
Abstract
Introduction
Glioblastoma multiform (GBM) is a neural stem cell (NSC)-derived malignant brain tumor with complex genetic alterations challenging clinical treatments. FAM72 is a NSC-specific protein comprised of four paralogous genes (FAM72 A-D) in the human genome, but its functional tumorigenic significance is unclear.
Methods
We conducted an in-depth expression and somatic mutation data analysis of FAM72 (A-D) in GBM using the comprehensive human clinical cancer study database cBioPortal [including The Cancer Genome Atlas (TCGA)].
Results
We established a FAM72 transcription profile across TCGA correlated with the expression of the proliferative marker MKI67 and a tissue-specific gene-mutation signature represented by pivotal genes involved in driving the cell cycle. FAM72 paralogs are overexpressed in cancer cells, specifically correlating with the mitotic cell cycle genes ASPM, KIF14, KIF23, CENPE, CENPE, CEP55, SGO1, and BUB1, thereby contributing to centrosome and mitotic spindle formation. FAM72 expression correlation identifies a novel GBM-specific gene set (SCN9A, MXRA5, ADAM29, KDR, LRP1B, and PIK3C2G) in the de novo pathway of primary GBM predestined as viable targets for therapeutics.
Conclusion
Our newly identified primary GBM-specific gene-mutation signature, along with FAM72, could thus provide a new basis for prognostic biomarkers for diagnostics of GBM and could serve as potential therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is a highly prevalent disease and a major cause of mortality worldwide. Globally, around 14 million people were diagnosed and 8.2 million perished due to cancer in 2012 [1]. Glioblastoma multiforme (GBM) is the most common malignant tumor of the central nervous system (CNS) arising from neural stem cells (NSCs) [2]. FAM72 is a NSC-specific gene usually not expressed in non-neuronal tissues [3]. Expression of FAM72A has been observed in the dentate gyrus of the hippocampus, where it is associated with maintenance of NSCs [4, 5]. FAM72A displays an on/off switch mechanism in NSCs, such that when FAM72 is silenced, the NSCs differentiate into post-mitotic neurons [4]. In humans, FAM72 consists of four human-specific paralogs (A-D) that are specifically associated as gene pairs with Slit-Robo Rho GTPase-activating protein 2 (SRGAP2) paralogs on chromosome 1 (chr1) (Supplementary Figs. S1 and S2), where they appear to be pivotal players as |-FAM72–SRGAP2-| master gene pairs of brain plasticity for control of higher brain functions [6,7,8,9].
Preliminary data indicate that FAM72 is overexpressed in cancerous tissues, but its exact role in oncogenesis has not yet been elucidated [5, 10,11,12]. We thus conducted an in-depth investigation of the human FAM72 (A-D) paralogs using the comprehensive public cBioPortal human cancer database in order to determine the specific role of FAM72 in oncogenesis, particularly in GBM. We analyzed the tissue-specific expression and mutation of the paralogs as well as the role of proto-oncogenes and tumor suppressor genes in tissues with high FAM72 expression. We determined the putative FAM72-promotor-specific transcription factor (TF)-binding sites (BS) (TFBS) using the Ensembl and JASPAR databases in order to define the regulators responsible for FAM72 expression.
Our study reveals that the expression of tumorigenic FAM72 (A-D) paralogs is activated by a cancer tissue-specific gene-mutation signature, represented by pivotal mutated cell cycle driver proto-oncogenes or tumor suppressors, and highly correlates with the expression of the proliferative marker MKI67 and genes associated with the M-phase of the cell cycle. These cell cycle genes and the FAM72 (A-D) paralogs share TFBS motifs, implying that they are co-regulated. Our study further indicates that FAM72 plays a major role in driving the cell through the mitotic cell cycle stage, especially in mitotic spindle function, and its expression profile reveals a new primary GBM-specific gene mutation signature.
Materials and methods
Human cancer patient data sources
Publicly available human cancer genome datasets (http://www.cbioportal.org/) [13] were analyzed for mutations and mRNA expression data. cBioPortal is a human cancer genomics database that contains 169 studies with 40,408 human subject samples (as of January 2018) covering 29 types of tissue. cBioPortal combines data from The Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/), the International Cancer Genome Consortium (ICGC; https://icgc.org/), the Wellcome Trust Sanger Institute’s (WTSI) Cancer Genome Project (http://www.sanger.ac.uk/research/projects/cancergenome/), and the Cancer Genomics Hub (CGHub; https://cghub.ucsc.edu/). TCGA is a collaborative effort between the National Cancer Institute (NCI; http://www.cancer.gov/) and the National Human Genome Research Institute (NHGRI; https://www.genome.gov/). FAM72A, B, C, and D were queried in the cBioPortal database (provisional data set) across all available tissues to screen for gene mutations. Changes in the protein sequences of the paralogs were analyzed for the presence of mutation hotspots.
Human cancer tissue-specific mRNA expression analysis of FAM72 (A-D) paralogs
mRNA expression z-scores (RNA sequencing (RNASeq V1/V2) or microarray) were locally computed on the foundation of raw expression data available on cBioPortal from all available TCGA studies across various human cancer tissues. Studies not containing expression data were excluded from analysis (Supplementary Table S1). TCGA studies on cBioPortal contain ‘provisional’ and ‘published’ data, where published data is a static subset of provisional data. We chose to use the provisional studies to maximize the number of samples. A z-score is a statistical measurement indicating how many standard deviations the element is from the mean. The formula is \({\text{z}}=({\text{X}} - {\text{m}})/\sigma\), where z is the z-score, X is the value of the element, m is the numerical mean of the population, and σ is the standard deviation [14]. The relative expression changes across cancer tissues compared to a normal control set were visualized using the Wanderer interactive viewer [15]. Control was either adjacent non-cancer tissues or leukocytes. mRNA expression data for FAM72C was unavailable for the tissues of interest on cBioPortal; hence, it could not be considered in the expression data analysis. Each tissue represented one TCGA study unless otherwise indicated. mRNA expression data was sorted by tissue, and the z-score was normalized for all samples so that they sum to zero. Linear regression was determined first between the FAM72 (A-D) paralogs and then between all available genes in the studies, for all available samples across all tissues. The regression curve analysis was visualized with the Python-based Bokeh online visualization tool [16].
Human cancer tissue-specific gene mutation—FAM72 (A-D) paralog mRNA expression correlation analysis—establishing the tissue-specific gene-mutation signature
Complete mutation data for all genes was retrieved from all available TCGA studies across various human cancer tissues. Each tissue was represented by one TCGA study, unless otherwise indicated. Data were sorted by the mRNA expression z-score of FAM72 (A-D) paralogs and then by the number of gene-specific mutations across individual tissue-specific studies. The five most frequently mutated genes in each tissue-specific study were presented as the cancer tissue-specific gene-mutation signature. Mutations in well-known non-oncogenic genes, as described by Lawrence et al. and Greenman et al., were not considered for analysis [17, 18]. Data were visualized with the Xena functional genomics explorer [19]. Null samples and some tissues (such as mesothelioma (MESO), colorectal adenocarcinoma (COADREAD), and uterine endometrial carcinoma (UCEC)) were excluded from the Xena visualization analysis due to mismatched or insufficient data on cBioPortal. As the Xena genomics explorer utilizes its own methods to process TCGA data, there is an absence of somatic mutation data for some genes in certain tissues such as cholangiocarcinoma (CHOL), head and neck squamous cell carcinoma (HNSC), kidney chromophobe (KICH), hepatocellular carcinoma (LIHC), pheochromocytoma and paraganglioma (PCPG), prostate adenocarcinoma (PRAD) and testicular germ cell cancer (TGCT), which is present in cBioPortal. In such cases of mismatches between Xena and cBioPortal somatic mutation data, we have relied on cBioPortal for our analysis. Clinical data from the TCGA GBM study (provisional data set) was retrieved from cBioPortal for patient-gene-specific analyses.
Human cancer tissue-specific gene mutation—FAM72 (A-D) paralog mRNA expression correlation analysis visualized by the bucket method
The mRNA expression z-scores for FAM72 (A-D) paralogs were grouped in buckets with a size of 0.7 z-score units and correlated with genes showing high numbers of tissue-specific gene mutations. The y-axis denotes the z-score buckets for the selected FAM72 gene. Genes whose mutation numbers are to be visualized lie on the x-axis. The data were visualized with the Python-based Bokeh interactive visualization tool [16]. Numbers of mutations in a gene in the samples within a bucket were denoted by a color code. Color intensity of the buckets is directly proportional to number of samples, while the colors visualize the relation of samples with a mutation to the total number of samples. Brighter colors indicate more samples in the bucket, while paler colors indicate fewer samples. Colors tending to the red side of the spectrum indicate increase in samples with a mutation in relation to the total number of samples in the bucket. Colors tending to the blue side of the spectrum indicate decreasing numbers of samples with a mutation in the bucket. Black bands denote absence of mutations or lack of expression data in the gene, while bright grey bands indicate absence of samples within the group. Bright pink boxes indicate that only one sample is present in the bucket that contains one mutation in the gene of interest.
Gene-specific survival analysis
The prognostic significance of selected genes from GBM was analyzed using available Kaplan–Meier curves from the cBioPortal database (provisional data set) and comparison was performed by log-rank test [20].
In-silico determination of the regulatory mechanism of human FAM72: Genomic transcription factor (TF)-binding site (TFBS) analysis for FAM72 using JASPAR
TFs are proteins that regulate gene transcription by binding to genome sequences usually located upstream of the coding sequence (CDS), thereby either enhancing or inhibiting the binding of RNA polymerase II and leading to a corresponding increase or decrease in gene transcription [21]. TFs are expected to bind upstream of the two genes FAM72 and SRGAP2 and thus to the intergenic region between the CDS of the specific FAM72 and SRGAP2 gene pair. The |-FAM72–SRGAP2-| gene pairs were extracted from NC_000001.11 (chr 1, genome reference consortium human build 38 patch release 13 (GRCh38.p13 primary assembly) taking the regions 206205970–206202026 (|-FAM72A–SRGAP2A-|), 121187446–121183490 (|-FAM72B–SRGAP2C-|), 143972638–143971142 (|-FAM72C–SRGAP2D-|) and 145092902–145096847 (|-FAM72D–SRGAP2B-|) from the National Center for Biotechnology Information (NCBI) database. These were used for TFBS prediction using the JASPAR database. Briefly, the JASPAR CORE collection is a high-quality database of curated profiles of experimentally validated eukaryotic TFBS derived from published data [22]. PHYLOFACTS is an online database created by the Berkeley phylogenomics group [23]. The database integrates structural, phylogenic, and alignment information for more than 15,000 protein families [23]. The intergenic regions between the |-FAM72–SRGAP2-| gene pairs are similar to each other [6]; hence, we used the |-SRGAP2A–FAM72A-| intergenic region as the model for TFBS analysis. The intergenic region between the CDS of the |-SRGAP2A–FAM72A-| gene pair was aligned with all motifs of the JASPAR CORE-collection and PHYLOFACTS-collection, labeled with respect to species “human”. For the alignment process, we relied on the motifs-module of BioPython [24]. The position-weighed matrices of each TF were assigned a score and only those TFs that showed a 100% match were selected.
Human FAM72 genomic TFBS prediction with the Ensembl database
Ensembl is a joint project between the European Bioinformatics Institute (EBI), an outstation of the European Molecular Biology Laboratory (EMBL), and the WTSI [25]. The Ensembl browser (March 2016 version) was used to visualize the various putative features present in the promoter region of |-SRGAP2–FAM72-|, which are the intergenic regions between SRGAP2 and FAM72 paralogs (206205970–206202026 for |-FAM72A–SRGAP2A-|, 121187446–121183490 for |-FAM72B–SRGAP2C-|, 143972638–143971142 for |-FAM72C–SRGAP2D-| and 145092902–145096847 for |-FAM72D–SRGAP2B-|).
Results
FAM72 (A-D) paralog mutations in human cancer tissues
We analyzed the distribution of all mutations in the human FAM72 (A-D) paralogs across all available human cancer tissues from 169 studies, comprising 29 tissues and 40,408 patient samples. A total of 34 non-synonymous mutations and three synonymous mutations in all four human FAM72 paralogs across all tissues were identified, with the highest number observed in FAM72A (Supplementary Table S2). The maximum numbers of mutations were observed in uterus and lung cancer tissues, with six cases in each tissue. Cholangiocarcinoma, skin, gallbladder, and pheochromocytoma and paraganglioma (PCPG) each showed only one case of FAM72 mutation (Supplementary Fig. S3).
We observed that missense mutations were predominant, representing 88% of all mutations across all tissues. In tissue-specific cases, the four tissues that showed mutations other than missense were PCPG (one case, nonsense mutation), liver (one case, frameshift insertion), kidney (one case, frameshift insertion), and stomach (one case, frameshift insertion) (Supplementary Fig. S4). In order to determine the significance of observed mutations in all four human FAM72 paralogs, we compared them with the mutations in an established proto-oncogene, the tumor protein p53 (TP53). Mutations in FAM72 paralogs were rare compared with those in TP53 (Supplementary Fig. S5).
Amino acid changes in FAM72 (A-D) paralogs in human cancer tissues
We analyzed the amino acid (AA) changes in mutated human FAM72 (A-D) paralogs across all human cancer tissues, both by individually affected AAs and by the frequency of mutation of an AA at a specific position (Supplementary Tables S3–S6). We found that, in 14% of cases, mutations occurred at serine residues (Supplementary Fig. S6). With regard to the frequency of mutations at a specific position, we found no bias toward a particular AA.
Human cancer tissue-specific mRNA expression changes in FAM72 (A-D) paralogs
Increased expression of FAM72A has been reported previously from breast, lung, ovary, kidney, and uterine cells [5, 10]. Our detailed analysis of FAM72 paralog expression revealed a significant increase in mean expression levels in human tumors as compared to control tissues (Supplementary Figs. S7a–S9b, Supplementary Tables S7–S9). The exceptions were kidney chromophobe (KICH), PCPG, and skin cutaneous melanoma (SKCM), where tumor samples show a decrease or equivalence in expression.
Correlation of FAM72 (A-D) paralog expression with expression of cell proliferation marker gene MKI67 across various human cancer tissues
We performed a comparative expression analysis of FAM72 (A-D) paralogs with the established marker of proliferation Ki-67 gene (MKI67) [26]. As seen in Supplementary Fig. S10, the expression of MKI67 is highly correlated with that of FAM72A, B, and D across nearly all human cancer tissues. This high correlation implies that MKI67 and FAM72 paralogs are co-expressed in proliferating cells, particularly cancer cells.
Correlation of FAM72 (A-D) paralog expression with expression of selected cell cycle-specific genes across various human cancer tissues
As FAM72 mRNA expression was elevated in all proliferating cells across various human cancer tissues, we determined whether the expression of FAM72A was correlated to the expression of specific cell cycle genes. Accordingly, we selected cell cycle genes across the entire cell cycle and correlated their expression across the selected tissues (Supplementary Fig. S11).
Correlation of FAM72 (A-D) paralog expression with expression of selected cell cycle-phase-specific genes across various human cancer tissues
Noting that FAM72 paralogs were co-expressed with genes expressed in the G2/M-phases of the cell cycle, we further narrowed the list of genes to those specifically expressed during the M-phase. We found that FAM72 (A-D) paralogs showed the highest correlation with each other and genes specifically involved in the M-phase of the cell cycle (Fig. 1), but not with genes involved in the G1-/S-phases (Supplementary Fig. S12). FAM72A showed the highest expression correlation with the kinesin family member 14 (KIF14) (Fig. 1).

Correlation of FAM72A expression with expression of marker of proliferation MKI67 and selected genes specifically involved in the M-phase of the cell cycle across various TCGA human cancer tissues. The x-axis represents mRNA expression z-scores for FAM72A, and the y-axis represents mRNA expression z-scores for MKI67 and selected cell cycle genes. The blue dots represent FAM72 sample values, and pink dots represent mutations in MKI67. The key explaining the color code for each linear regression graph is on the bottom left. The mRNA expression correlations between FAM72A and selected genes are consistently high across all selected cancer tissues. The highest mRNA expression correlation is observed with KIF14, indicating FAM72A mRNA co-expression with KIF14 during the M-phase of the cell cycle, in particular the prophase to metaphase stages. Sample size, standard error, slope, and p-values for the graphs are provided in Supplementary Table S12
Correlation of FAM72 paralog expression with proto-oncogenes and tumor suppressor genes frequently mutated in GBM: the GBM-specific gene-mutation signature
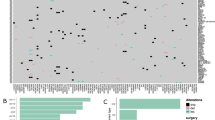
With FAM72 specifically expressed in brain NSCs, we looked at the aggressive cancer GBM in the brain (Fig. 2). The largest number of mutations was observed for the proto-oncogene epidermal growth factor receptor (EGFR) and the tumor suppressor TP53 across all samples in the TCGA GBM study, yet the mRNA expression level of FAM72 paralogs was not correlated with these mutations (Fig. 2a). Figure 2b corroborates this observation, where mutations in TP53, phosphatase and tensin homolog (PTEN), NF1, SPTA1 and EGFR are spread through the samples, with no clear single driver oncogene that causes cellular proliferation. An accumulation of mutations across various genes may be responsible for GBM [27]. The MKI67-FAM72 mRNA expression correlation graph clearly demonstrates that FAM72 is highly expressed in proliferating GBM cells (Fig. 2c). We compared the list of frequently mutated genes overall with genes which were mutated only in deceased patients. We found six genes, SCN9A, MXRA5, ADAM29, KDR, LRP1B, and PI3KC2G, which showed a significant number of mutations and thereby defining a novel GBM-specific gene mutation signature (Fig. 2g). We also observed a potential gender effect: mutations in ADAM29 were observed mainly in men, with a ratio of eight male patients to one female patient (8:1). Mutations in SCN9A also tends to affect rather male patients (8:3; Supplementary data file 2), which hints at a gender-specific role of these genes in GBM. Patient data and list of genes mutated solely in deceased patients is provided in Supplementary data file 2.

GBM-specific gene-mutation—FAM72-expression signature. Correlation between mRNA expression of human FAM72 (A-D) paralogs and proto-oncogenes/tumor suppressor genes frequently mutated in the TCGA GBM study, comprised of 165 samples containing mRNA expression and mutation data. a Comparison between the tumor samples sorted by sample in descending order of FAM72A expression (on the left hand), and the GBM-specific gene-mutation signature represented by the five most frequently mutated genes (EGFR, TP53, PTEN, NF1, SPTA1) in the same GBM samples sorted by number of mutations (on the right hand; sorted from left to right). The FAM72A expression and gene-mutation signature comparison was visualized with the Xena interactive browser [19]. Red bands indicate increased expression, green bands indicate decreased expression, and black bands indicate no change in expression. Blue dots (a right hand) represent missense or in-frame mutations in the indicated gene in a sample, while red dots represent nonsense or frameshift indel mutations in that gene in the sample. EGFR is the most frequently mutated gene in GBM. The data demonstrate that accumulation of mutations in cancer-driver genes, such as EGFR, PTEN, TP53, and neurofibromin 1 (NF1), leads to oncogenesis. b Bucket-wise distribution of mutations in the five most frequently mutated genes in GBM, sorted by FAM72A expression. The grey areas in the heat maps and bucketed diagrams indicate lack of data. c High gene expression correlation between FAM72A and the proliferative marker MKI67 as well as other M-phase-specific cell cycle genes indicates that FAM72A is highly expressed in proliferating GBM cells. Standard error, slope, and p-values for the graphs are provided in Supplementary Table S13. d Mutations in selected genes across all 287 patients with mutation data in the GBM study. e Most frequently mutated genes in 59 survivors with mutation data. f Most frequently mutated genes in 223 deceased patients with mutation data. g Somatic mutations observed uniquely in deceased patients highlights a novel GBM-specific gene-mutation signature. h OncoPrint data from the GBM study on cBioPortal for visualization of the relationship between somatic mutations in genes from g and survival of patient. d reflects genes mentioned in a–c. Mutations in the genes stated in d–f are well-established oncogenic drivers. However, the genes mentioned in g are—thus far—not reported to be oncogenic drivers, but may assist in metastasis. OncoPrint data in h clearly shows that mutations occurred in separate patients, all of whom are deceased, and did not overlap, implying that all genes from this novel gene-mutation signature from g could have each played a pivotal lethal role as primary driver oncogene in the de novo pathway of primary GBM or in metastasis in conjunction with other driver oncogenes
Correlation of FAM72 (A-D) paralog mRNA expression and genes frequently mutated in other human cancer tissues—other tissue-specific gene-mutation signatures
Since mutations in a single driver gene or a combination of genes (the tissue-specific gene-mutation signature) appeared to be responsible for triggering high FAM72 expression that correlates with cancer cell proliferation, we also investigated the mRNA co-expression of FAM72 with the proliferative marker MKI67 and mitotic cell cycle genes in other tissues and correlated the level of FAM72 expression with the frequency of tissue-specific gene mutations. A combination of well-known cancer drivers was observed in tissues such as bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), lymphoid neoplasm diffuse large B-cell lymphoma (DLBC), head and neck squamous cell carcinoma (HNSC), acute myeloid leukemia (LAML), low grade glioma (LGG), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), pancreatic adenocarcinoma (PAAD), prostate adenocarcinoma (PRAD), sarcoma (SARC), testicular germ cell cancer (TGCT), and uveal melanoma (UVM) (Supplementary Figs. S13–S15, S17, S19, S23–26, S29, S31, S32, S35, and S39), while a single gene was the primary oncogenic driver in tissues such as esophageal carcinoma (ESCA), kidney chromophobe (KICH), renal clear cell carcinoma (KIRC), lung squamous cell carcinoma (LUSC), ovarian serous adenocarcinoma (OV), skin cutaneous melanoma (SKCM), stomach adenocarcinoma (STAD), papillary thyroid carcinoma (THCA), thymoma (THYM), and uterine carcinosarcinoma (UCS) (Supplementary Figs. S18, S20, S21, S27, S28, S33, S34, S36, S37 and S38). No obvious single driver genes could be conclusively identified in cancerous tissues such as cholangiocarcinoma (CHOL), renal papillary cell carcinoma (KIRP), and pheochromocytoma and paraganglioma (PCPG) (Supplementary Figs. S16, S22, and S30).
Gene-specific survival analysis in GBM
Kaplan–Meier survival curves for SCN9A, MXRA5, ADAM29, KDR, LRP1B, and PI3KC2G showed that patients with mutations in SCN9A and PIK3C2G had a worse prognosis compared to all other GBM patients, including those with EGFR and TP53 mutations (Fig. 3). However, similarly to EGFR, mutations in MXRA5, ADAM29, LRP1B and KDR did not significantly alter prognosis. Although mortality in GBM patients is high, with the average survival being about 15 months [28], mutations in the new gene set (Fig. 2g) seem to be particularly lethal (Fig. 2h) and inducing the de novo pathway leading to primary GBM, independent of mutations in any of the other well-established GBM-associated oncogenes such as EGFR, TP53 or PTEN.

Survival plots showing the prognosis of GBM patients with somatic mutations in SCN9A, MXRA5, ADAM29, KDR, LRP1B and PIK3C2G, respectively. Interestingly, SCN9A, MXRA5, ADAM29, KDR, LRP1B and PIK3C2G are the genes with highest number of somatic mutations observed only in deceased GBM patients. Patients with mutations in SCN9A and PIK3C2G show worse prognosis compared to all other GBM patients. EGFR and TP53 survival plots are shown as control comparison. Red line: cases with alterations in query gene, blue line: cases without alterations in query gene
In-silico determination of the regulatory mechanism controlling FAM72 expression: An |-SRGAP2–FAM72-| master-gene intergenic region-TFBS analysis
Since the promoter regions, i.e., the intergenic region among the four paralogous |-SRGAP2–FAM72-| master-gene pairs, are highly similar [6], it could be assumed that the same putative TFBS should be present in the intergenic regions of all four |-SRGAP2–FAM72-| master-gene pairs. Accordingly, we selected the |-SRGAP2A–FAM72A-| intergenic region for our TFBS analysis.
Ensembl provided us potential TFBS in the promoter region of all FAM72 (A-D) paralogs. For further detailed analysis, we aligned putative human TFBS along the |-SRGAP2A–FAM72A-| intergenic region using the JASPAR database. This revealed multiple regions where TFs could bind: multiple spleen focus forming virus (SFFV), pro-viral integration oncogene (SPI1), and myeloid zinc finger 1 (MZF1)-binding sites were detected, as were sites for SP1 TF (SP1), GATA-binding protein 2 (GATA2), nuclear factor I C (NFIC), and ETS proto-oncogene 1 (ETS1) (Supplementary Fig. S40). A common TFBS site was determined for the TFs MAX dimerization protein and T-box genes (MGA/TBX1/TBX4/TBX15), which could mean they share a motif. Most TBX family members bind to the AGGTGTGA ‘‘half-site’’ palindromes, and this motif is shared by MGA [29].
Most strikingly, the BS motifs for the TF GATA-binding protein 2 (GATA2) are common for all human FAM72 paralogs and those M-phase-specific cell cycle genes that demonstrated high expression correlation with those paralogs (Supplementary Table S14). GATA2 expression is critical for hematopoietic cell development, and ectopic expression of GATA2 is associated with megakaryocyte proliferation [30].
Discussion
NSC-specific FAM72 gene expression can be localized to the hippocampal dentate gyrus, one of the two sites of neurogenesis in the adult brain, where it is associated with maintenance of NSCs as part of the |-SRGAP2–FAM72-| master-gene responsible for brain plasticity [4,5,6,7,8]. Our group reported high protein expression correlation between FAM72A and protein kinase C (PRKC) in cancer cells, such as neuroblastoma and breast adenocarcinoma (MCF-7 and MDA-MB-231 cells), and we established that in cancer cells FAM72A acts downstream of PRKC [5]. We also described FAM72A interaction with epigenetically modified tumor suppressors along with influence of TP53 signaling pathways [3]. Other studies also point at FAM72A as being linked to tumorigenic effects in breast, colon, and lung cell lines [3, 5, 11]. In the present study, we established a cancer tissue-specific FAM72 expression profile further clarifying the biological significance of FAM72 expression in cancer and unravelling a novel primary GBM-specific gene-mutation signature.
Figure 4 shows the various integrated cancer gene-mutation signature-activated cell cycle pathways, with the potential cell cycle-specific role of FAM72 being highlighted.

Integrated cancer tissue gene mutation signature-activated cell signaling pathways with schematic hypothesis of FAM72 action during the cell cycle in proliferating NSCs or cancer cells. Mutations in proto-oncogenes or tumor suppressors (gene X) lead to activation of cell proliferation pathways, bypass of cell cycle checkpoints, and inhibition of the pro-apoptotic pathway. This leads to increased expression of FAM72, especially during the prophase to metaphase stages of the M-phase of the cell cycle. An accumulation of such gene mutations or a single driver-gene mutation triggers the cell cycle, leading to high FAM72 expression. The tumor cell formed as a result continues to proliferate, and FAM72 is expressed repeatedly once the cell enters the M-phase, especially during the prophase to metaphase stages. Metastasis and angiogenesis may be enhanced by mutations in genes such as KMT2A or TG (Supplementary Fig. S36), which are not oncogenic themselves (gene Y). A loss of function mutation in FAM72 could disrupt the M-phase, thereby inhibiting spindle formation and mediating cell aging, senescence, differentiation, or mitotic cell death. Targeting FAM72 could thus be a viable treatment method for GBM and other cancer types outside the CNS. The cell cycle is divided into several phases: G0, quiescent or differentiated stage; G1, Gap1 phase; G2, Gap2 phase; M, mitotic phase; S, synthesis phase; CCNA1, cyclin A; CCNB1, cyclin B; CCND1, cyclin D; CCNE1, cyclin E; CDK, cyclin-dependent kinase
In most human cancer tissues, however, a single mutated gene may not be responsible. Proto-oncogenic mutations in two (or more) driver genes, thus characterizing the tissue-specific gene-mutation signature, may lead to cell cycle activation, cell transformation, and cell proliferation [31]. The small number of non-synonymous mutations in FAM72 across all cancer tissues indicates that such mutations could not cause FAM72 to become an oncogenic driver gene (Supplementary Fig. S3). FAM72 (A-D) paralogs themselves are mutated at a very low level across all cancer tissues and not at a specific site. This lack of mutations in a gene functionally associated with cellular proliferation (in NSCs) [4,5,6,7] is surprising. One reason could be that the cells in which FAM72 becomes mutated are fated to differentiate, to age or to die (as assumed for neural FAM72 activity in non-neuronal tissue) and thus cannot be detected. It has been hypothesized that the two genes within the |-SRGAP2–FAM72-| master-gene act in an antagonistic manner in NSCs: activation of SRGAP2 (and concurrent inactivation of FAM72) when the cell decides to mature and differentiate and activation of FAM72 (and simultaneous inactivation of SRGAP2) when the cell decides to proliferate [4, 7, 9]. However, this dual role for the neural-specific |-SRGAP2–FAM72-| master-gene is currently restricted to neural tissue/NSCs.
Our current analysis shows that any master gene activity in non-neuronal tissues may cause cell differentiation/death (SRGAP2 active) or cancer (FAM72 active, e.g., due to an upstream cell cycle driver oncogene). This finding is underscored by the high correlation of human FAM72 expression with expression of the proliferative marker MKI67, indicating that neural FAM72 (A-D) paralogs are co-expressed with MKI67 during cellular proliferation also outside neural tissue, i.e., in cancer cells. Moreover, our study clearly demonstrates that genes with the highest correlation of expression with FAM72 paralogs are involved in the late G2- to M-stages of the cell cycle (Fig. 1 and Supplementary Fig. S11) and could associate FAM72 action with genes acting during the M-phase of the cell cycle, in particular the prophase to metaphase stages (Fig. 4). High correlation between two genes may indicate that they act together as a complex for a particular cellular function or are involved in the same biological process [32]. Based on our results, FAM72 paralogs are specifically co-expressed with genes involved in the prophase/metaphase stages of the cell cycle. These genes include ASPM, CENPE, CENPF, KIF14, KIF23, BUB1, SGO1, NUF2, CEP55, and NEK2. This may mean that the human FAM72 (A-D) paralogs also act in the prophase to metaphase stages, possibly being involved with the centromere-kinetochore complex or mitotic spindle formation. These genes also share the same TFBS motif, GATA2, indicating that their expression is co-regulated in concert with the FAM72 paralogs and thus implying a common temporal and spatial function [33]. GATA2 has differing functions during hematopoietic cell development, and GATA2 overexpression has been shown to increase cellular quiescence [34]. However, GATA2 is required for RAS-pathway driven non-small cell lung cancers [35] and leads to poor prognosis in leukemia [36]. GATA2 regulation of FAM72 paralogs thus suits their proliferative function.
GBM develops either de novo from NSCs into GBM stem cells and primary GBM or develops through a multistep process into secondary GBM arising from NSCs by means of aggregated mutations, different from the triggering mutations in GBM stem cells of primary GBM [2]. The differentiation of these two distinct forms, primary and secondary GBM, depends on the presence of different triggering mutations in pivotal genes such EGFR or CDKN2A in primary GBM and PDGFRA or PTEN (and others) in secondary GBM [2]. Our identified primary GBM-specific gene-mutation signature is comprised of the five genes EGFR, TP53, PTEN, NF1, and SPTA1. The first four are well-studied oncogenes associated with GBM while somatic mutations in the cell motility protein SPTA1 have been reported from glioblastoma as well as other cancers [27]. The gene set with somatic mutations observed solely in deceased GBM patients (SCN9A, MXRA5, ADAM29, KDR, PIK3C2G and LRP1B) (Fig. 2g, h) is intriguing, as they could be oncogenic by themselves or act fatally in combination with other oncogenic driver genes (Fig. 2d). Sodium voltage-gated channel alpha subunit 9 (SCN9A) codes for an ion channel subunit protein, which is highly expressed in primary sensory and sympathetic ganglion neurons. Mutations in SCN9A have been reported to be the causative factor behind primary erythromelagia [37], but thus far have no known role in cancer. Matrix remodeling associated 5 (MXRA5) codes for a proteoglycan and is possibly involved in cell adhesion. In addition to its probable role in matrix remodeling and cell adhesion, MXRA5 function is regulated by transforming growth factor -β1, implying an anti-inflammatory role [38]. Mutations in MXRA5 have been reported from non-small cell lung and pleural cancers [39]. ADAM metallopeptidase domain 29 (ADAM29) belongs to the ADAM disintegrin and metalloprotease family. The ADAM family members are transmembrane and secreted proteins, most of whom encode functional proteases. The functional role of ADAM29 is not clarified, but increased expression was observed on breast cancer cells in vitro and mutations in ADAM29 led to increased proliferation of breast cancer cells [40]. ADAM29 is also highly mutated in melanoma and may have a role in integrin-dependent cell adhesion and increasing cell migration ability [41]. Kinase insert domain receptor (KDR) encodes for a VEGF receptor (VEGFR2). VEGFR2 phosphorylation activates the MAPK pathway, stimulates the RAS pathway as well ensuring cellular survival via activation of the protein kinase B (PKB), thus mediating VEGF-induced endothelial proliferation [42]. Phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 gamma (PIK3C2G) is a class II PI3K, primarily expressed in pancreas, liver and prostate tissues. Little is known about its exact function, but other members of the class II PI3Ks such as PI3KC2A and PIK3C2B are involved in angiogenesis, cell cycle progression and endocytosis [43]. LRP1B mutations have been previously reported from glioblastoma and deletions in LRP1B are associated with worse prognosis [44], which matches with our data. It is clear that mutations in these genes are significant for GBM metastasis as they may assist the tumor cells in proliferation, migration and angiogenesis. This novel FAM72 expression profile-associated gene set (SCN9A, MXRA5, ADAM29, KDR, LRP1B and PIK3C2G) is thus a critical gene-mutation signature for GBM prognosis and—as cell membrane receptors (SCN9A, ADAM29, LRP1B and KDR)—these genes are preordained targets for therapeutics. Although EGFR inhibitors are widely used chemotherapeutics, evidence suggests that resistance to EGFR inhibitors is high. mTOR inhibitors are also ineffective due to incomplete inhibition of the targets [28]. Thus in this context our novel gene set emerges as new potential drug targets. Since mutations in these genes show very poor prognosis and seems to be lethal as associated with the de novo pathway of primary GBM (irrespective of mutations in EGFR or TP53), targeting mutant variants of this new gene set in combination with established drugs could significantly extend patients’ lifespan.
Human FAM72 paralogs are driven and overexpressed in GBM by these genes, confirming FAM72’s important role in proliferating GBM cells (Supplementary Figs. S7a, S8a, and S9a). An inactivating mutation in the FAM72 paralogs may not have any effect in the master gene |-SRGAP2–FAM72-| on SRGAP2 expression as long as the SRGAP2 promotor itself is not affected. A mutation-induced inactivation of FAM72, however, may cause a defect in mitotic spindle formation and cellular apoptosis. In the case of SRGAP2 activation, however, as SRGAP2 expression is required for neuronal NSCs differentiation and for functional neurons (synaptic activities) but not in glial cells [7, 9], the glial cells will arrest the cell cycle, differentiate, or undergo apoptosis. Accordingly, not all such cells will be isolated and detected by sequencing methods in large-scale cancer cell studies. Still, as |-SRGAP2–FAM72-| is a master gene, we also analyzed the change in expression in SRGAP2 in GBM, similar to that in FAM72 paralogs. SRGAP2 shows no change in expression in GBM, as compared to normal tissue, indicating that SRGAP2 is inactive in GBM (Supplementary Fig. S41). SRGAP2 is reported to be a tumor suppressor and its expression may lead to apoptosis of the cancerous cell [45]. In development and progression of GBM, SRGAP2 itself may not play a causative role as its expression will cause the cells to undergo apoptosis or to differentiate. This stands in direct contrast with FAM72, and further serves to reinforce the proliferative function of FAM72 in cancer. Only those FAM72 mutations that have no effect on the cell function of FAM72 would be detected, which could be a reason why only a small number of mutations are observed in human FAM72 (A-D) paralogs in all cancer samples across all tissues (Supplementary Table S2).
Moreover, we observed genomic hypomethylation within the FAM72A promoter region in GBM, correlating with increased FAM72A expression in tumor tissues (Supplementary Fig. S42). This raises the possibility that epigenetic regulation via methylation could also be a factor in the differential expression of FAM72 paralogs in other cancer tissues, though our analysis could not confirm this hypothesis. Although non-CpG methylation occurs inside the first intron of SRGAP2A, cancer-specific methylation has been reported to occur at so-called ‘shores’, i.e., regions of 2–4 kb distance from CpG islands [46]. Non-CpG methylation has also been reported from neurons and is associated with repression of gene activity [47]. This aligns with the role of FAM72A in NSCs, as its expression should decrease in differentiated neurons [4]. Investigation into the methylation of FAM72 paralogs in non-neuronal cancer tissues is underway.
Expression levels of FAM72 (A-D) paralogs depend on the cell cycle stage. If the cell was in G1-/S-phase, expression of FAM72 would be low. This was verified by determining if FAM72 co-expresses with certain genes specifically active during the G1- or S-phase of the cell cycle (Supplementary Figs. S11, 12 and Supplementary Table S15). In contrast, all the genes that show high expression correlation with FAM72 paralogs could be associated with the G2-/M-phase (prophase to metaphase stages of the M-phase) and have been reported as prognostic markers in cancer or as being upregulated during oncogenesis. These include ASPM, BUB1, CENPE, CENPF, CEP55, KIF14, KIF23, NEK2, NUF2, and SGO1 [48, 49]. During mitosis, chromosome segregation is a critical process regulated by the mitotic spindle, and the centrosome is the organelle controlling mitotic spindle formation. Upregulation of mitosis-specific cell cycle genes has been reported from cancer tissues. KIF14, ASPM, KIF23, BUB1, NEK2, and CEP55 are involved either with spindle formation or with regulation, while CENPE, CENPF, NUF2, and SGO1 are involved in the centromere-kinetochore complex. Co-expression of FAM72 with these genes strongly suggests functional FAM72 involvement either with the mitotic spindle or with the kinetochore-centromere complex. The aforementioned ‘guilt-by-association’ (GBA) principle is attractive and has been used to characterize gene function in a variety of organisms [33]; however, assigning a biological cell function to FAM72 based on transcriptomic co-expression alone would be fraught with risk. Additional proteomic profiling, as described by Wang et al. [50], could be performed to corroborate co-expression-based gene function prediction. This will require comprehensive FAM72 proteomic data to construct a co-expression network. The transcriptomic and proteomic networks could then be compared to predict co-expression-based gene function.
Conclusion
Our study highlights a novel FAM72 expression profile-associated primary GBM-specific gene-mutation signature (SCN9A, MXRA5, ADAM29, KDR, LRP1B and PIK3C2G) predestinated as viable targets for therapeutics. It further indicates that an increase in mitotic FAM72 expression in cancer cells is triggered by upstream mutations in primary proto-oncogenes or tumor suppressor genes, such as EGFR, BRAF, RAS, or TP53, which leads to increased cellular proliferation. Silencing NSC-specific FAM72 could prove critical in preventing cancer cells from proliferating. It could therefore prove a highly promising therapeutic target for treatment of cancer, particularly outside the CNS, as knock-out neural-specific FAM72 gene function may cause spindle assembly defects and mitotic catastrophe, followed by cell death in all non-neuronal cancer tissues.
References
Ferlay J, Soerjomataram I, Dikshit R et al (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–E386. https://doi.org/10.1002/ijc.29210
Stoyanov GS, Dzhenkov D, Ghenev P et al (2018) Cell biology of glioblastoma multiforme: from basic science to diagnosis and treatment. Med Oncol 35:27. https://doi.org/10.1007/s12032-018-1083-x
Heese K (2013) The protein p17 signaling pathways in cancer. Tumour Biol 34:4081–4087. https://doi.org/10.1007/s13277-013-0999-1
Benayoun BA, Pollina EA, Ucar D et al (2014) H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158:673–688. https://doi.org/10.1016/j.cell.2014.06.027
Nehar S, Mishra M, Heese K (2009) Identification and characterisation of the novel amyloid-beta peptide-induced protein p17. FEBS Lett 583:3247–3253. https://doi.org/10.1016/j.febslet.2009.09.018
Kutzner A, Pramanik S, Kim PS, Heese K (2015) All-or-(N)One—an epistemological characterization of the human tumorigenic neuronal paralogous FAM72 gene loci. Genomics 106:278–285. https://doi.org/10.1016/j.ygeno.2015.07.003
Ho NTT, Kutzner A, Heese K (2017) Brain plasticity, cognitive functions and neural stem cells: a pivotal role for the brain-specific neural master gene |-SRGAP2-FAM72-|. Biol Chem 399:55–61. https://doi.org/10.1515/hsz-2017-0190
Ho NT, Kim PS, Kutzner A, Heese K (2017) Cognitive functions: human vs. animal—4:1 advantage |-FAM72-SRGAP2-|. J Mol Neurosci 61:603–606. https://doi.org/10.1007/s12031-017-0901-5
Charrier C, Joshi K, Coutinho-Budd J et al (2012) Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell 149:923–935. https://doi.org/10.1016/j.cell.2012.03.034
Guo C, Zhang X, Fink SP et al (2008) Ugene, a newly identified protein that is commonly overexpressed in cancer and binds uracil DNA glycosylase. Cancer Res 68:6118–6126. https://doi.org/10.1158/0008-5472.CAN-08-1259
Wang LT, Lin CS, Chai CY et al (2011) Functional interaction of Ugene and EBV infection mediates tumorigenic effects. Oncogene 30:2921–2932. https://doi.org/10.1038/onc.2011.16
Rajan P, Stockley J, Sudbery IM et al (2014) Identification of a candidate prognostic gene signature by transcriptome analysis of matched pre- and post-treatment prostatic biopsies from patients with advanced prostate cancer. BMC Cancer 14:977. https://doi.org/10.1186/1471-2407-14-977
Gao J, Aksoy BA, Dogrusoz U et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1. https://doi.org/10.1126/scisignal.2004088
Iverson GL (2011) Z scores. In: Kreutzer JS, DeLuca J, Caplan B (eds) Encyclopedia of clinical neuropsychology. Springer New York, New York, pp 2739–2740
Diez-Villanueva A, Mallona I, Peinado MA (2015) Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenetics Chromatin 8:22. https://doi.org/10.1186/s13072-015-0014-8
Team BD (2014) Bokeh: python library for interactive visualization. http://www.bokeh.pydata.org
Lawrence MS, Stojanov P, Polak P et al (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214–218. https://doi.org/10.1038/nature12213
Greenman C, Stephens P, Smith R et al (2007) Patterns of somatic mutation in human cancer genomes. Nature 446:153–158. https://doi.org/10.1038/nature05610
Goldman M, Craft B, Zhu J, Haussler D (2017) Abstract 2584: the UCSC Xena system for cancer genomics data visualization and interpretation. Cancer Res 77:2584–2584. https://doi.org/10.1158/1538-7445.am2017-2584
Bland JM, Altman DG (2004) The logrank test. BMJ 328:1073. https://doi.org/10.1136/bmj.328.7447.1073
Levo M, Segal E (2014) In pursuit of design principles of regulatory sequences. Nat Rev Genet 15:453–468. https://doi.org/10.1038/nrg3684
Mathelier A, Fornes O, Arenillas DJ et al (2016) JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 44:D110–D115. https://doi.org/10.1093/nar/gkv1176
Krishnamurthy N, Brown DP, Kirshner D, Sjölander K (2006) PhyloFacts: an online structural phylogenomic encyclopedia for protein functional and structural classification. Genome Biol 7:R83. https://doi.org/10.1186/gb-2006-7-9-r83
Cock PJ, Antao T, Chang JT et al (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25:1422–1423. https://doi.org/10.1093/bioinformatics/btp163
Yates A, Akanni W, Amode MR et al (2016) Ensembl 2016. Nucleic Acids Res 44:D710–D716. https://doi.org/10.1093/nar/gkv1157
Whitfield ML, George LK, Grant GD, Perou CM (2006) Common markers of proliferation. Nat Rev Cancer 6:99–106. https://doi.org/10.1038/nrc1802
Brennan CW, Verhaak RG, McKenna A et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. https://doi.org/10.1016/j.cell.2013.09.034
Cloughesy TF, Cavenee WK, Mischel PS (2014) Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol 9:1–25. https://doi.org/10.1146/annurev-pathol-011110-130324
Castellanos R, Xie Q, Zheng D, Cvekl A, Morrow BE (2014) Mammalian TBX1 preferentially binds and regulates downstream targets via a tandem T-site repeat. PLoS One 9:e95151. https://doi.org/10.1371/journal.pone.0095151
Zheng R, Blobel GA (2010) GATA transcription factors and cancer. Genes Cancer 1:1178–1188. https://doi.org/10.1177/1947601911404223
Pereira B, Chin SF, Rueda OM et al (2016) The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 7:11479. https://doi.org/10.1038/ncomms11479
Usadel B, Obayashi T, Mutwil M et al (2009) Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ 32:1633–1651. https://doi.org/10.1111/j.1365-3040.2009.02040.x
Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P (2004) Coexpression analysis of human genes across many microarray data sets. Genome Res 14:1085–1094. https://doi.org/10.1101/gr.1910904
Tipping AJ, Pina C, Castor A et al (2009) High GATA-2 expression inhibits human hematopoietic stem and progenitor cell function by effects on cell cycle. Blood 113:2661–2672. https://doi.org/10.1182/blood-2008-06-161117
Kumar MS, Hancock DC, Molina-Arcas M et al (2012) The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell 149:642–655
Vicente C, Vazquez I, Conchillo A et al (2012) Overexpression of GATA2 predicts an adverse prognosis for patients with acute myeloid leukemia and it is associated with distinct molecular abnormalities. Leukemia 26:550–554. https://doi.org/10.1038/leu.2011.235
Rush AM, Dib-Hajj SD, Liu S et al (2006) A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci USA 103:8245–8250. https://doi.org/10.1073/pnas.0602813103
Poveda J, Sanz AB, Fernandez-Fernandez B et al (2017) MXRA5 is a TGF-beta1-regulated human protein with anti-inflammatory and anti-fibrotic properties. J Cell Mol Med 21:154–164. https://doi.org/10.1111/jcmm.12953
He Y, Chen X, Liu H et al (2015) Matrix-remodeling associated 5 as a novel tissue biomarker predicts poor prognosis in non-small cell lung cancers. Cancer Biomark 15:645–651. https://doi.org/10.3233/CBM-150504
Zhao M, Jia W, Jiang WG et al (2016) ADAM29 expression in human breast cancer and its effects on breast cancer cells in vitro. Anticancer Res 36:1251–1258
Wei X, Moncada-Pazos A, Cal S et al (2011) Analysis of the disintegrin-metalloproteinases family reveals ADAM29 and ADAM7 are often mutated in melanoma. Hum Mutat 32:E2148–E2175. https://doi.org/10.1002/humu.21477
Liu Z, Qi L, Li Y, Zhao X, Sun B (2017) VEGFR2 regulates endothelial differentiation of colon cancer cells. BMC Cancer 17:593. https://doi.org/10.1186/s12885-017-3578-9
Falasca M, Maffucci T (2012) Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem J 443:587–601. https://doi.org/10.1042/BJ20120008
Tabouret E, Labussiere M, Alentorn A et al (2015) LRP1B deletion is associated with poor outcome for glioblastoma patients. J Neurol Sci 358:440–443. https://doi.org/10.1016/j.jns.2015.09.345
Marko TA, Shamsan GA, Edwards EN et al (2016) Slit-Robo GTPase-activating protein 2 as a metastasis suppressor in osteosarcoma. Sci Rep 6:39059. https://doi.org/10.1038/srep39059
Irizarry RA, Ladd-Acosta C, Wen B et al (2009) The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41:178–186. https://doi.org/10.1038/ng.298
Guo JU, Su Y, Shin JH et al (2014) Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci 17:215–222. https://doi.org/10.1038/nn.3607
Abba MC, Lacunza E, Butti M, Aldaz CM (2010) Breast cancer biomarker discovery in the functional genomic age: a systematic review of 42 gene expression signatures. Biomark Insights 5:103–118. https://doi.org/10.4137/BMI.S5740
Lee M, Rivera-Rivera Y, Moreno CS, Saavedra HI (2017) The E2F activators control multiple mitotic regulators and maintain genomic integrity through Sgo1 and BubR1. Oncotarget 8:77649–77672. https://doi.org/10.18632/oncotarget.20765
Wang J, Ma Z, Carr SA et al (2017) Proteome profiling outperforms transcriptome profiling for coexpression based gene function prediction. Mol Cell Proteomics 16:121–134. https://doi.org/10.1074/mcp.M116.060301
Funding
This study was supported by Hanyang University by providing a scholarship to C.S.R. and by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2015R1D1A1A01057243 and 2016R1D1A1B03932599).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rahane, C.S., Kutzner, A. & Heese, K. A cancer tissue-specific FAM72 expression profile defines a novel glioblastoma multiform (GBM) gene-mutation signature. J Neurooncol 141, 57–70 (2019). https://doi.org/10.1007/s11060-018-03029-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-018-03029-3