res 20:93–104
Glien M, Brandt C, Potschka H, Löscher W (2002) Effects of the novel antiepileptic drug levetiracetam on spontaneous recurrent seizures in the rat pilocarpine model of temporal lobe epilepsy. Epilepsia 43:350–357
Bankstahl M, Bankstahl JP, Löscher W (2012) Inter-individual variation in the anticonvulsant effect of phenobarbital in the pilocarpine rat model of temporal lobe epilepsy. Exp Neurol 234:70–84
Mazarati AM, Thompson KW, Suchomelova L, Sankar R, Shirasaka Y, Nissinen J, Pitkänen A, Bertram E, Wasterlain C (2006) Status epilepticus: electrical stimulation models. In: Pitkänen M, Schwartzkroin PA, Moshé SL (eds.) Models of seizures and epilepsy. Elsevier, Amsterdam, pp 449–464
Brandt C, Volk HA, Löscher W (2004) Striking differences in individual anticonvulsant response to phenobarbital in rats with spontaneous seizures after status epilepticus. Epilepsia 45:1488–1497
Bethmann K, Brandt C, Löscher W (2007) Resistance to phenobarbital extends to phenytoin in a rat model of temporal lobe epilepsy. Epilepsia 48:816–826
Stables JP, Bertram E, Dudek FE, Holmes G, Mathern G, Pitkänen A, White HS (2003) Therapy discovery for pharmacoresistant epilepsy and for disease-modifying therapeutics: summary of the NIH/NINDS/AES models II workshop. Epilepsia 44:1472–1478
Brandt C, Bethmann K, Gastens AM, Löscher W (2006) The multidrug transporter hypothesis of drug resistance in epilepsy: proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis 24:202–211
Feldmann M, Asselin MC, Liu J, Wang S, McMahon A, Anton-Rodriguez J, Walker M, Symms M, Brown G, Hinz R, Matthews J, Bauer M, Langer O, Thom M, Jones T, Vollmar C, Duncan JS, Sisodiya SM, Koepp MJ (2013) P-glycoprotein expression and function in patients with temporal lobe epilepsy: a case-control study. Lancet Neurol 12:777–785
Blanco MM, Dos SJ Jr, Perez-Mendes, P., Kohek SR, Cavarsan CF, Hummel M, Albuquerque C, Mello LE (2009) Assessment of seizure susceptibility in pilocarpine epileptic and nonepileptic Wistar rats and of seizure reinduction with pentylenetetrazole and electroshock models. Epilepsia 50:824–831
Töllner K, Twele F, Löscher W (2016) Evaluation of the pentylenetetrazole seizure threshold test in epileptic mice as surrogate model for drug testing against pharmacoresistant seizures. Epilepsy Behav 57:95–104
Bankstahl M, Bankstahl JP, Löscher W (2013) Pilocarpine-induced epilepsy in mice alters seizure thresholds and the efficacy of antiepileptic drugs in the 6-Hz psychomotor seizure model. Epilepsy Res 107:205–216
Leclercq K, Kaminski RM (2015) Status epilepticus induction has prolonged effects on the efficacy of antiepileptic drugs in the 6-Hz seizure model. Epilepsy Behav 49:55–60
Erker T, Brandt C, Töllner K, Schreppel P, Twele F, Schidlitzki A, Löscher W (2016) The bumetanide prodrug BUM5, but not bumetanide, potentiates the antiseizure effect of phenobarbital in adult epileptic mice. Epilepsia 57:698–705
Löscher W (2017) The search for new screening models of pharmacoresistant epilepsy: is induction of acute seizures in epileptic rodents a suitable approach? Neurochem Res (in press)
Annegers JF, Rocca WA, Hauser WA (1996) Causes of epilepsy: contributions of the Rochester epidemiology project. Mayo Clin Proc 71:570–575
Semah F, Picot MC, Adam C, Broglin D, Arzimanoglou A, Bazin B, Cavalcanti D, Baulac M (1998) Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 51:1256–1262
Stephen LJ, Kwan P, Brodie MJ (2001) Does the cause of localisation-related epilepsy influence the response to antiepileptic drug treatment? Epilepsia 42:357–362
Mohanraj R, Brodie MJ (2005) Outcomes in newly diagnosed localization-related epilepsies. Seizure 14:318–323
Pitkänen A, Kharatishvili I, Karhunen H, Lukasiuk K, Immonen R, Nairismagi J, Grohn O, Nissinen J (2007) Epileptogenesis in experimental models. Epilepsia 48(Suppl 2):13–20
Pitkänen A, Bolkvadze T, Immonen R (2011) Anti-epileptogenesis in rodent post-traumatic epilepsy models. Neurosci Lett 497:163–171
Eastman CL, Verley DR, Fender JS, Temkin NR, D’Ambrosio R (2010) ECoG studies of valproate, carbamazepine and halothane in frontal-lobe epilepsy induced by head injury in the rat. Exp Neurol 224:369–388
Eastman CL, Verley DR, Fender JS, Stewart TH, Nov E, Curia G, D’Ambrosio R (2011) Antiepileptic and antiepileptogenic performance of carisbamate after head injury in the rat: blind and randomized studies. J Pharmacol Exp Ther 336:779–790
Libbey JE, Fujinami RS (2011) Neurotropic viral infections leading to epilepsy: focus on Theiler’s murine encephalomyelitis virus. Future Virol 6:1339–1350
Wilcox K, Vanagas F, Underwood T, Patel D, Metcalf C (2016) Evaluation of prototype antiseizure drugs in the theiler’s murine encephalomyelitis virus-induced model of temporal lobe epilepsy. American Epilepsy Society 70th annual meeting abstracts online abst. 1.259
Bender RA, Baram TZ (2007) Epileptogenesis in the developing brain: what can we learn from animal models? Epilepsia 48(Suppl 5):2–6
Wasterlain CG, Gloss DS, Niquet J, Wasterlain AS (2013) Epileptogenesis in the developing brain. Handb Clin Neurol 111:427–439
Auvin S, Pineda E, Shin D, Gressens P, Mazarati A (2012) Novel animal models of pediatric epilepsy. Neurotherapeutics 9:245–261
Kandratavicius L, Balista PA, Lopes-Aguiar C, Ruggiero RN, Umeoka EH, Garcia-Cairasco N, Bueno-Junior LS, Leite JP (2014) Animal models of epilepsy: use and limitations. Neuropsychiatr Dis Treat 10:1693–1705
Galanopoulou AS, Moshe SL (2015) Pathogenesis and new candidate treatments for infantile spasms and early life epileptic encephalopathies: a view from preclinical studies. Neurobiol Dis 79:135–149
Löscher W (1984) Genetic animal models of epilepsy as a unique resource for the evaluation of anticonvulsant drugs. A review. Methods Findings Experiment. Clin Pharmacol 6:531–547
Vergnes M, Marescaux C, Micheletti G, Reis J, Depaulis A, Rumbach L, Warter JM (1982) Spontaneous paroxysmal electroclinical patterns in rat: a model of generalized non-convulsive epilepsy. Neurosci Lett 33:97–101
Depaulis A, David O, Charpier S (2016) The genetic absence epilepsy rat from Strasbourg as a model to decipher the neuronal and network mechanisms of generalized idiopathic epilepsies. J Neurosci Methods 260:159–174
Van Luijtelaar G, Zobeiri M (2014) Progress and outlooks in a genetic absence epilepsy model (WAG/Rij). Curr Med Chem 21:704–721
Guerrini R, Marini C, Mantegazza M (2014) Genetic epilepsy syndromes without structural brain abnormalities: clinical features and experimental models. Neurother 11:269–285
Blumenfeld H, Klein JP, Schridde U, Vestal M, Rice T, Khera DS, Bashyal C, Giblin K, Paul-Laughinghouse C, Wang F, Phadke A, Mission J, Agarwal RK, Englot DJ, Motelow J, Nersesyan H, Waxman SG, Levin AR (2008) Early treatment suppresses the development of spike-wave epilepsy in a rat model. Epilepsia 49:400–409
Russo E, Citraro R, Scicchitano F, De Fazio S, Di Paola ED, Constanti A, De Sarro G (2010) Comparison of the antiepileptogenic effects of an early long-term treatment with ethosuximide or levetiracetam in a genetic animal model of absence epilepsy. Epilepsia 51:1560–1569
Russo E, Citraro R, Scicchitano F, De Fazio S, Perrotta I, Di Paola ED, Constanti A, De Sarro G (2011) Effects of early long-term treatment with antiepileptic drugs on development of seizures and depressive-like behavior in a rat genetic absence epilepsy model. Epilepsia 52:1341–1350
Dezsi G, Ozturk E, Stanic D, Powell KL, Blumenfeld H, O’Brien TJ, Jones NC (2013) Ethosuximide reduces epileptogenesis and behavioral comorbidity in the GAERS model of genetic generalized epilepsy. Epilepsia 54:635–643
Griffin A, Hamling KR, Knupp K, Hong S, Lee LP, Baraban SC (2017) Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 140:669–683
Denayer T, Stöhr T, van Roy M (2014) Animal models in translational medicine: validation and prediction. New Hor Transl Med 2:5–11
Wartha K, Herting F, Hasmann M (2014) Fit-for purpose use of mouse models to improve predictivity of cancer therapeutics evaluation. Pharmacol Ther 142:351–361
Willner P, Belzung C (2015) Treatment-resistant depression: are animal models of depression fit for purpose? Psychopharmacology 232:3473–3495
Acknowledgements
I thank my colleagues and friends Harvey Kupferberg and Steve White for critically reading a first version of this review and John Kehne for providing Fig. 4. WL is a member of the External Consultant Board (ECB) of the ETSP and thanks the other ECB members and all colleagues from the NINDS Epilepsy Branch and ETSP contract site in Utah for fruitful discussions on how to improve ASD discovery. The author’s own studies have been supported by grants from the German Research Foundation (DFG, Bonn, Germany; Grant # LO 274/1 - LO 274/16), the National Institutes of Health (NIH; Bethesda, MD, USA: Grant # R21 NS049592-01), the European Union’s Seventh Framework Programme (FP7) under grant agreements 201380 (EURIPIDES) and 602102 (EPITARGET), and the Niedersachsen-Research Network on Neuroinfectiology (N-RENNT) of the Ministry of Science and Culture of Lower Saxony in Germany.
Author information
Authors and Affiliations
Corresponding author
Additional information
This review is dedicated to my colleague and friend Dr. H. Steve White to acknowledge his outstanding contributions in anti-seizure drug discovery and development of novel models of drug-resistant seizures.
Rights and permissions
About this article

Cite this article
Löscher, W. Animal Models of Seizures and Epilepsy: Past, Present, and Future Role for the Discovery of Antiseizure Drugs. Neurochem Res 42, 1873–1888 (2017). https://doi.org/10.1007/s11064-017-2222-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-017-2222-z