
Abstract
Research in genetics of epilepsy represents an area of great interest both for clinical purposes and for understanding the basic mechanisms of epilepsy. Most mutations in epilepsies without structural brain abnormalities have been identified in ion channel genes, but an increasing number of genes involved in a diversity of functional and developmental processes are being recognized through whole exome or genome sequencing. Targeted molecular diagnosis is now available for different forms of epilepsy. The identification of epileptogenic mutations in patients before epilepsy onset and the possibility of developing therapeutic strategies tested in experimental models may facilitate experimental approaches that prevent epilepsy or decrease its severity. Functional analysis is essential for better understanding pathogenic mechanisms and gene interactions. In vitro experimental systems are either cells that usually do not express the protein of interest or neurons in primary cultures. In vivo/ex vivo systems are organisms or preparations obtained from them (e.g., brain slices), which should better model the complexity of brain circuits and actual pathophysiological conditions. Neurons differentiated from induced pluripotent stem cells generated from the skin fibroblasts of patients have recently allowed the study of mutations in human neurons having the genetic background of a given patient. However, there is remarkable complexity underlying epileptogenesis in the clinical dimension, as reflected by the fact that experimental models have not provided yet results having clinical translation and that, with a few exceptions concerning rare conditions, no new curative treatment has emerged from any genetic finding in epilepsy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The role of genetic determinism in epilepsy has long been emphasized. Although an increasing number of genetic epilepsy syndromes or disorders have recently been identified, in many cases both genetic and acquired factors can contribute to the determinism of epilepsy. Epilepsies of unknown cause have classically been divided into two main etiologic categories: presumed symptomatic epilepsies are those in which the cause is suspected to be induced by a pathology that is below the limit of detection of the available diagnostic tests; “idiopathic” epilepsies are instead thought to be caused by a genetic predisposition. Symptomatic epilepsies are caused by an obvious brain abnormality, which can, in turn, be determined genetically or by external factors acting prenatally or after birth. Defining genetically determined epilepsies as “symptomatic” is easy, for example for neuronal migration disorders, or neurocutaneous diseases, such as tuberous sclerosis, or a syndromic form of mental retardation. The same applies to many forms of progressive myoclonus epilepsy, the clinical expression of which is relatively homogeneous in relation to the genetically determined degenerative or metabolic causes. The task is much more complex when the genetic cause is unknown and will only be recognizable or become highly likely later during the course, when a given electroclinical pattern or syndrome will become obvious. This is, for example, the case of several early-onset epileptic encephalopathies (EEs) such as those caused by alterations of the CDKL5, STXBP1, or ARX genes. For this reason, the category of “genetic epilepsy”, which has recently been proposed [1] is controversial and, perhaps, somewhat uncertain, as what is presumed to be symptomatic today may become genetic after molecular screening, or remain as such if screening is unavailable.
About 30% of all epilepsies are idiopathic [2], and research in genetics of epilepsy represents an area of great interest both for clinical purposes and for understanding of the basic mechanisms of epilepsy. The idiopathic epilepsies are characterized by age-related onset, normal neurological and cognitive development, and absence of brain damage. The study of electroencephalogram (EEG) anomalies while awake and during sleep is useful for characterizing the different forms. An underlying genetic defect has been clearly demonstrated for some forms with Mendelian inheritance [3]. In particular, genetic studies of families and twins contributed to the definition of genetic epilepsy, and especially to the evaluation of the risk of familial recurrence [4, 5]. After the first report in 1988 of the chromosomal mapping of juvenile myoclonic epilepsy [6], several additional loci were demonstrated to be implicated in syndromic epilepsies. In 1995, the first mutation in the gene coding for the nicotinic acetylcholine receptor was identified in a family with autosomal dominant nocturnal frontal lobe epilepsy [7]. The subsequent identification of new epilepsy genes has greatly favored research into experimental models and new therapeutic strategies. Notably, most mutations in epilepsies without structural brain abnormalities have been identified in ion channel genes because they are known to be involved in neuronal excitability, but some mutations have also been identified in genes that do not code for ion channels [8]. The use of nontargeted genetic approaches (e.g., whole exome or genome sequencing) will probably increase their number in the future, although it is possible that several genes will be identified that play a role in ion channel regulation, targeting, or expression.
A targeted molecular diagnosis is now available for different forms of epilepsy [9]. The identification of epileptogenic mutations in patients before epilepsy onset and the possibility of developing therapeutic strategies tested in experimental models may allow experimental approaches towards interventions that inhibit epileptogenesis, prevent epilepsy, or decrease its severity. However, ethical problems may arise, especially in asymptomatic mutation carriers or in individuals and families in whom—after mutations of specific genes have been identified—the severity of the associated phenotype remains unpredictable. In fact, most idiopathic epilepsies do not follow a simple Mendelian pattern of inheritance and, even in families where involvement of a single gene is suspected, a high degree of phenotypic heterogeneity is possible.
Genetic mutations are defined as modifications in the sequence of a gene that is clearly identified as the cause of the disease; thus, the term should be used for Mendelian monogenic disorders, whereas genetic variants are modifications that contribute to disease susceptibility. In polygenic epilepsies, a specific epilepsy phenotype can be generated by the combination of less penetrant alleles with large effect, of polygenic alleles with small effect, and of interactions with environmental factors. In these cases, single modifications of gene sequences contribute to susceptibility to epilepsy, and their implication in disease is often inferred from the fact that the variants are mainly found in patients and that they induce functional effects. In some families with Mendelian inheritance, phenotypic variability has been ascribed to genetic modifiers (polymorphisms or other genetic variants that can modulate the effect of the genetic mutation) or environmental factors that influence phenotypic expression [10].
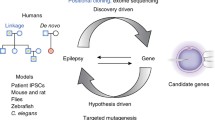
Functional analysis is essential for better understanding pathogenic mechanisms and gene interactions [8]. In vitro experimental systems (Fig. 1) are either cells that usually do not express the protein of interest (oocytes of the clawed frog Xenopus laevis or human cell lines, e.g., human embryonic kidney cells), which simplify functional analysis, or neurons in primary cultures, which provide neuronal cellular background and variety. In vivo/ex vivo systems are organisms or preparations obtained from them (e.g., brain slices), which should better model the complexity of brain circuits and actual pathophysiological conditions. Neurons differentiated from induced pluripotent stem cells generated from the skin fibroblasts of patients have recently allowed the study of mutations in human neurons that have the genetic background of a given patient. It is important that the results of functional studies are interpreted considering the characteristics of the experimental system used [8].

Experimental models used for functional analyses of epileptogenic mutations/variants, in particular for ion channel genes. cRNA cloned RNA containing the sequence of the gene of interest and the mutation/variant under study; cDNA cloned DNA containing the sequence of the gene of interest and the mutation/variant under study; iPS induced pluripotent stem cells
We will focus our review on the main genetic epilepsy syndromes in which no structural brain abnormalities are currently apparent.
The Spectrum of SCN1A Gene-related Epilepsies and EEs
Dravet Syndrome or Severe Myoclonic Epilepsy of Infancy
Dravet syndrome manifests at about 6 months of age in previously healthy infants, typically with prolonged generalized or hemiclonic febrile seizures (FS) [11]. Between 1 and 4 years, other types of seizures usually appear, including myoclonic, partial, and absence seizures. Hyperthermia, such as having a fever or sitting in a warm bath, often precipitates seizures [12]. EEG may be normal until the age of 2 years when generalized spike wave activity is seen; approximately 10% of patients are photosensitive. Magnetic resonance imaging is either normal or shows nonspecific features [13, 14]. Development in the first year of life is normal, but subsequently slows, and may regress leading to severe cognitive impairment. Therefore, Dravet syndrome is considered an EE, that is a disorder in which epileptic seizures and epileptiform electrical activity impair brain function, although this causal link has not yet been clearly demonstrated.
Dravet syndrome is related, in at least 85% of cases, to abnormalities of the SCN1A Na+ channel (Table 1). Neuronal Na+ channels are essential for excitability, and consist of a principal α subunit and accessory β subunits (Fig. 2); SCN1A codes for the Nav1.1 α subunit. The majority of patients exhibiting mutations of this gene carry de novo mutations (90%); about 40% of these mutations are truncation and 40% are missense mutations. Ten percent of patients who are mutation-negative on sequence-based analysis have copy number variations, including exonic deletions or duplications that can involve several exons or the whole gene [15, 16]. Some rare patients have a mutation in the GABRG2 gene, gamma-aminobutyric acid (GABA) receptor γ2 subunit [3], or in SCN1B sodium channel β1 subunit [17] (Fig. 2; Table 1). SCN1A mutations are also commonly found in the borderline variant of Dravet syndrome, the separation of which from the core syndrome may be arbitrary [18]. Mutations are less commonly found in patients that have been categorized within different subgroups exhibiting various elements of Dravet syndrome [19–21]. Germline mosaicism may result in siblings with Dravet syndrome born from an unaffected or mildly unaffected parent carrying a low level of mosaicism for the mutation [22, 23].

Basic structure of main voltage- and ligand-gated ion channel proteins involved in genetic epilepsy. The structure of the subunits targeted by mutations/variants identified in genetic forms of epilepsy are shown. The names of the genes and the forms of epilepsy in which they are involved are indicated below the diagram of the protein. Nav voltage-gated sodium channels; Cav3.2 voltage-gated calcium channels, T-type-1H; Kv7 voltage-gated potassium channels, M-type; KCa4.1 sodium-activated potassium (KNa) channel, SLACK-SLO2.2 type; GABA-A gamma-aminobutyric acid receptor, type A; Ach nicotinic acetylcholine receptor; NMDA glutamate receptor, N-methyl-D-aspartate (NMDA) type; DS Dravet syndrome; GEFS+ generalized (genetic) epilepsy with febrile seizures plus; EE epileptic encephalopathies; BNIFS benign neonatal-infantile familial seizures; BNIS benign neonatal familial seizures; IGE idiopathic generalized epilepsies; MMPSI malignant migrating partial seizures of infancy; ADNFLE Autosomal dominant nocturnal frontal lobe epilepsy; EAS epilepsy–aphasia syndromes
In vitro functional studies of missense SCN1A mutations in cell lines or Xenopus oocytes have initially shown controversial results, revealing both gain-of-function and loss-of-function effects, but loss of function without negative dominance seems to be the predominant mechanism of action of both truncations and missense mutations [8, 24]. Two mouse models for SCN1A, in which loss of function mutations were introduced into the endogenous mouse gene, a knockout line and a knockin of a human nonsense mutation, exhibited spontaneous seizures, cognitive impairment, and reduced sodium current selectively in GABAergic inhibitory interneurons [25–27]. These findings suggest that Dravet syndrome, and probably the other SCN1A-related seizure disorders, are caused by a decreased excitability of GABAergic interneurons owing to SCN1A haploinsufficiency [25, 26]. Recent functional studies carried out using human neurons differentiated form induced pluripotent stem cells have generated controversial results [28–30], similarly to the first functional studies in transfected cells. Evidently, this promising technique still needs improvements for faithfully modeling pathophysiological conditions and functional effects. Phenotypes with some clinical features resembling Dravet syndrome have also been associated with protocadherin 19 (PCDH19) mutations in a few female patients. See the description of the spectrum of PCDH19-related epilepsies below.
Generalized (Genetic) Epilepsy with Febrile Seizures Plus
Generalized (genetic) epilepsy with febrile seizures plus (GEFS+) is a familial epilepsy syndrome diagnosed on the basis of at least 2 individuals with GEFS+ phenotypes in a family. The GEFS+ spectrum denotes phenotypic heterogeneity in families, including FS and FS plus. Overlap with classical idiopathic generalized epilepsy (IGE) is also seen. The course and response to antiepileptic drugs may vary considerably within the same family: some patients experience rare FS or nonfebrile seizures that remit after a few years, while other individuals, even within the same family, have drug-resistant epilepsy, with Dravet syndrome as the extreme of the spectrum. GEFS+ was originally recognized because of remarkably large autosomal dominant pedigrees with 60–70% penetrance. However, it is likely that most cases occur in small families or are sporadic [31].
GEFS+ has been associated with mutations in the SCN1A (Nav1.1) or SCN1B (β1) genes (Table 1) [32, 33]. Missense mutations in SCN1A are the most commonly identified molecular abnormality, found in about 10% of families [34]. In some families, mutations in the gene encoding the γ2 subunit of the GABAA receptor (GABRG2) have been identified (Table 1) [35–37]. The phenotypic variability observed in GEFS+ could be linked to the combined action of mutations in different genes. Some SCN1A mutations can cause phenotypes extending from different types of epilepsy to familial hemiplegic migraine [38]. It has also been shown that a SCN1A missense mutant, identified in families with extreme phenotypes, including Dravet syndrome, causes loss of function because of folding defects, which can be rescued by molecular interactions with associated proteins or pharmacological chaperones [39]. These results have been confirmed and extended to typical GEFS+ families and Dravet syndrome patients with de novo mutations [39–42]. This mechanism may generate phenotypic variability and also possibly be used in the development of therapeutic approaches. As recently shown for a missense SCN1A mutant identified in a family with pure familial hemiplegic migraine [43], rescue of folding defects may also transform nonfunctional loss-of-function mutants, an effect that is consistent with severe epilepsy, into gain-of-function ones, an effect that is consistent with familial hemiplegic migraine [44]. Animal models have confirmed that GEFS+ mutations cause loss of function of SCN1A and reduced excitability of GABAergic neurons [45]. GABA receptors have long been suspected in epileptogenesis. Functional expression of some GABRG2 mutations, identified in patients with GEFS+ revealed a pronounced loss of function by altered gating or defective trafficking, and reduced surface expression as a common pathogenic mechanism [8, 37]. Knock-in mouse models of the R43Q GABRG2 mutation shows generalized seizures and reduced GABAergic inhibition [46]. Hence, these mutations reduce the main mechanism for neuronal inhibition in the brain, similarly to SCN1A mutations, which can explain the occurrence of seizures.
Spectrum of Benign Epilepsies in Newborns and Infants
Benign epilepsies of the first year of life represent a group of syndromes that are defined as “benign” because their clinical manifestations, which occur in otherwise asymptomatic babies, regress and eventually disappear spontaneously. These forms are relatively rare and are transmitted with an autosomal dominant pattern of inheritance. Molecular diagnosis, where possible, is important in order to avoid unnecessary invasive testing and support genetic counseling [47]. The clinical manifestations of these forms of epilepsy overlap considerably, while age of onset may be slightly variable [48]. Some rare mutations in the same genes can also cause severe forms that manifest as EEs. Thus, these forms should be considered as part of a spectrum, likewise SCN1A-gene related epilepsies.
Benign Familial Neonatal Seizures
Benign familial neonatal seizures (BFNS) are characterized by clusters of seizures that appear from the first days of life up to the third month and spontaneously disappear after weeks to months. Seizures have focal onset, often with hemitonic or hemiclonic symptoms, or apneic spells, or can appear clinically as generalized. Interictal EEG is usually normal. The rare available ictal EEGs show both focal and generalized discharges. The risk of seizures recurring later in life is about 15% [48]. Although psychomotor development is usually normal, an increasing number of cases with learning disability have been described (see below) [49]. BFNS are inherited as an autosomal dominant trait with 85% penetrance. Most patients have mutations in the gene encoding the voltage-dependent K+ channel, KQT-like subtype member (KCNQ2), and deletions/duplications involving one or more exons of KCNQ2 (Table 1) [50–53]. A small proportion of families carry mutations in the associated gene voltage-dependent K+ channel, KQT-like subtype, member 3 (KCNQ3) [54]. KCNQ2 and KCNQ3 form a heteromeric K+ channel (Fig. 2), which is particularly important in the axon initial segment and nodes of Ranvier of glutamatergic neurons, determines the M-current, and influences subthreshold properties and membrane potential at rest [55]. Functional studies in in vitro systems coexpressing heteromeric wildtype and mutant KCNQ2⁄3 channels revealed a reduction of about 20–30% in the resulting potassium current, which is apparently sufficient to cause BFNS [56, 57]. Although the reduction of the potassium current can cause epileptic seizures by subthreshold membrane depolarization, which increases neuronal firing, it is not fully understood why seizures preferentially occur in neonates [58]. It is possible that the neonatal brain is more vulnerable to even small changes in neuronal excitability. Alternatively, KCNQ2 and KCNQ3 channels, when mutated, might be replaced by other K+ channels that become functional after the first months of life.
Transgenic and knockin BFNS mice have been generated. Transgenic mice expressing a dominant negative KCNQ2 mutant [59] show spontaneous partial and generalized tonic–clonic seizures, but also pronounced hyperactivity and cell loss in the hippocampus, with impaired hippocampus-related memory, which are not consistent with the typical BNFS human phenotype. Knockin mice of KCNQ2 A306T and KV7.3 G311V mutations [60] show spontaneous tonic–clonic seizures and, consistent with BNFS, no hippocampal neurodegeneration; however, only homozygous mice manifest epilepsy, and they also tend to have seizures in adulthood.
Benign Familial Neonatal–Infantile Seizures
Benign familial neonatal–infantile seizures (BFNIS) are characterized by seizures similar to those observed in children with BFNS. However, age at seizure onset ranges from the neonatal period to infancy in different family members, with a mean onset age of 3 months. Mutations in SCN2A (Nav1.2 sodium channel α subunit) have been identified in most families (Table 1) [61]. Although remission occurs by 12 months, with a very low risk of later seizures [61, 62], Nav1.2-related EEs have been reported in some instances (see below). Nav1.2 (Fig. 2) is particularly important for the excitability of the axon initial segment in glutamatergic neurons early in development. Mutations causing BFNIS have been studied in both transfected neocortical neurons and cell lines identifying gain of function effects [63, 64] consistent with hyperexcitability of excitatory neurons. Remission may depend on a developmental switch between Nav1.2 and Nav1.6 in myelinated axons that occur at early developmental stages [63, 64]. No animal models reproducing a BFNIS mutation have yet been developed.
Benign Familial Infantile Seizures
Benign familial infantile seizures are characterized by seizures similar to those observed in BNFS, with an age of onset around 6 months [65], sometimes in children who also develop paroxysmal dyskinesia [66]. Mutations of the PRRT2 gene, at the pericentromeric region of chromosome 16, have been associated with familial infantile convulsions, paroxysmal kinesigenic or exercise-induced dyskinesia, migraine, or hemiplegic migraine, or in various combinations (Table 1) [67–70]. PRRT2 mutations appear to have a high prevalence in patients with benign familial infantile seizures (or epilepsy), either alone or in association with paroxysmal kinesigenic or exercise-induced dyskinesia (from 40% to 90%), but are only rarely detected in sporadic cases and only rarely cause other types of epilepsy. PRRT2 mutations identified so far include a recurrent and frequent frameshift mutation (c.649_650insC, p.R217Pfs*8), found in a large number of patients from different ethnic backgrounds, as well as a considerable number of loss-of-function and missense amino acid-changing mutations. The location and type of mutation within PRRT2 do not appear to predict the clinical phenotype [69]. Yeast 2-hybrid studies suggest that PRRT2 interacts with synaptosomal-associated protein 25 kDA, a presynaptic Q-soluble N-ethylmaleimide sensitive fusion attachment protein receptor protein involved in the fusion of synaptic vesicles to the plasma membrane and calcium-triggered exocytosis [71]. Confirmation of PRRT2 mutations in infants with infantile seizures, paroxysmal dyskinesia, or both, provides families and clinicians with reassurance that seizures are likely to be self-limited, with an excellent prognosis [69]. Linkage studies have identified an additional locus for benign familial infantile seizures on chromosome 19 [72]. Several families with mutations in the ATP1A2 gene have benign infantile seizures in conjunction with hemiplegic migraine [73]. Rare families with mutations in SCN2A or KCNQ2 have also been described in which only infantile seizures occur [74, 75].
EEs Caused by Mutations of Ion Channel Genes Involved in Benign Epilepsies of the First Year of Life
As observed for SCN1A mutations, KCNQ2 and SCN2A mutations can cause a wide phenotypic spectrum that includes severe EEs. In particular, KCNQ2 mutations have been identified in severe neonatal EEs associated with intellectual disability and motor impairment, with a burst–suppression EEG pattern or multifocal epileptiform activity, but also milder forms (Table 1) [76, 77]. Functional studies of these mutations have recently been performed in Xenopus oocytes, showing that they can have more severe loss of function than BFNS [78] or, in some cases, cause negative dominance inhibiting function of wildtype KCNQ2 [79]. A transgenic mouse model expressing a KCNQ2 dominant negative mutant may model these forms [59].
Similarly, de novo SCN2A truncating and missense mutations have been identified in early-onset intractable childhood epilepsies, with features ranging from Ohtahara syndrome to Dravet syndrome [80–82]. Contrary to BNFIS mutants, functional analysis of some of these mutants using in vitro expression systems has shown loss of function [82], although it is not yet clear how loss of function in a sodium channel predominantly expressed in excitatory neurons can lead to network hyperexcitability. Notably, a loss-of-function Scn2a knockout mouse does not show an overt epileptic phenotype, although this has not been studied in detail [83].
EEs Caused by Non-ion Channel Genes
A genetic basis is also being recognized for an increasing number of monogenic epilepsies that are often manifested as severe forms, often having early seizure onset and developmental delay, but diverse, sometime distinctive, phenotypes.
PCDH19
PCDH19 mutations have been associated with epilepsy and mental retardation [84], an X-linked disorder affecting only girls and sparing transmitting boys, but also often appearing de novo (Table 1) [85]. It is characterized by a variable clinical presentation, including delayed development from birth, normal early development followed by regression starting after seizure onset, and normal development without regression [86]. The epilepsy spectrum associated with PCDH19 mutations is, in turn, variable and includes mild focal epilepsy starting in infancy or epilepsy with recurrent episodes of focal or generalized seizures in series or status epilepticus triggered by fever [86]. PCDH19 is part of the protocadherin delta-2 subclass of the cadherin super family. Protocadherin members are expressed predominantly in the nervous system, where they have a role in establishing neuronal connections and in signal transduction at synaptic membranes [84]. Men who are hemizygous for PCDH19 mutations have a normal cognitive level and no epilepsy. Cellular interference has been proposed to explain the discrepancy between the clinical manifestations of heterozygous girls and hemizygous boys [84, 87]. This model suggests that for a pathological phenotype to occur an individual must have two distinct populations of protocadherin cells (mutated and nonmutated). A normal woman or a transmitting man have only one protocadherin population of cells, protocadherin wildtype or protocadherin mutant cells, which would prevent the pathological phenotype from being manifested. The development of genetically modified animal models will, hopefully, allow for the better exploration of this puzzling disease mechanism.
Aristaless-Related Homeobox
The Aristaless-related homeobox (ARX) gene, on chromosome Xp22, is a transcription factor that belongs to a family of paired class homeobox genes, and plays an important role in embryogenesis, especially in the development of the central nervous system [88]. To date, ARX mutations have been identified in about 10 different clinical conditions, with or without brain malformations [89–91]. Malformation phenotypes, including X-linked lissencephaly with abnormal genitalia (XLAG), X-linked lissencephaly with abnormal genitalia with severe hydrocephalus, and Proud syndrome (agenesis of the corpus callosum with abnormal genitalia) are associated with protein truncation mutations and missense mutations in the homeobox [89, 92]. Nonmalformation phenotypes are associated with missense mutations outside of the homeobox or expansion of the second polyA tract, and include X-linked infantile spasms (ISSX)/West syndrome, Partington syndrome (mental retardation with mild distal dystonia), and nonspecific X-linked mental retardation [89, 92]. Expansions in the first polyA tract cause ISSX/West syndrome (tonic spasms with clustering, severe psychomotor delay, and hypsarrhythmia on EEG) [93, 94], a severe epileptic–dyskinetic encephalopathy [90], tonic seizures and dystonia without infantile spasms [95], and Ohtahara syndrome (Table 1) [96]. No neuropathologic studies of the brain of patients with “nonmalformation” phenotypes are available to confirm that structural abnormalities are, in fact, absent at the microscopic level.
Cyclin-Dependent, Kinase-Like 5
Mutations in the X-linked gene cyclin-dependent, kinase-like 5 (CDKL5) cause early-onset intractable seizures, severe developmental delay, and, often, subsequent appearance of stereotypic hand movements, absent speech development, and severely impaired communication skills—a combination of features that had initially been considered consistent with the Rett syndrome spectrum (Table 1) [97]. CDKL5 mutations also cause ISSX [98], a form of myoclonic encephalopathy [99], and severe encephalopathy with refractory seizures (Table 1) [100]. Mutations in CDKL5 are found mainly in girls, suggesting gestational lethality in boys [101]. Parental germline mosaicism has been reported [102]. In affected girls, a seemingly normal early development, followed by onset of intractable seizures between the first days and fourth month of life are early key diagnostic criteria. Severe developmental delay with regression becomes apparent after seizures onset. Seizures are usually manifested as infantile spasms, or prolonged tonic seizures followed by spasms and myoclonus, with a peculiar electroclinical pattern [103], variably associated with migrating focal seizures during the course. In one series, 16% of girls with early-onset intractable epilepsy, with or without infantile spasms, exhibited either mutations or genomic deletions involving CDKL5 [104].
Syntaxin Binding Protein
Mutations in the syntaxin binding protein 1 (STXBP1) gene have been associated with Ohtahara syndrome or early infantile EE (EIEE), with early-onset intractable tonic spasms, the characteristic suppression–burst EEG pattern, and extremely poor developmental outcome (Table 1) [105, 106]. Mutations in STXBP1 have also been found in some children with infantile spasms [107, 108]. A transition from EIEE to West syndrome occurs in 75% of children with EIEE [105, 106]. The STXBP1 protein plays an essential role in synaptic vesicle release and secretion of neurotransmitters [109]. The Ohtahara syndrome–infantile spasms spectrum can also be caused by mutations of the ARX gene [90, 96]. Available information on the causative genes for the Ohtahara syndrome–infantile spasms spectrum suggests a mutation rate of about 6% for ARX polyalanine expansions in boys with isolated or X-linked infantile spasms and no brain lesions [90], and up to 37% for STXBP1 in infants with Ohtahara syndrome [109]. However, larger series are needed to fully elucidate the phenotypic spectrum and better understand the causative role of these genes in this particular syndrome spectrum.
Epilepsy–Aphasia Syndromes
Epilepsy–aphasia syndromes including Landau–Kleffner syndrome and continuous spike-waves in slow sleep are a group of rare, severe EEs with a characteristic EEG pattern and developmental regression affecting language, in particular. The lack of evident brain magnetic resonance imaging abnormalities, in addition to a frequent positive family history of epilepsy or EEG abnormalities, has always raised the suspicion of a genetic etiology. Mutations in the GRIN2A gene encoding the α2 subunit of the N-methyl-D-aspartate glutamate receptor (also known as GluNR2A; Fig. 2) have been identified in 5% of patients with centro-temporal spikes, and 20% of nonlesional continuous spike-waves in slow sleep and Landau–Kleffner syndrome; functional analysis using in vitro systems has shown gain of function (Table 1) [110–112]. Mutations in the GRIN2B gene encoding the β2 subunit of the N-methyl-D-aspartate glutamate receptor have also been recently identified in 3 patients. The phenotypes include West syndrome and focal epilepsy with intellectual disability, and appear to be very different from that of GRIN2A mutations (Table 2) [113].
Whole Exome Sequencing Studies
The current and very exciting era of the whole exome sequencing (WES) is generating a great amount of genetic information, thus widening our understanding of the pathophysiology of epilepsies with genetic etiology, especially in the area of the rare EEs. Most of the mutations that have been interpreted as causative arise as de novo mutations or are inherited in an autosomal recessive fashion, often as compound heterozygous mutations.
The hypothesis that mutations in a “few and common genes” might cause phenotypes with overlapping clinical features needs to be revised. Indeed, genetic findings emerging from WES studies of large cohorts of patients, realized by consortia of laboratories, suggest that many “private” mutations in many genes are at play [114]. The function of such genes might be localized within a so-called network system. Mutations of such new epilepsy genes, associated with EE with variable phenotypes, and occasionally resembling known syndromes, including Dravet syndrome, are found in a very limited number of patients [115, 116]. For example, mutations in the CHD2 gene have been associated with a Dravet syndrome-like phenotype in 10 patients and mutations in the SYNGAP1 gene in 5 patients with EE. Genes coding for ion channels are still very much genes to hunt as a cause of epilepsy; indeed, SCN8A (Fig. 2), the fifth sodium channel gene to be mutated in epilepsy, is associated with early-onset EE [114, 117]. WES studies or targeted sequencing of panels of selected genes of probands and their parents, or of small families with affected siblings, also show that genes previously associated with milder nonlesional phenotypes, including GABRA1 and, as highlighted above, SCN2A and KCNQ2, might also cause EE in some patients [115]. De novo gain-of-function mutations in the KCNT1 gene, on chromosome 9q34, have been identified in patients with malignant migrating partial seizures of infancy, a rare syndrome with infantile onset intractable and migrating focal seizures with severe impairment of psychomotor development [118]. Likewise, most of the currently known epilepsy genes and mutations in KCNT1 gene have been associated with additional and very different epilepsy phenotypes, including autosomal dominant nocturnal frontal lobe epilepsy [119].
Exome sequencing of affected individuals and their parents therefore provides the most powerful strategy for gene discovery. Table 2 shows the most interesting findings arising from large WES and targeted sequencing.
Familial Focal Epilepsies
Autosomal Dominant Nocturnal Frontal Lobe Epilepsy
Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) has a penetrance of about 70–80% and includes frequent, usually brief, seizures with onset, on average, at around 10 years of age, with hyperkinetic or tonic manifestations, typically in clusters at night during slow-wave sleep. Paroxysmal arousals, dystonia-like attacks, and epileptic nocturnal wanderings are also part of the phenotype [120]. Ictal video-EEG studies have revealed partial seizures originating from the frontal lobe, but also from parts of the insula and temporal lobe, suggesting a defect of a broader network [121, 122]. A mutation in the gene CHRNA4, encoding the α4-subunit of a neuronal nicotinic acetylcholine receptor (nAchR), was the first ion channel mutation found in an inherited form of epilepsy [7]. About 15 mutations in CHRNA4, 5 in CHRNB2 (which encodes the β2-subunit of nAchR), and 1 in CHRNA2 (encoding the neuronal nAchR α2-subunit) have been reported so far [123–130]. These receptors are heteropentameric, consisting of various combinations of subunits. The α4–β2 combination (Figs. 2 and 3) is the most common in the thalamus and cerebral cortex. All the identified mutations are located in the pore-forming M2 transmembrane segments.

(a) General structure of a single subunit of the nicotinic receptor. (b) Nicotinic receptors are formed by 5 subunits (pentamers), symmetrically arranged to delimit a pore through which cations flow, with Na+ and Ca2+ incoming, and K+ outgoing. Nine different types of α-subunits and 4 different types of β-subunits are known. Isoforms for subunits δ, ε, and ϒ are not yet known. Numerous receptor subtypes that have specific anatomic locations (e.g., muscle or neuronal) are generated by multiple combinations of different types of these subunits. In the central nervous system, the pentameric structures of the receptor are composed of α-subunit homodimers or of α-subunits/β-subunits combinations. (c) Top view of the structure of a neuronal nicotinic receptor composed by 2 α4-subunits (CHRNA4) and 3β2-subunits (CHRNB2). Abnormalities in this receptor, caused by mutations of its subunits, cause nocturnal frontal lobe epilepsy
Functional studies of nAchR have produced controversial results, which makes the underlying pathogenic mechanisms unclear. Expression of α4 mutants in human embryonic kidney cells or Xenopus oocytes had various effects, consistent with either gain or loss of function: increased sensitivity to acetylcholine (gain of function), or decreased Ca2+ potentiation or accelerated desensitization (loss of function) [131, 132]. Whereas β2 mutations showed gain of function by increased sensitivity to acetylcholine or slower desensitization [125], the α2 mutation showed gain of function by increased sensitivity to acetylcholine [128]. The α4 mutations S252F and +L264 engineered in knockin mice [133] induced spontaneous seizures of various types, in some cases similar to those of the human phenotype, but no paroxysmal arousal and dystonia-like manifestations that are typical of ADNFLE; α4 subunit knockout mice showed no spontaneous seizures, but had nicotine-induced dystonic attacks [134]. α4-subunit S284L transgenic rats [135] show a more complete ADNFLE phenotype, with spontaneous attacks during slow-wave sleep, comprising paroxysmal arousals (frightened behavior), dystonic activity, and epileptic wandering. Notably, the pathogenic mechanisms are also different in these models: upon application of nicotine, GABAergic inhibition is increased in frontal cortex of S252F and +L264 knockin mice [133], whereas it is reduced in somatosensory cortex of S284L transgenic rats [135].
A recent study reported 4 unrelated families with a severe form of ADNFLE, in which patients had an earlier mean age of onset (6 years) than other ADNFLE forms, and frequently showed psychiatric features and intellectual disability [119]. They carried missense mutations in the sodium-gated potassium channel gene KCNT1, but no functional analysis was performed. Thus, the phenotype has features of EE and, interestingly, mutations of KCNT1 have also been identified in the EE malignant migrating partial seizures in infancy (see above).
Autosomal Dominant Temporal Lobe Epilepsy
Autosomal dominant temporal lobe epilepsy (ADTLE) is a form of autosomal dominant partial epilepsy associated with auditory symptoms and audiogenic seizures. The first clinical manifestations, usually occurring during childhood or adolescence, are auditory hallucinations, sometimes accompanied by vision or olfactory manifestations, or dizziness [136, 137]. Mutations in the leucine-rich, glioma-inactivated 1 (LGI1) gene have been associated with ADTLE in several families (Table 1) [138, 139].
The pathogenetic mechanism related to LGI1 mutations remains to be clarified. Functional inactivation of 1 allele leads to ADTLE, whereas silencing of both alleles has been observed in several high-grade gliomas [140]. The functions of the LGI1 protein (epitempin) are still poorly understood. LGI1 harbors a domain consisting of a 7-fold repeat of 44 amino acids, the epilepsy-associated repeat domain [141], which is common to MASS1, a large G-protein coupled receptor, mutated in the Frings murine model of audiogenic epilepsy [142]. It has been shown that LGI1 can be secreted, and that several ADLTE mutations cause secretion defects [143] and enhance glutamate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor functions by interacting extracellularly with the transmembrane protein ADAM22 [144], but it is not clear how lack of this function may cause epilepsy. However, it has also been shown that LGI1 might have presynaptic functions, reducing the inactivation of type A K+ current through intracellular interactions with Kv1.1K+ channels; ADLTE mutants do not exert this effect, resulting in fast inactivating A-type currents, an effect that could induce increased excitability, synaptic transmission, or both [143, 145]. Three independent LGI1 knock-out mouse models and a rat model (carrying the L385R LGI1 mutation produced by N-ethyl-N-nitrosourea – ENU - mutagenesis) have been generated that show a robust phenotype consisting of spontaneous and severe seizures [146]. Data obtained from animal models are more consistent with a direct presynaptic epileptogenic mechanism, whereas postsynaptic effects may be more involved in glutamatergic synapse maturation and dendritic pruning [147, 148].
Familial Focal Epilepsy with Variable Foci
In this form, affected members within a family can have seizures generated in different cortical regions (frontal, temporal, frontotemporal, parietal, or occipital), with onset that varies from infancy to adulthood. Affected individuals do not show cognitive deficits, although some family members exhibit intellectual disability. Recent studies have identified in some of these families mutations in Dishevelled, Egl-10 and Pleckstrin domain-containing protein, the function of which is still unclear, but it might be involved in membrane trafficking, G protein signaling and/or modulation of the mammalian target of rapamycin complex 1 (Table 1) [149–151]. Most mutations resulted in a truncated protein and are consistent with loss of function.
IGEs with Complex Inheritance
The IGEs represent 20–30% of all epilepsies and include a group of syndromes characterized by absence seizures, myoclonus, and generalized tonic–clonic seizures. There is partial overlap in age of onset, type and frequency of seizures, prognosis, and response to treatment. Among these disorders there are several subsyndromes, including childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy, and epilepsy with generalized tonic–clonic seizures alone. Generalized spike-wave activity and normal background are observed in EEGs. Among epilepsies with complex inheritance, IGEs have long been considered to be particularly suitable for genetic studies because they are common, have a relatively well-defined phenotype, and often occur in familial clusters. Close relatives of IGE probands have 4–10% risk of developing epilepsy [152]. Higher risk is seen in siblings and offspring, and is lower in second-degree relatives. As expected with polygenic inheritance, there is a rapid decrease of the risk in relatives as the distance from the affected individuals increases [153]. Twin studies have shown higher concordance for IGE in monozygotic than in dizygotic twins (0.76 vs 0.33), which is also consistent with polygenic inheritance [154]. Therefore, IGEs are thought to be genetic epilepsies with complex inheritance. It has been suggested that they might result from the interaction of two or more genes [155]. However, a high degree of complexity is operating: large-scale exome sequencing of ion channels reveals that rare missense variation in known Mendelian disease genes are equally prevalent in healthy individuals and in those with idiopathic generalized epilepsy, revealing that even deleterious ion channel mutations confer an uncertain risk to an individual depending on the other variants with which they are combined [156]. Although several chromosomal loci for different forms of IGEs have been identified, they have not often been replicated. Linkage studies on a large number of families with IGE have identified several susceptibility loci (18q, 2q, 3q, and 14q) [157, 158]. In rare families, pathogenic mutations in single genes have been reported. Mutations in the GABRG2 gene were identified in families with febrile seizures and childhood absence epilepsy [35, 36], a mutation in the GABRA1 gene was identified in a family with dominantly inherited juvenile myoclonic epilepsy [159], and mutations in CLCN2 in families with heterogeneous IGE phenotypes, including childhood absences epilepsy [160]. Rare variants in CACNA1H have been identified in childhood absence epilepsy and other generalized epilepsy phenotypes (Table 1) [161, 162]. It has consistently been shown that the Cacna1h variant R1584P is a susceptibility factor in a spontaneous rat model of absences [163]. Finally, the study of variants has provided some evidence that GABRD [164], ME2 [165], BRD2 [166], and NEDD4L [167] are susceptibility genes for IGE.
What We Have Learned from Experimental Models
Clinical and genetic studies cannot always fully elucidate genotype–phenotype correlations because they do not allow for inferring exhaustively the functional effects of a mutation and its pathogenic mechanism(s). Thus, functional analysis is important for clarifying genotype–phenotype correlations, which can facilitate diagnosis, genetic counseling, and studies aimed at developing effective therapies. Experimental models have helped to identify pathogenic mechanisms of some epileptic syndromes (e.g., SCN1A- or KCNQ2/KCNQ3-related), although for other syndromes there is still uncertainty after years of studies (e.g., for nAchR or LGI1 related).
Notably, experimental models of genetic epilepsy have made it possible to identify an early pre-epileptic period in which specific dysfunctions of neuronal network are present without generating behavioral seizures, which will eventually develop at a later stage [168]. This period can be considered of epileptogenesis, and the “presymptomatic” dysfunctions can therefore be targeted using therapeutic approaches that may inhibit or delay the transition to active epilepsy. These age-related periods may suggest specific developmental roles of the mutant protein, but might also depend on age-related factors that modulate the functional effects of mutations and possibly sustain disease progression. Studying the pathogenic mechanisms operating in the presymptomatic period might provide a window for therapeutic approaches that prevent seizures and the associated developmental defects. Such an early intervention would be particularly important because seizures can trigger pathological modifications that sustain and aggravate the epileptic state or interfere with brain functions, and that may be only indirectly related to the initial epileptogenic cause. Animal models can be used to test these hypotheses, for example using conditional knockin mice as the GABAA receptor γ2 subunit R43Q mouse model of GEFS+, in which seizures in adult mice are reduced when the expression of the mutant allele is suppressed during development [46]. A critical period for phenotype development has also been identified in transgenic mice expressing a dominant negative KCNQ2 mutant, which may model KCNQ2 EE [59].
Although performing high-throughput screening with mouse models may prove difficult, the recent development of simpler animal models (e.g., zebrafish) has identified possible effective compounds testing hundreds of small molecules [169], but these compounds have still to be validated in mammalian models
How Genetic and Functional Information Have Been Translated to Clinical Practice
Genetic testing can be offered for single-gene or Mendelian epilepsy syndromes, or epilepsy-associated disorders, if the gene has been identified. If not, empirical counseling can be offered, based on the type of epilepsy, mode of inheritance, and penetrance. Although we can now carry out preclinical and prenatal diagnosis in many cases, the difficulty in predicting the severity and prognosis of epilepsy in specific individuals, particularly in those with nonlesional epilepsies, uncertainties in genotype–phenotype relationships, reduced penetrance, and complex models of inheritance, complicate their use as diagnostic and predictive tools. Moreover, many treating clinicians are not yet familiar with the available tests, how to access them, and how to decide when and whether they should be used. In fact, not everybody is enthusiastic about their utility [170]. The International League Against Epilepsy Genetics Commission has recently proposed a framework for evaluating the utility of genetic testing [9, 171], concluding that it has high utility in some well-defined clinical contexts.
The correlations between phenotype and genotype in genetic epilepsies is rapidly changing in relation to new findings emerging from exome sequencing and the use of diagnostic panels as unexpected phenotypes become associated with mutations of specific genes and vice versa [109, 110, 172]. A constantly updated database will be essential to establish all the known gene mutations and polymorphisms, and their clinical correlates, so that genotype–phenotype correlations can be determined.
In some cases, genetic tests are becoming useful tools for achieving a more accurate diagnosis and, at least for some epilepsy syndromes, facilitating early diagnosis and helping treatment choices. For instance, mutations that truncate Nav1.1 have thus far a selective causative link to Dravet syndrome, and genetic tests can disclose these mutations in patients, allowing early diagnosis; in fact, genotyping is becoming a prerequisite in the diagnosis of Dravet syndrome. The exploitation of the information obtained by genetic tests can help treatment choices (e.g., excluding potentially harmful drugs in Dravet syndrome, in particular some Na+ channel blockers) [173] and guide genetic counseling and management.
However, to date, no new curative treatment has emerged from any genetic finding in epilepsy (with the possible exception of the ketogenic diet in GLUT1 deficiency syndrome), nor have the many pharmacogenomic studies in epilepsy yielded any widely applicable treatment advances (with the possible exception of HLA-B*1502 screening for carbamazepine toxicity). Moreover, functional studies performed with experimental models have not yet provided results that have been directly translated to the clinical setting. This is a disappointing lack of progress, reflecting as it does on the complex nature of epilepsies and their treatment. Counseling needs to consider these issues.
Conclusions
Most of the epilepsy syndromes we have reviewed are characterized by marked phenotypic and genetic heterogeneity; thus, it may be difficult to distinguish some of these epilepsies based on the genetic cause alone. This may be explained by pleiotropic expression of a single-gene mutation, modifying genes, or by several genes producing a similar phenotype, at times because they affect the same developmental or functional pathway. It will be essential to compare clinical and experimental studies in order to better disclose pathogenic mechanisms that lead to a form of epilepsy.
Another major area for future investigation concerns the interaction of genetic and developmental mechanisms of epilepsy in determining the age-dependent expression of many of the nonlesional syndromes and cognitive deterioration in EEs. These areas have been difficult to explore based on clinical grounds alone and have often remained the object of speculation. However, important insights are likely to be generated in the near future with the help of contemporary animal models in which predefined human gene mutations can be selectively expressed at different stages of brain development. The secondary changes prompted by early seizure activity in the physiology and anatomy of the affected neural circuits can now be analyzed and may represent a potential target for therapy. Also, growing evidence that genes regulating synaptic function, not just ion channel activity, are implicated in early-onset epilepsies, may help in the near future to better explore the role of synaptic plasticity in determining the age-related expression of specific epilepsy syndromes.
The next steps include identifying additional genes both in monogenic epilepsies and epilepsies with complex inheritance, genotype–phenotype correlations, and functional studies of the abnormal proteins in experimental models that reproduce real pathophysiological conditions. These studies may have practical applications for diagnosis, genetic counseling, and treatment.
It is hoped that a constantly updated database will be established for all the known gene mutations and polymorphisms, and their clinical correlates, so that genotype–phenotype correlations can be determined. This is the objective of the Human Variome project [174]. For example, over 800 mutations in SCN1A associated with the Dravet syndrome spectrum have now been identified, and the severity of the related phenotype can be predicted early with reasonable accuracy for some recurrent mutations for which sufficient numbers of clinical observations are available.
References
Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2011;51:676–685.
Heron SE, Scheffer IE, Berkovic SF, Dibbens LM, Mulley JC. Channelopathies in idiopathic epilepsy. Neurotherapeutics 2007;4:295–304.
Helbig I, Scheffer IE, Mulley JC, Berkovic SF. Navigating the channels and beyond: unravelling the genetics of the epilepsies. Lancet Neurol 2008;7:231–245.
Marini C, Scheffer IE, Crossland KM, et al. Genetic architecture of idiopathic generalized epilepsy: clinical genetic analysis of 55 multiplex families. Epilepsia 2004;45:467–478.
Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins. In: Wolf P (ed.) Epileptic seizures and syndromes. John Libbey, London, 1994, pp. 157–164.
Greenberg DA, Delgado-Escueta AV, Widelitz H, et al. Juvenile myoclonic epilepsy (JME) may be linked to the BF and HLA loci on human chromosome 6. Am J Med Genet 1988;31:185–192.
Steinlein OK, Mulley JC, Propping P, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor a4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 1995;11:201–203.
Mantegazza M, Rusconi R, Scalmani P, Avanzini G, Franceschetti S. Epileptogenic ion channel mutations: from bedside to bench and, hopefully, back again. Epilepsy Res 2010;92:1–29.
Hirose S, Scheffer IE, Marini C, et al. SCN1A testing for epilepsy: application in clinical practice. Epilepsia 2013;54:946–952.
Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet 2007;16:2892–2899.
Dravet C, Bureau M, Roger J. Severe myoclonic epilepsy in infants. In: Roger J, Dravet C, Bureau M, Dreifuss FE, Wolf P (eds) Epileptic syndromes in infancy, childhood and adolescence. John Libbey Eurotext, London, 1985, pp. 58–104.
Guerrini R, Dravet Ch. Severe epileptic encephatopathies of infancy. In: Engel J and Pedley TA (eds) Epilepsy. Raven Press, New York, 1997, pp. 2285–2302.
Guerrini R, Striano P, Catarino C, Sisodiya SM. Neuroimaging and neuropathology of Dravet syndrome. Epilepsia 2011;52(Suppl. 2):30–34.
Catarino CB, Liu JY, Liagkouras I, et al. Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology. Brain 2011;134:2982–3010.
Suls A, Claeys KG, Goossens D, et al. Microdeletions involving the SCN1A gene may be common in SCN1A-mutation-negative SMEI patients. Hum Mutat 2006;27:914–920.
Marini C, Scheffer IE, Nabbout R, et al. SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia 2009;50:1670–1678.
Patino GA, Claes LR, Lopez-Santiago LF, et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 2009;29:10764–10778.
Guerrini R, Oguni H. Borderline Dravet syndrome: a useful diagnostic category? Epilepsia 2011;52(Suppl. 2):10–12.
Fujiwara T, Sugawara T, Mazaki-Miyazaki E, et al. Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 2003;126:531–546.
Wallace RH, Scheffer IE, Barnett S, et al. Neuronal sodium channel a1 subunit (SCN1A) mutations in generalised epilepsy with febrile seizures plus. Am J Hum Genet 2001;68:859–865.
Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007;130:843–852.
Depienne C, Arzimanoglou A, Trouillard O, et al. Parental mosaicism can cause recurrent transmission of SCN1A mutations associated with severe myoclonic epilepsy of infancy. Hum Mutat 2006;27:389.
Marini C, Mei D, Helen Cross J, Guerrini R. Mosaic SCN1A mutation in familial severe myoclonic epilepsy of infancy. Epilepsia 2006;47:1737–1740.
Bechi G, Scalmani P, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Pure haploinsufficiency for Dravet syndrome Na(V)1.1 (SCN1A) sodium channel truncating mutations. Epilepsia 2011;53:87–100.
Ogiwara I, Iwasato T, Miyamoto H, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet 2007;22:4784–4804.
Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006;9:1142–1149.
Han S, Tai C, Westenbroek RE, et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012;489:385–390.
Higurashi N, Uchida T, Lossin C, et al. A human Dravet syndrome model from patient induced pluripotent stem cells. Mol Brain 2013;6:19.
Jiao J, Yang Y, Shi Y, et al. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum Mol Genet 2013;22:4241–4252.
Liu Y, Lopez-Santiago LF, Yuan Y, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol 2013;74:128–139.
Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120:479–490.
Wallace RH, Wang DW, Singh R, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na +-channel beta1 subunit gene SCN1B. Nat Genet 1998;19:366–370.
Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000;24:343–345.
Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007;48:1678–1685.
Wallace RH, Marini C, Petrou S, et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet 2001;28:49–52.
Baulac S, Huberfeld G, Gourfinkel-An I, et al. First genetic evidence of GABAA receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet 2001;28:46–48.
Tian M, Mei D, Freri E, et al. Impaired surface alphabetagamma GABA(A) receptor expression in familial epilepsy due to a GABRG2 frameshift mutation. Neurobiol Dis 2013;50:135–141.
Cestele S, Labate A, Rusconi R, et al. Divergent effects of the T1174S SCN1A mutation associated with seizures and hemiplegic migraine. Epilepsia 2013;54:927–935.
Rusconi R, Scalmani P, Cassulini RR, et al. Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant. J Neurosci 2007;27:11037–11046.
Rusconi R, Combi R, Cestele S, et al. A rescuable folding defective Nav1.1 (SCN1A) sodium channel mutant causes GEFS+: common mechanism in Nav1.1 related epilepsies? Hum Mutat 2009;30:E747–760.
Thompson CH, Porter JC, Kahlig KM, Daniels MA, George AL, Jr. Nontruncating SCN1A mutations associated with severe myoclonic epilepsy of infancy impair cell surface expression. J Biol Chem 2012;287:42001–42008.
Sugiura Y, Ogiwara I, Hoshi A, Yamakawa K, Ugawa Y. Different degrees of loss of function between GEFS+ and SMEI Nav 1.1 missense mutants at the same residue induced by rescuable folding defects. Epilepsia 2012;53:e111–114.
Cestele S, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Nonfunctional NaV1.1 familial hemiplegic migraine mutant transformed into gain of function by partial rescue of folding defects. Proc Natl Acad Sci U S A 2013;110:17546–17551.
Cestele S, Scalmani P, Rusconi R, Terragni B, Franceschetti S, Mantegazza M. Self-limited hyperexcitability: functional effect of a familial hemiplegic migraine mutation of the Nav1.1 (SCN1A) Na+ channel. J Neurosci 2008;28:7273–7283.
Tang B, Dutt K, Papale L, et al. A BAC transgenic mouse model reveals neuron subtype-specific effects of a generalized epilepsy with febrile seizures plus (GEFS+) mutation. Neurobiol Dis 2009;35:91–102.
Chiu C, Reid CA, Tan HO, et al. Developmental impact of a familial GABAA receptor epilepsy mutation. Ann Neurol 2008;64:284–293.
Zara F, Specchio N, Striano P, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia 2013;54:425–436.
Plouin P. Benign familial neonatal convulsions. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R (eds) Idiopathic generalized epilepsies; clinical, experimental and genetic aspects. John Libbey, London, 1994, pp. 39–44.
Steinlein OK, Conrad C, Weidner B. Benign familial neonatal convulsions: always benign? Epilepsy Res 2007;73:245–249.
Biervert C, Schroeder BC, Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy. Science 1998;279:403–406.
Charlier C, Singh NA, Ryan SG, et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet 1998;18:53–55.
Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet 1998;18:25–29.
Heron SE, Cox K, Grinton BE, et al. Deletions or duplications in KCNQ2 can cause benign familial neonatal seizures. J Med Genet 2007;44:791–796.
Singh NA, Westenskow P, Charlier C, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain 2003;126:2726–2737.
Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 2005;6:850–862.
Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 1998;396:687–690.
Maljevic S, Wuttke TV, Seebohm G, Lerche H. KV7 channelopathies. Pflugers Arch 2010;460:277–288.
Weber YG, Lerche H. Genetic mechanisms in idiopathic epilepsies. Dev Med Child Neurol 2008;50:648–654.
Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci 2005;8:51–60.
Singh NA, Otto JF, Dahle EJ, et al. Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J Physiol 2008;586:3405–3423.
Berkovic SF, Heron SE, Giordano L, et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol 2004;55:550–557.
Heron SE, Crossland KM, Andermann E, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 2002;360:851–852.
Scalmani P, Rusconi R, Armatura E, et al. Effects in neocortical neurons of mutations of the Na(v)1.2 Na+ channel causing benign familial neonatal-infantile seizures. J Neurosci 2006;26:10100–10109.
Liao Y, Deprez L, Maljevic S, et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 2010;133:1403–1414.
Vigevano F. Benign familial infantile seizures. Brain Dev 2005;27:172–177.
Szepetowski P, Rochette J, Berquin P, Piussan C, Lathrop GM, Monaco AP. Familial infantile convulsions and paroxysmal choreoathetosis: a new neurological syndrome linked to the pericentromeric region of human chromosome 16. Am J Hum Genet 1997;61:889–898.
Marini C, Conti V, Mei D, et al. PRRT2 mutations in familial infantile seizures, paroxysmal dyskinesia, and hemiplegic migraine. Neurology 2012;79:2109–2114.
Scheffer IE, Grinton BE, Heron SE, et al. PRRT2 phenotypic spectrum includes sporadic and fever-related infantile seizures. Neurology 2012;79:2104–2108.
Guerrini R, Mink JW. Paroxysmal disorders associated with PRRT2 mutations shake up expectations on ion channel genes. Neurology 2012;79:2086–2088.
Chen WJ, Lin Y, Xiong ZQ, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet 2011;43:1252–1255.
Stelzl U, Worm U, Lalowski M, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005;122:957–968.
Guipponi M, Rivier F, Vigevano F, et al. Linkage mapping of benign familial infantile convulsions (BFIC) to chromosome 19q. Hum Mol Genet 1997;6:473-477.
Vanmolkot KR, Kors EE, Hottenga JJ, et al. Novel mutations in the Na+, K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol 2003;54:360–366.
Zhou X, Ma A, Liu X, et al. Infantile seizures and other epileptic phenotypes in a Chinese family with a missense mutation of KCNQ2. Eur J Pediatr 2006;165:691–695.
Striano P, Bordo L, Lispi ML, et al. A novel SCN2A mutation in family with benign familial infantile seizures. Epilepsia 2006;47:218–220.
Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 2012;71:15–25.
Weckhuysen S, Ivanovic V, Hendrickx R, et al. Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology 2013;81:1697–1703.
Miceli F, Soldovieri MV, Ambrosino P, et al. Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc Natl Acad Sci U S A 2013;110:4386–4391.
Orhan G, Bock M, Schepers D, et al. Dominant-negative Effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol 2013 Dec 7 [Epub ahead of print].
Nakamura K, Kato M, Osaka H, et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 2013;81:992–998.
Touma M, Joshi M, Connolly MC, et al. Whole genome sequencing identifies SCN2A mutation in monozygotic twins with Ohtahara syndrome and unique neuropathologic findings. Epilepsia 2013;54:e81-85.
Lossin C, Shi X, Rogawski MA, Hirose S. Compromised function in the Na(v)1.2 Dravet syndrome mutation R1312T. Neurobiol Dis 2012;47:378–384.
Planells-Cases R, Caprini M, Zhang J, et al. Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice. Biophys J 2000;78:2878–2891.
Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–781.
Marini C, Mei D, Parmeggiani L, et al. Protocadherin 19 mutations in girls with infantile-onset epilepsy. Neurology 2010;75:646–653.
Marini C, Darra F, Specchio N, et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 2012;53:2111–2119.
Depienne C, Bouteiller D, Keren B, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 2009;5(2):e1000381.
Ohira R, Zhang YH, Guo W, et al. Human ARX gene: genomic characterization and expression. Mol Genet Metab 2002;77:179–188.
Gecz J, Cloosterman D, Partington M. ARX: a gene for all seasons. Curr Opin Genet Dev 2006;16:308–316.
Guerrini R, Moro F, Kato M, et al. Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology 2007;69:427–433.
Marsh E, Fulp C, Gomez E, et al. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain 2009;132:1563–1576.
Kato M, Das S, Petras K, et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum Mutat 2004;23:147–159.
Stromme P, Mangelsdorf ME, Scheffer IE, Gecz J. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev 2002;24:266–268.
Wallerstein R, Sugalski R, Cohn L, Jawetz R, Friez M. Expansion of the ARX spectrum. Clin Neurol Neurosurg 2008;110:631–634.
Shinozaki Y, Osawa M, Sakuma H, et al. Expansion of the first polyalanine tract of the ARX gene in a boy presenting with generalized dystonia in the absence of infantile spasms. Brain Dev 2009;31:469–472.
Kato M, Saitoh S, Kamei A, et al. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am J Hum Genet 2007;81:361–366.
Mari F, Azimonti S, Bertani I, et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet 2005;14:1935–1946.
Kalscheuer VM, Tao J, Donnelly A, et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet 2003;72:1401–1411.
Buoni S, Zannolli R, Colamaria V, et al. Myoclonic encephalopathy in the CDKL5 gene mutation. Clin Neurophysiol 2006;117:223–227.
Archer HL, Evans J, Edwards S, et al. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet 2006;43:729–734.
Elia M, Falco M, Ferri R, et al. CDKL5 mutations in boys with severe encephalopathy and early-onset intractable epilepsy. Neurology 2008;71:997–999.
Weaving LS, Christodoulou J, Williamson SL, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 2004;75:1079–1093.
Melani F, Mei D, Pisano T, et al. CDKL5 gene-related epileptic encephalopathy: electroclinical findings in the first year of life. Dev Med Child Neurol 2011;53:354–360.
Mei D, Marini C, Novara F, et al. Xp22.3 genomic deletions involving the CDKL5 gene in girls with early onset epileptic encephalopathy. Epilepsia 2010;51:647–654.
Ohtahara S, Yamatogi Y. Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res 2006;70(Suppl. 1):S58-67.
Djukic A, Lado FA, Shinnar S, Moshe SL. Are early myoclonic encephalopathy (EME) and the Ohtahara syndrome (EIEE) independent of each other? Epilepsy Res 2006;70(Suppl. 1):S68–76.
Mignot C, Moutard ML, Trouillard O, et al. STXBP1-related encephalopathy presenting as infantile spasms and generalized tremor in three patients. Epilepsia 2011;52:1820–1827.
Milh M, Villeneuve N, Chouchane M, et al. Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia 2011;52:1828–1834.
Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008;40:782–788.
Lemke JR, Lal D, Reinthaler EM, et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet 2013;45:1067–1072.
Lesca G, Rudolf G, Bruneau N, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet 2013;45:1061–1066.
Carvill GL, Regan BM, Yendle SC, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet 2013;45:1073–1076.
Lemke JR, Hendrickx R, Geider K, et al. GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy. Ann Neurol 2014;75:147–154.
Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221.
Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013;45:825–830.
Suls A, Jaehn JA, Kecskes A, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 2013;93:967–975.
Veeramah KR, O’Brien JE, Meisler MH, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 2012;90:502–510.
Barcia G, Fleming MR, Deligniere A, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 2012;44:1255–1259.
Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 2012;44:1188–1190.
Scheffer IE, Bhatia KP, Lopes-Cendes I, et al. Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain 1995;118:61–73.
Picard F, Baulac S, Kahane P, et al. Dominant partial epilepsies. A clinical, electrophysiological and genetic study of 19 European families. Brain 2000;123:1247–1262.
Ryvlin P, Minotti L, Demarquay G, et al. Nocturnal hypermotor seizures, suggesting frontal lobe epilepsy, can originate in the insula. Epilepsia 2006;47:755–765.
Marini C, Guerrini R. The role of the nicotinic acetylcholine receptors in sleep-related epilepsy. Biochem Pharmacol 2007;74:1308–1314.
Steinlein OK, Magnusson A, Stoodt J, et al. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum Mol Genet 1997;6:943–948.
De Fusco M, Becchetti A, Patrignani A, et al. The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet 2000;26:275–276.
Liu H, Lu C, Li Z, et al. The identification of a novel mutation of nicotinic acetylcholine receptor gene CHRNB2 in a Chinese patient: Its possible implication in non-familial nocturnal frontal lobe epilepsy. Epilepsy Res 2011;95:94–99.
Chen Y, Wu L, Fang Y, et al. A novel mutation of the nicotinic acetylcholine receptor gene CHRNA4 in sporadic nocturnal frontal lobe epilepsy. Epilepsy Res 2009;83:152–156.
Aridon P, Marini C, Di Resta C, et al. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am J Hum Genet 2006;79:342–350.
Phillips HA, Favre I, Kirkpatrick M, et al. CHRNB2 Is the Second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet 2001;68:225–231.
Steinlein OK, Hoda JC, Bertrand S, Bertrand D. Mutations in familial nocturnal frontal lobe epilepsy might be associated with distinct neurological phenotypes. Seizure 2012;21:118–123.
Phillips HA, Scheffer IE, Crossland KM, et al. Autosomal dominant nocturnal frontal lobe epilepsy: genetic heterogeneity and a probable second locus at 15q24. Am J Hum Genet 1998;63:1108–1116.
Bertrand D, Picard F, Le Hellard S, et al. How mutations in the nAChRs can cause ADNFLE epilepsy. Epilepsia 2002;43(Suppl. 5):112–122.
Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A 2006;103:19152–19157.
Teper Y, Whyte D, Cahir E, et al. Nicotine-induced dystonic arousal complex in a mouse line harboring a human autosomal-dominant nocturnal frontal lobe epilepsy mutation. J Neurosci 2007;27:10128–10142.
Zhu G, Okada M, Yoshida S, et al. Rats harboring S284L Chrna4 mutation show attenuation of synaptic and extrasynaptic GABAergic transmission and exhibit the nocturnal frontal lobe epilepsy phenotype. J Neurosci 2008;28:12465–12476.
Ottman R, Risch N, Hauser WA, et al. Localization of a gene for partial epilepsy to chromosome 10q. Nat Genet 1995;10:56–60.
Michelucci R, Poza JJ, Sofia V, et al. Autosomal dominant lateral temporal epilepsy: clinical spectrum, new epitempin mutations, and genetic heterogeneity in seven European families. Epilepsia 2003;44:1289–1297.
Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 2002;30:335–341.
Ottman R, Winawer MR, Kalachikov S, et al. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology 2004;62:1120–1126.
Chernova OB, Somerville RP, Cowell JK. A novel gene, LGI1, from 10q24 is rearranged and downregulated in malignant brain tumors. Oncogene 1998;17:2873–2881.
Scheel H, Tomiuk S, Hofmann K. A common protein interaction domain links two recently identified epilepsy genes. Hum Mol Genet 2002;11:1757–1762.
Skradski SL, Clark AM, Jiang H, White HS, Fu YH, Ptacek LJ. A novel gene causing a mendelian audiogenic mouse epilepsy. Neuron 2001;31:537–544.
Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet 2005;14:1613–1620.
Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science 2006;313:1792–1795.
Schulte U, Thumfart JO, Klocker N, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron 2006;49:697–706.
Baulac S, Ishida S, Mashimo T, et al. A rat model for LGI1-related epilepsies. Hum Mol Genet 2012;21:3546–3557.
Zhou YD, Lee S, Jin Z, Wright M, Smith SE, Anderson MP. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med 2009;15:1208–1214.
Yu YE, Wen L, Silva J, et al. Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum Mol Genet 2010;19:1702–1711.
Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 2013;45:546–551.
Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 2013;45:552–555.
Bar-Peled L, Chantranupong L, Cherniack AD, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013;340:1100–1106.
Annegers JF, Hauser WA, Anderson VE, Kurland LT. The risks of seizure disorders among relatives of patients with childhood onset epilepsy. Neurology 1982;32:174–179.
Risch N. Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet 1990;46:222–228.
Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol 1998;43:435–445.
Berkovic SF, Scheffer IE. Genetics of the epilepsies. Epilepsia 2001;42(Suppl. 5):16–23.
Klassen T, Davis C, Goldman A, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 2011;145:1036–1048.
Durner M, Keddache MA, Tomasini L, et al. Genome scan of idiopathic generalized epilepsy: evidence for major susceptibility gene and modifying genes influencing the seizure type. Ann Neurol 2001;49:328–335.
Sander T, Schulz H, Saar K, et al. Genome search for susceptibility loci of common idiopathic generalised epilepsies. Hum Mol Genet 2000;9:1465–1472.
Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet 2002;31:184–189.
D’Agostino D, Bertelli M, Gallo S, et al. Mutations and polymorphisms of the CLCN2 gene in idiopathic epilepsy. Neurology 2004;63:1500–1502.
Chen Y, Lu J, Pan H, al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol 2003;54:239–243.
Heron SE, Phillips HA, Mulley JC, et al. Genetic variation of CACNA1H in idiopathic generalized epilepsy. Ann Neurol 2004;55:595–596.
Powell KL, Cain SM, Ng C, et al. A Cav3.2 T-type calcium channel point mutation has splice-variant-specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J Neurosci 2009;29:371–380.
Dibbens LM, Feng HJ, Richards MC, et al. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet 2004;13:1315–1319.
Greenberg DA, Cayanis E, Strug L, et al. Malic enzyme 2 may underlie susceptibility to adolescent-onset idiopathic generalized epilepsy. Am J Hum Genet 2005;76:139–146.
Pal DK, Evgrafov OV, Tabares P, Zhang F, Durner M, Greenberg DA. BRD2 (RING3) is a probable major susceptibility gene for common juvenile myoclonic epilepsy. Am J Hum Genet 2003;73:261–270.
Dibbens LM, Ekberg J, Taylor I, et al. NEDD4-2 as a potential candidate susceptibility gene for epileptic photosensitivity. Genes Brain Behav 2007;6:750–755.
Liautard C, Scalmani P, Carriero G, de Curtis M, Franceschetti S, Mantegazza M. Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome. Epilepsia 2013;54:1251–1261.
Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun 2013;4:2410.
Delgado-Escueta AV, Bourgeois BF. Debate: Does genetic information in humans help us treat patients? PRO—genetic information in humans helps us treat patients. CON—genetic information does not help at all. Epilepsia 2008;49(Suppl. 9):13–24.
Ottman R, Hirose S, Jain S, et al. Genetic testing in the epilepsies—report of the ILAE Genetics Commission. Epilepsia 2010;51:655–670.
Lemke JR, Riesch E, Scheurenbrand T, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012;53:1387–1398.
Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia 1998;39(Suppl.):508–512.
Cotton RG, Auerbach AD, Axton M, et al. GENETICS. The Human Variome Project. Science 2008;322:861–862.
Acknowledgement
Research in Renzo Guerrini’s and Carla Marini’s team and laboratories is funded by Europen Union FP7 Grant no. 602531 (“DESIRE”), and Italian Ministry of Health and Tuscany Region Grant RF 2009–1525669. Research in Massimo Mantegazza’s laboratory is funded by LabEx “ICST”, CNRS-PICS “NavRole”, and Europen Union FP7 Grant no. 602531 (“DESIRE”).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 338 kb)
Rights and permissions
About this article
Cite this article
Guerrini, R., Marini, C. & Mantegazza, M. Genetic Epilepsy Syndromes Without Structural Brain Abnormalities: Clinical Features and Experimental Models. Neurotherapeutics 11, 269–285 (2014). https://doi.org/10.1007/s13311-014-0267-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-014-0267-0