
Abstract
Background
Familial Mediterranean fever (FMF) is an autosomal recessive autoinflammatory disease primarily affecting individuals of Turkish, Armenian, Arab, and non-Ashkenazi Jewish descent, caused by mutations in the MEFV gene. The aim of this study was to review the common genotype distributions of MEFV variants and mutations in the Turkish population and evaluate rare mutations.
Methods and results
The study included 2984 patients who applied to Ankara University Ibni Sina Hospital Immunology Laboratory with clinical suspicion of FMF between 2004 and 2014. The data of patients from different regions of the country who were followed up in the immunology-rheumatology clinic with clinical suspicion and presumptive diagnosis of FMF were evaluated retrospectively. Patients were tested for all mutations in Exon 2 and Exon 10, including M694V, M680I, M694I, V726A, E148Q and R202Q. There were 2504 patients with FMF variant. According to genotyping, R202Q (n = 1567, 39.2%) was the most common mutation. The most common co-variant was the R202Q/M694V genotype (n = 507, 16.98%). Allele frequencies for MEFV mutations were as follows: R202Q (n = 1567, 39.2%), M694V (n = 1004, 25.1%), E148Q (n = 463, 11.5%), M680I (n = 354, 8.8%), V726A (n = 319, 7.9%), A744S (n = 51, 1.2%), R761H (N = 41, 1.0%), P706P (N = 25, 0.6%), E167D (N = 23, 0.5%), M694I (N = 23, 0.5%), and K695R (N = 20, 0.5%).
Conclusion
This research revealed the prevalence of both common and rare MEFV gene mutations in Turkish FMF patients in various age groups. R202Q was the most prevalent mutation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Familial Mediterranean Fever (FMF) is an autosomal recessive inflammatory disorder caused by mutations in the MEFV gene [1,2,3]. The term “Familial Mediterranean Fever” is derived from its genetic inheritance and its high occurrence in Mediterranean areas, particularly notable in Armenian, Arab, Jewish, and Turkish populations. Although FMF is not common in Spain, Italy, Iran, and Greece, it has acquired worldwide awareness because of increasing immigration in recent decades [1, 4,5,6]. Advancements in molecular biology and genetics has led to a more detailed understanding of FMF, including aspects such as penetrance, pathogenesis, mutation types (gain-of-function or loss-of-function), and heredity [7, 8].
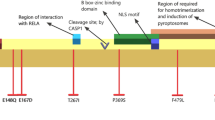
The responsible gene, MEFV, located on the short arm of chromosome 16p13.3, was identified three decades ago [9] and comprises 10 exons and 781 codons [2, 10]. Mutations within this gene lead to the synthesis of a dysfunctional protein known as “pyrin,” which plays a crucial role in both the innate immune system and the regulation of inflammation [7, 11]. Consequently, these mutations can result in uncontrolled and excessive inflammation through the synthesis of interleukin-1 (IL-1) [2, 4, 7].
MEFV mutations predominantly occur in exons 2 and 10, with 390 different variants identified, of which approximately 59% are located in these exons [6, 7, 12]. Among the various mutations identified in the MEFV gene, M694V is the most common and pathogenic worldwide, particularly among the Mediterranean regions. Other commonly observed pathogenic mutations include M680I, V726A, M694I, and E148Q. Collectively, these five mutations account for approximately 80% of genotypes in FMF patients from Mediterranean populations [7, 13, 14]. Out of the reported 29 MEFV mutations, 26 are missense mutations, one is a nonsense mutation (Y688X), and two are minor deletions (I692del, M694del) [2, 15].
FMF patients often have recurrent attacks characterized by fever and serositis with intense chest, abdominal, or joint pain, as well as accompanying symptoms like abdominal discomfort, pleuritis, arthritis, and skin manifestations [3, 5]. The unpredictable nature of these episodes poses clinical challenges, necessitating a comprehensive understanding of the disease spectrum for accurate diagnosis [16]. Despite the availability of genetic testing for FMF, challenges persist due to the diversity of mutations and the potential for atypical presentations in clinical practice [7, 17]. In this context, Türkiye stands out as a focal point for FMF research. With a population of approximately 82 million, Türkiye carries a considerable burden of FMF, estimated at 1 in 1000 individuals in [7, 17]. This places Türkiye among the countries most affected globally, highlighting the urgent necessity for thorough genetic investigations and improved diagnostic methodologies [7, 18].
In this study, our aim was to investigate the genotype distribution of common and rare FMF variants in the Turkish population.
Materials and methods
Patient groups
Over a ten years period (between January 2004 and December 2014), patient’s histories were retrospectively investigated. This research included a total of 2984 patients who were suspected of having FMF. All patients exhibited one or more symptoms typically associated with FMF, predominantly including abdominal pain, fever, arthralgia, and erysipelas-like erythema. Diagnosis was based on the Tel-Hashomer criteria or a detailed clinical assessment. Informed consent was obtained from all patients or their parents before undergoing genetic testing. This work was approved by the Ankara University Human Research Ethics Committee. (decision no: I06-356-22).
Sanger sequencing
DNA sequence analysis was conducted using two different instruments. Blood samples were collected in vacuum tubes containing ethylenediaminetetraacetic acid (EDTA) according to the manufacturer’s instructions for the Blood Genomic DNA Isolation Kit (Norgen Biotek, Canada), enabling DNA extraction from peripheral blood. PCR amplification of the MEFV gene was performed using forward and reverse primers targeting exons 2 and 10. The PCR conditions were as follows: initial denaturation at 94 °C for 5 min, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 45 s, extension at 72 °C for 1 min, and a final extension at 72 °C for 5 min. The PCR products were observed on a 2% agarose gel through electrophoresis.
For the first instrument, the amplification products were purified using the Genomic DNA Purification Kit (Fermentas, USA) before sequence analysis with an automated fluorescence-based sequencer system (Beckman Coulter CEQ 8000). For the second instrument, the PCR products underwent sequencing using the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit according to the manufacturer’s instructions (Applied Biosystems, Foster City, USA). Sequence analysis was conducted with an automated fluorescence-based sequencer system (ABI PRISM 3130, Applied Biosystems).
Statistical analysis
Descriptive statistics were presented as mean ± standard deviation for the variables whereas they were presented as number and percentage (%) for nominal variables. Categorical variables were evaluated using Pearson’s chi-square test or Fisher’s exact test as appropriate. Ap value of less than 0.05 was considered statistically significant and the analyses were conducted using the Statistical Package for Social Sciences (SPSS, Version 15.0, Chicago, IL).
Results
MEFV variations (one or more MEFV mutations per patient) located in Exon 2 and Exon 10 were detected in 2504 of the 2984 patients (83.9%). 480 of the 2984 patients (16.1%) had no detected variant despite clinical symptoms. A total of 44 different FMF variants were identified in 2504 patients included in the study, with 28 variants were found in the exon 2 region and 16 in exon 10. The study group comprised 1499 (59.9%) female and 1005 (40.1%) male patients, with an average age of 31.91 (± 14.34) years.
The most common FMF variants/mutations are R202Q (n = 1567, %39,2), M694V (n = 1004, %25,1), E148Q (n = 463, %11,5), M680I (n = 354, %8,8), V726A (n = 319, by 7.9%), A744S (n = 51, %1,2), R761H (N = 41, %1,0), P706P (n = 25, by 0.6%), E167D (N = 23 0.5%), M694I (N = 23 0.5%) K695R (N = 20, 0.5), respectively. Gender-specific distributions of mutation prevalence is shown in Table 1, while Fig. 1 demonstrates the distribution of the most common mutations observed in our study population. This common mutation constitutes 92,5% of the entire patient population. Eighteen variants are present in only one patient. Among the 14 consensus variants, our patient group did not have the pathogenic variant I692del in exon 10.

Most common mutations in the study group
According to zygosity, patients were classified as heterozygous (n = 1172), homozygous (n = 157), compound heterozygous (n = 591), compound homozygous (n = 174), or compound heterozygous/homozygous (complex) (n = 410). The three most common homozygous mutations found in patients are as follows: in Exon 2 R202Q (n = 334, 93.8%), E148Q (n = 17, 4.7%), and E167D (n = 7, 0.23%). In Exon 10, the most common variations are M694V (n = 111, 3.72%), V726A (n = 100, 3.35%), and M680I (n = 94, 3.15%). Combined mutations found in patients; Heterozygous/Heterozygous: R202Q/M694V (n = 243, 8.14%), R202Q/E148Q (n = 56, 1.88%), M680I/V726A (n = 46, 1.54%), M694V/E148Q (n = 38, 1.27%) Homozygous/Homozygous: R202Q/M694V (n = 169, 5.66%). Homozygous/Heterozygous: R202Q/M694V (n = 71, 2.38%), Heterozygous/Homozygous: R202Q/M694V (n = 24, 0.8%). The complex genotype was R202Q/M694V/M680I (n = 108, 3.62%). The distribution of MEFV genotypes found in patients is given in Table 2.
The ranking of the three most prevalent mutations in different age groups is as follows: for individuals aged 12 and under, M680I (n = 205, 47.0%), R202Q (n = 111, 25.4%), and M694V (n = 58, 13.3%). In the age group of 13–19 years old, R202Q (n = 189, %39,7), M694V (n = 106, %22,2), E148Q (n = 71, %14,9). For individuals aged 20 and above, the top three mutations were R202Q (n = 1254, 38.8%), M694V (n = 828, 25.6%), E148Q (n = 358, 11.0%). Notably, in individuals aged 12 and under, the most frequent mutation observed was M680I, distinguishing this group from others. Furthermore, the incidence of mutation is significantly greater in individuals above the age of 20 compared to other age groups (p < 0.001). Figure 2 shows the frequency of the first 10 mutations according to age groups. In the patient group in our study women more than men mutations has been identified. Our study also revealed a higher prevalence of mutations in females compared to males within the patient group. Specifically, R202Q (n = 944, 40.62%), M694V (n = 543, 23.36%), and E148Q (n = 271, 11.75%) mutations were more frequently observed in females. Conversely, in males, the mutation frequencies were R202Q (n = 623, 37.28%), M694V (n = 461, 27.59%), and E148Q (n = 190, 11.37%). Our analysis found a higher prevalence of mutations in women compared to males within the patient group. However, there was no statistically significant difference between the frequency of mutations in males and females (p < 0.076). Figure 3 shows the distribution of the top 10 mutations in men and women.

Most common variants in the study population according to age groups

Most common variants in both males and female
Discussion
FMF, a hereditary autoinflammatory disease prevalent in Turkish, Arab, Jewish, and Armenian populations, is characterized by recurrent episodes of abdominal pain, serositis, and fever [6]. Approximately 1 in 1000 individuals in the Turkish population are estimated to be affected by FMF, with around 20% of the population carrying the disease [19]. While clinical diagnosis is sufficient to identify FMF, nearly 80% of diagnosed patients exhibit MEFV gene mutations [3, 20]. However, genetic analysis is not mandatory, but it is important for confirming or excluding the diagnosis, particularly in cases with atypical clinical phenotypes [17, 21]. Early diagnosis and treatment of FMF are necessary to prevent amyloidosis, the most common and worse complication of the disease, and to improve patients’ quality of life [3, 22, 23]. Detection of MEFV gene variants provides benefits for the diagnosis and treatment of FMF. Shinar et al. recommended screening for 14 selected MEFV gene variants, of which 9 are classified as known pathogenic variants (M694V, M680I, V726A, M694I, A744S, R761H, I692del, E167D, T267I), while 5 are categorized as variants of uncertain significance (K695R, E148Q, P369S, F479L, I591T) [24].
Regarding polymorphisms associated with FMF, we identified single nucleotide changes with allele frequencies of A165A 2406 (21.8%), G138G 2383 (21.5%), and D102D 2255 (20.4%) in our patient population. These synonymous nucleotide changes in the Exon 2 region of the MEFV gene do not alter amino acid structure but modify the codon specifying an amino acid [25]. Akar et al. noted a significant difference in the ala138gly polymorphism between patients diagnosed with amyloidosis and FMF and those without amyloidosis, suggesting an association between the g138g polymorphic allele and amyloidosis development [26]. However, Öksuz et al. found no significant association between these polymorphisms and clinical features in FMF patients [27]. Consequently, these polymorphisms were excluded from our study’s evaluation. Further research is warranted to explore all variations in the MEFV gene and evaluate the impact of these genetic variants on linkage disequilibrium.
In our study, Table 1 demonstrates that the most prevalent variant is R202Q, accounting for 1567 cases (39.22%). Among these, 84 cases (2.81%) were homozygous for the R202Q variant, while 561 cases (18.80%) were heterozygous. Arpacı et al. conducted a study in the Hatay region involving 2639 patients, reporting the R202Q mutation as the most common, with 1319 cases (19.55%) being homozygous [13]. They found that clinical findings among patients with homozygous and heterozygous R202Q mutations were similar, and all patients responded to colchicine treatment.
In a study by Kırnaz et al. involving 3230 patients and using NGS, the most frequent mutation identified was R202Q, with 1097 cases (37.48%) reported [12]. Sönmezgöz et al. highlighted the prevalence of M694V and R202Q mutations in children with FMF in the Black Sea region [28]. They observed that patients with M694V and R202Q mutations commonly presented with recurrent abdominal pain and arthritis/arthralgia. Additionally, they stated that the R202Q mutation commonly causes chest pain [28]. Kandur et al. reported that R202Q mutation was associated with the inflammatory phenotype of FMF, and typical clinical findings of arthritis were observed in patients with M694V/R202Q heterozygous mutation [29].
Furthermore, Dundar et al. found that 85.8% of patients diagnosed with amyloidosis carried the R202Q mutation [7]. Recent investigations have highlighted the prevalence of the R202Q variant among FMF patients within the Turkish population [7, 12, 13, 29]. The lack of recognition of the R202Q mutation in prior studies may have resulted from its exclusion in analyses focusing on the 14 accepted variants considered as mutations. This omission could be the cause of why the R202Q variant was overlooked in previous analyses. Taken together, these findings, in conjunction with our study, suggest that the R202Q mutation may play a significant role in the pathogenesis of the disease.
In a study comprising 1719 FMF patients in Turkey, researchers noted the most frequently observed pathogenic variants among Turkish FMF patients from 15 rheumatology clinics across various regions to be M694V, M680I, V726A, E148Q, and M694I, respectively. Notably, the study did not report the presence of the R202Q mutation within this patient cohort [20]. Additionally, Balta et al. reported in a study involving 1028 patients that the first three homozygous variants M694V, R202Q, E148Q, as well as the most common M694V/R202Q genotype, were identified among their patients [19].
In a study involving 3230 patients, Kırnaz et al. reported that the most common mutations in sequence were R202Q, E148Q, M694V, V726A, and M680I, collectively constituting 80% of the patient group [12]. According to research conducted by a comprehensive national genetic consortium, including over 27,000 patients, indicated that the dominant MEFV gene variants in Turkish society are M694V, E148Q, R202Q, M680I, and V726A, respectively [7]. In our study with 2984 patients, the most common mutations observed, in sequence, were R202Q, M694V, E148Q, M680I, and V726A. Notably, most of our patient population are from the Central Anatolia region. The variance observed in the research conducted by the National Genetic Consortium may be attributed to the inclusion of patients from all regions of Turkey, including the Turkish Republic of Northern Cyprus. Additionally, this discrepancy could be associated with the genetic diversity within our nation, stemming from prolonged interactions among various ethnic groups residing in geographically distinct areas.
The major complication of FMF is renal amyloidosis, present in 12% of patients. In populations where the disease is common, such as in our country, it is essential to diagnose and treat the condition early to prevent amyloidosis and enhance quality of life [3, 7, 22, 23]. Homozygous M694V is associated with renal amyloidosis, which is considered the most important genotype [7, 20, 30,31,32]. Studies by Dundar et al. reported the prevalence of M694V as 29.47%, while Yaşar bilge et al. reported it as 24%, Sari et al. reported it as 54.9%, and Yildirim et al. reported it as 39%, making it the most common mutation in Turkish patients [7, 20, 33, 34].
In our investigation, we found that the M694V mutation ranks as the second most prevalent mutation, with an allele frequency of 25.13% (n = 1004). Homozygous M694V was found in 0.34% (n = 10) of individuals, while heterozygous M694V was found in 3.72% (n = 111) of individuals. The most common co-variant reported in studies is the R202Q/M694V genotype [13, 14, 21, 32]. The INFEVERS genetic database reported that the G allele of the R202Q (c.605G > A) mutation was in linkage disequilibrium with M694V(http://fmf.igh.cnrs.fr/ISSAID/infevers/search.php) [35]. Sayın Kocakap et al. showed that M694V mutation (55.8%) was in LD with R202Q mutation [17]. Kandur et al. have shown that the R202Q/M694V mutation association causes an increase in disease symptoms [29]. Additionally, both M694V and R202Q have been reported as mutations associated with amyloidosis in studies [7, 13]. In our study, the heterozygous R202Q/M694V mutation was observed in 8.14% (n = 243) of individuals, while the homozygous R202Q/M694V mutation was observed in 5.66% (n = 169) of individuals. Our findings are in accord with previous research, highlighting the prevalence of the R202Q/M694V co-variant.
Regarding the E148Q mutation, classified as a mutation of unknown significance in FMF diagnosis, it emerged as the third most common mutation in our study. It exhibited an allele frequency of 11.59% (n = 463), with 0.37% (n = 11) homozygous and 6.63% (n = 198) heterozygous occurrences. Notably, E148Q has been identified as the most frequent mutation in certain ethnic populations, such as Japanese and Egyptian patients [36, 37]. Research indicates that both homozygous and heterozygous E148Q variants are associated with a mild FMF phenotype [13, 38, 39]. It is among the most common mutations in studies conducted in different regions of Türkiye [12, 13, 17, 20, 38]. In our study, it was found to be the 3rd most common mutation. The observed regional disparities in mutation frequencies may stem from differences in study methodologies or ethnic compositions.
Despite clinical presentations, no variants were observed in 480 individuals (16.1%). This frequency contrasts with the 4.5% reported by Balta et al., 26.3% by Dundar et al., and 9% by Yaşar bilge et al. [7, 19, 20]. Studies have indicated that some FMF patients exhibit no detectable mutations. It is possible that our patients belong to this subset without mutations. Notably, our study only screened mutations in the Exon2 and Exon10 regions, potentially overlooking mutations in Exon 1, 3, and 5. In our study, we identified nucleotide changes not described in INFEVERS [36]. Although some of the patients in whom these changes were detected received a diagnosis and treatment for FMF, they could not be classified as new genotypes because they may have pathogenic mutations in other exons (e.g. A279E, P132P, T262T). Additionally, no patients in our study exhibited the I692del mutation, which is occasionally observed in other Mediterranean populations.
Numerous studies have consistently reported a higher proportion of female FMF patients compared to male patients [7, 12, 17, 19, 20]. In our study, the rate of female patients was 59.9%, indicating a potential epigenetic effect in FMF. Figure 3 demonstrates that there was no statistically significant difference in the most common mutations observed between men and women.
Regarding different age groups, the highest mutation rate of 78% was observed in individuals over 20 years of age (p < 0.001). Mutations were identified in 11.5% of individuals aged 13–19 years and 10.5% of individuals under 12 years of age. It is established that the initial onset of FMF symptoms occurs before the age of 10 in approximately 60% of patients, before the age of 20 in 90%, and before the age of 40 in most of the remaining cases. Gursoy et al. reported the mean age at diagnosis as 29.05 ± 13.59 years [40] .Yaşar bilge et al. reported a similar age at diagnosis of 26.6 ± 13.59 years [20]. Dündar et al. noted that 50% of the variants were detected in patients older than 20 years [7]. In our study, 78% of the variants were observed in individuals over 20 years of age. Moreover, the M680I mutation was more frequently detected in individuals aged 12 years and younger compared to other age groups in our study. This finding suggests a potential for early-onset disease mutation in children. The M680I mutation has been associated with abdominal and chest pain in previous studies [7, 18, 38]. Therefore, it is crucial to request genetic testing at the onset of symptoms and promptly confirm the diagnosis to avoid delays in diagnosing FMF in patients.
Routine blood tests to diagnose FMF are non-specific and limited. In addition, they cannot eliminate the need for genetic testing. It is accepted that we need genetic test results for a definitive diagnosis of FMF, and only clinical criteria can be considered for a possible diagnosis and trial of colchicine treatment. Currently, MEFV gene testing plays a critical role in the diagnosis of FMF to confirm the presence of the disease and determine the appropriate treatment plan [41]. Many of the known mutations in the MEFV gene are associated with different symptom profiles and disease severity [42]. Therefore, determining the genotype allows the development of personalised treatment approaches in the management of the disease. The use of genetic testing appears to be an essential element in improving patients’ quality of life and minimising disease-related complications.
Our study has several limitations. Firstly, due to the retrospective nature of the data analysis spanning a period of ten years, clinical information for most patients was unavailable. Consequently, the relationship between patient genotypes and phenotypes could not be fully elucidated. Additionally, the sequence analysis of all exons in the MEFV gene is crucial for accurately diagnosing and treating patients. This comprehensive analysis may offer additional insights into diagnosing patients with varying phenotypes. Understanding which MEFV variant corresponds to specific phenotypes or ameliorates certain symptoms could facilitate early treatment initiation and prevent life-threatening complications.
Conclusion
In conclusion, variants/mutations in the MEFV gene were evaluated in 2984 patients. In our study group, the genotype distributions of both common and rare mutations in the exon 2 and exon 10 regions were revealed. Currently, the most common mutation in exon 2 in the Turkish population is known as E148Q. Compared to large studies from Turkey, our study shows that the R202Q mutation is found in about 40% of patients and is more common in our society. In addition, the M680I variant was found to be the most common variant in patients aged 12 years and younger. The clinical symptoms of FMF are diverse and vary in severity. As it is not a disease with simple genotype-phenotype characteristics, it is important to know all possible genotypes in the community. For this reason, our study also shows the distribution of MEFV mutations found in patients in our community. The data from our study will contribute to future genotype-phenotype studies of FMF disease.
Data availability
No datasets were generated or analysed during the current study.
References
Koehler AW (2024) Unraveling the genome: Familial Mediterranean fever. J Am Assoc Nurse Pract. ;36(1):3–5. https://doi.org/10.1097/JXX.0000000000000959. PMID: 38165779
Bernot A, da Silva C, Petit JL, Cruaud C, Caloustian C, Castet V, Ahmed-Arab M et al (1998) Non-founder mutations in the MEFV gene establish this gene as the cause of familial Mediterranean fever (FMF). Hum Mol Genet. 7(8):1317-25. https://doi.org/10.1093/hmg/7.8.1317. PMID: 9668175
Gullu UU, Balaban İ, Kara SS, Yaralı O, Türkyılmaz A, İpek S, Güllü ŞD, Çalışkan OF (2023) Frequency of familial Mediterranean Fever Gene Mutation in patients presenting with Joint Pain and diagnosed with Acute Rheumatic Fever. Cureus 15(8):e43001. https://doi.org/10.7759/cureus.43001. PMID: 37671203; PMCID: PMC10476970
Soriano A, Soriano M, Espinosa G, Manna R, Emmi G, Cantarini L, Hernández-Rodríguez J (2020) Front Immunol 11:865. https://doi.org/10.3389/fimmu.2020.00865. PMID: 32655539; PMCID: PMC7325944 Current Therapeutic Options for the Main Monogenic Autoinflammatory Diseases and PFAPA Syndrome: Evidence-Based Approach and Proposal of a Practical Guide
Heller H, Sohar E, Sherf L (1958) Familial Mediterranean fever. AMA Arch Intern Med. ;102(1):50–71. https://doi.org/10.1001/archinte.1958.00260190052007https://doi.org/10.1001/archinte.1958.00260190052007. PMID: 13558745
Lancieri M, Bustaffa M, Palmeri S, Prigione I, Penco F, Papa R, Volpi S, Caorsi R, Gattorno M (2023) An update on familial Mediterranean Fever. Int J Mol Sci 24(11):9584. https://doi.org/10.3390/ijms24119584. PMID: 37298536; PMCID: PMC10253709
Dundar M, Fahrioglu U, Yildiz SH, Bakir-Gungor B, National Genetics Consortium Study et al (2022) Clinical and molecular evaluation of MEFV gene variants in the Turkish population: a study by the National Genetics Consortium. Funct Integr Genomics 22(3):291–315. https://doi.org/10.1007/s10142-021-00819-3. Epub 2022 Jan 31. PMID: 35098403
Booth DR, Gillmore JD, Lachmann HJ, Booth SE, Bybee A, Soytürk M, Akar S, Pepys MB, Tunca M, Hawkins PN (2000) The genetic basis of autosomal dominant familial Mediterranean fever. QJM. ;93(4):217 – 21. https://doi.org/10.1093/qjmed/93.4.217. PMID: 10787449
Onen F (2006) Familial Mediterranean fever. Rheumatol Int. ;26(6):489 – 96. https://https://doi.org/10.1007/s00296-005-0074-3. Epub 2005 Nov 10. PMID: 16283319
Touitou I (2001) The spectrum of Familial Mediterranean Fever (FMF) mutations. Eur J Hum Genet. ;9(7):473 – 83. https://doi.org/10.1038/sj.ejhg.5200658. PMID: 11464238
Dogan H, Akdemir F, Tasdemir S, Atis O, Diyarbakir E, Yildirim R, Emet M, Ikbal M (2014) A novel insertion mutation identified in exon 10 of the MEFV gene associated with familial Mediterranean Fever. BMC Med Genet 15:74. https://doi.org/10.1186/1471-2350-15-74. PMID: 24980720; PMCID: PMC4094690
Kırnaz B, Gezgin Y, Berdeli A (2022) MEFV gene allele frequency and genotype distribution in 3230 patients’ analyses by next generation sequencing methods. Gene 827:146447. https://doi.org/10.1016/j.gene.2022.146447. Epub 2022 Mar 28. PMID: 35358658
Arpacı A, Doğan S, Erdoğan HF, El Ç, Cura SE (2021) Presentation of a new mutation in FMF and evaluating the frequency of distribution of the MEFV gene mutation in our region with clinical findings. Mol Biol Rep 48(3):2025–2033. https://doi.org/10.1007/s11033-020-06040-y. Epub 2021 Mar 18. PMID: 33738724; PMCID: PMC8060170
Alghamdi M (2017) Familial Mediterranean fever, review of the literature. Clin Rheumatol. ;36(8):1707–1713. https://doi.org/10.1007/s10067-017-3715-5. Epub 2017 Jun 18. PMID: 28624931
Gershoni-Baruch R, Brik R, Shinawi M, Livneh A (2002) The differential contribution of MEFV mutant alleles to the clinical profile of familial Mediterranean fever. Eur J Hum Genet. ;10(2):145-9. https://doi.org/10.1038/sj.ejhg.5200776. PMID: 11938447
Aldea A, Casademont J, Aróstegui JI, Rius J, Masó M, Vives J, Yagüe J (2002) I591T MEFV mutation in a Spanish kindred: is it a mild mutation, a benign polymorphism, or a variant influenced by another modifier? Hum Mutat. ;20(2):148 – 50. https://doi.org/10.1002/humu.10103. PMID: 12124996
Sayın Kocakap DB, Günel-Özcan A, Çabuk F, Ensari C (2014) The frequency of familial Mediterranean fever gene mutations and genotypes at Kirikkale and comparison with the mean of regional MEFV mutation frequency of Turkey. Mol Biol Rep 41(3):1419–1426. https://doi.org/10.1007/s11033-013-2986-4. Epub 2014 Jan 1. PMID: 24381109
Caglayan AO, Demiryilmaz F, Ozyazgan I, Gumus H (2010) MEFV gene compound heterozygous mutations in familial Mediterranean fever phenotype: a retrospective clinical and molecular study. Nephrol Dial Transpl 25(8):2520–2523. https://doi.org/10.1093/ndt/gfp632. Epub 2009 Nov 23. PMID: 19934083
Balta B, Erdogan M, Kiraz A, Akalın T, Baştug F, Bayram A (2020) A comprehensive molecular analysis and genotype-phenotype correlation in patients with familial mediterranean fever. Mol Biol Rep 47(3):1835–1843. https://doi.org/10.1007/s11033-020-05277-x. Epub 2020 Jan 27. PMID: 31989427
Yaşar Bilge Ş, Sarı İ, Solmaz D, Şenel S, Emmungil H et al (2019) The distribution of MEFV mutations in Turkish FMF patients: multicenter study representing results of Anatolia. Turk J Med Sci 49(2):472–477. https://doi.org/10.3906/sag-1809-100. PMID: 30887796; PMCID: PMC7018361
Wang HH (2023) MEFV gene mutation spectrum in patients with familial mediterranean fever. Pediatr Neonatol 64(2):107–108. https://doi.org/10.1016/j.pedneo.2023.02.001. Epub 2023 Feb 23. PMID: 36889987
Accetturo M, D’Uggento AM, Portincasa P, Stella A (2020) Improvement of MEFV gene variants classification to aid treatment decision making in familial Mediterranean fever. Rheumatology (Oxford) 59(4):754–761. https://doi.org/10.1093/rheumatology/kez332. PMID: 31411330; PMCID: PMC7188344
El Hawary R, El-Baioumy M, Meshaal S, Elanwary S, El-Guindy N et al (2022) MEFV gene sequencing for unresolved molecular diagnosis in Egyptian familial Mediterranean fever patients; role of R202Q variant. Gene Rep 27:101620. https://doi.org/10.1016/j.genrep.2022.101620
Shinar Y, Obici L, Aksentijevich I, Bennetts B, Austrup F, Ceccherini I, Costa JM, De Leener A et al (2012) European Molecular Genetics Quality Network. Guidelines for the genetic diagnosis of hereditary recurrent fevers. Ann Rheum Dis 71(10):1599–1605. https://doi.org/10.1136/annrheumdis-2011-201271. Epub 2012 Jun 1. PMID: 22661645; PMCID: PMC3500529
Gürkan H, Algüneş Ç, Özkayın Neşe E (2010) MEFV Gene exon 2 and exon 10 Gene Region mutations of familial Mediterranean Fever patients in Trakya Population. Balkan Med J 27:37–43
Akar E, Yalcinkaya F, Akar N (2001) Is the Ala138Gly alteration of MEFV gene important for amyloidosis? Hum Mutat. ;17(1):71. https://doi.org/10.1002/1098-1004(2001)17:1%3C71::AID-HUMU8%3E3.0.CO;2-3. PMID: 11139244
Öksuz MF, Karkucak M, Görukmez O, Ocakoğlu G, Yıldız A, Ture M, Yakut T, Dilek K (2017 Nov-Dec) Investigation of MEFV gene polymorphisms (G138G and A165A) in adult patients with familial Mediterranean fever. Rev Bras Reumatol Engl Ed 57(6):501–506 English, Portuguese. https://doi.org/10.1016/j.rbre.2016.02.004
Sönmezgöz E, Özer S, Gül A, Yılmaz R, Kasap T, Takcı Ş, Gümüşer R, Demir O (2019) Clinical and Demographic Evaluation According to MEFV Genes in Patients with Familial Mediterranean Fever. Biochem Genet. ;57(2):289–300. https://https://doi.org/10.1007/s10528-018-9889-y. Epub 2018 Oct 3. PMID: 30284126
Kandur Y, Kocakap DBS, Alpcan A, Tursun S (2022) Clinical significance of MEFV gene variation R202Q. Clin Rheumatol 41(1):271–274. https://doi.org/10.1007/s10067-021-05906-1. Epub 2021 Sep 7. PMID: 34491459
Kisaoglu H, Baba O, Kalyoncu M (2023) Genotype-phenotype associations of Children with Familial Mediterranean Fever in a Cohort Consisting of M694V Mutation and implications for Colchicine-Resistant Disease. J Clin Rheumatol 29(4):207–213. https://doi.org/10.1097/RHU.0000000000001953. Epub 2023 Mar 6. PMID: 36870084
Akpolat T, Özkaya O, Özen S (2012) Homozygous M694V as a risk factor for amyloidosis in Turkish FMF patients. Gene 492(1):285–289. https://doi.org/10.1016/j.gene.2011.10.012. Epub 2011 Oct 13. PMID: 22037353
Ayaz NA, Tanatar A, Karadağ ŞG, Çakan M, Keskindemirci G, Sönmez HE (2021) Comorbidities and phenotype-genotype correlation in children with familial Mediterranean fever. Rheumatol Int 41(1):113–120. https://doi.org/10.1007/s00296-020-04592-7. Epub 2020 Apr 28. PMID: 32347339
Yildirim ME, Kurtulgan HK, Ozdemir O, Kilicgun H, Aydemir DS, Baser B, Sezgin I (2019 Nov-Dec) Prevalence of MEFV gene mutations in a large cohort of patients with suspected familial Mediterranean fever in Central Anatolia. Ann Saudi Med 39(6):382–387. https://doi.org/10.5144/0256-4947.2019.382. Epub 2019 Dec 5. PMID: 31804137; PMCID: PMC6894460
Babaoglu H, Armagan B, Bodakci E, Satis H, Atas N, Sari A, Yasar Bilge NS, Bilici Salman R et al Factors associated with damage in patients with familial Mediterranean fever. Clin Exp Rheumatol 2020 Sep-Oct;38 Suppl 127(5):42–48. Epub 2020 Jun 18. PMID: 32573410.
Infevers - Tabular list [Internet] (2024) Feb 8. https://infevers.umai-montpellier.fr/web/search.php?n=1
Kishida D, Nakamura A, Yazaki M, Tsuchiya-Suzuki A, Matsuda M, Ikeda S (2014) Genotype-phenotype correlation in Japanese patients with familial Mediterranean fever: differences in genotype and clinical features between Japanese and Mediterranean populations. Arthritis Res Ther 16(5):439. https://doi.org/10.1186/s13075-014-0439-7. PMID: 25261100; PMCID: PMC4201677
Mansour AR, El-Shayeb A, El Habachi N, Khodair MA, Elwazzan D, Abdeen N, Said M, Ebaid R, ElShahawy N, Seif A, Zaki N (2019) Molecular patterns of MEFV Gene mutations in Egyptian patients with familial Mediterranean Fever: a retrospective cohort study. Int J Inflam 2019:2578760. https://doi.org/10.1155/2019/2578760. PMID: 30915208; PMCID: PMC6399540
Fujimoto K, Hidaka Y, Koga T, Kaieda S, Yamasaki S, Nakashima M, Hoshino T, Yamamoto K, Nishikomori R, Ida H (2021) MEFV E148Q variant is more associated with familial Mediterranean fever when combined with other non-exon 10 MEFV variants in Japanese patients with recurrent fever. Mod Rheumatol 31(6):1208–1214. https://doi.org/10.1080/14397595.2021.1880534. Epub 2021 Mar 16. PMID: 33497256
Tanatar A, Karadağ ŞG, Sönmez HE, Çakan M, Ayaz NA (2021) Comparison of Pediatric Familial Mediterranean Fever Patients Carrying Only E148Q Variant With the Ones Carrying Homozygous Pathogenic Mutations. J Clin Rheumatol. ;27(5):182–186. https://doi.org/10.1097/RHU.0000000000001261. PMID: 31972733
Gursoy DE, Gezer HH, Oz N, Ozer A, Kasman SA, Duruoz MT (2023) Clinical features, functional status, and quality of life in patients with late-onset familial Mediterranean fever. North Clin Istanb 10(4):451–457. https://doi.org/10.14744/nci.2022.76736. PMID: 37719256; PMCID: PMC10500247
Ben-Chetrit E (2024) Old paradigms and new concepts in familial Mediterranean fever (FMF): an update 2023. Rheumatology (Oxford). ;63(2):309–318. https://doi.org/10.1093/rheumatology/kead439. PMID: 37725337
Chaaban A, Salman Z, Karam L, Kobeissy PH, Ibrahim JN (2024) Updates on the role of epigenetics in familial mediterranean fever (FMF). Orphanet J Rare Dis 19(1):90. https://doi.org/10.1186/s13023-024-03098-w. PMID: 38409042; PMCID: PMC10898143
Funding
The authors have no funding to declare.
Author information
Authors and Affiliations
Contributions
The concept and design of the study was carried out by RA, EU and TMT. Investigation, Data collection by RA and DFA. Data analysis by RA and ED.Review and editing by RA, EU and TMT. The article was written by RA and DFA. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
Informed consent was obtained from all patients or their parents before undergoing genetic. This work was approved by the Ankara University Human Research Ethics Committee. (decision no: I06-356-22).
Consent for publication
The final content of this paper was read and approved by all of the authors.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aksoy, R., Us, E., Aksoy, D.F. et al. Molecular analyses of MEFV gene mutation variants in Turkish population. Mol Biol Rep 51, 844 (2024). https://doi.org/10.1007/s11033-024-09786-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09786-x