
Abstract
Familial Mediterranean fever (FMF) is an autoinflammatory disease caused by mutations in the MEFV gene and characterized by recurrent episodes of fever and polyserositis. To date, over 317 MEFV mutations have been reported, only nine of which account for almost all Japanese patients with FMF. Therefore, the prevalence of rare MEFV variants and their clinical characteristics remains unclear. This study identified MEFV mutations previously unreported in the Japanese population and described their clinical features. We performed MEFV genetic testing in 488 Japanese patients with clinically suspected FMF. Of these patients, we retrospectively analyzed three patients with novel or very uncommon MEFV mutations. In all patients, the clinical diagnosis of FMF was made according to Tel-Hashomer’s criteria. One novel missense mutation (N679H) and two rare mutations (T681I and R410H) were identified in the MEFV gene. These mutations were found in compound heterozygous or complex genotypes with other known mutations in exons 1 or 2. According to clinical images, all three patients exhibited typical FMF symptoms. A number of patients with FMF caused by novel or uncommon MEFV variants might exist in the Japanese population; therefore, careful genetic testing is required for accurate diagnosis of this curable genetic disorder.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Familial Mediterranean fever (FMF) is the most common autoinflammatory disease characterized by recurrent self-limiting attacks of fever and polyserositis [1]. FMF is caused by mutations in the MEFV gene, which is located on chromosome 16p13.3 and encodes a protein of 781 amino acids called pyrin [2]. More than 317 MEFV mutations or polymorphisms have been identified in the Infevers database (http://fmf.igh.cnrs.fr/ISSAID/infevers/) [3]. Among them, four mutations in exon 10 (M694V, V726A, M680I, and M694I) and one mutation in in exon 2 (E148Q) account for 74% of MEFV mutations in Mediterranean regions [4]. However, nine MEFV mutations account for almost all instances of FMF in Japanese patients. These nine mutations include E148Q, L110P, R202Q, G304R (in exon 2), M694I (in exon 10), P369S, R408Q (in exon 3), E84K (in exon 1), and S503C (in exon 5) [5]. Although FMF was previously recognized as a rare genetic disease in Japan, a large number of Japanese patients with FMF have recently been identified [6, 7]; however, whether additional rare MEFV variants exist in the Japanese population in addition to the nine major variants remains to be determined, as do any corresponding clinical features.
In this study, we identified three Japanese patients with FMF and carrying one novel mutation (N679H) and two uncommon mutations (T681I and R410H) of the MEFV gene. Previously, T681I was reported in one Iranian female patient [8], and R410H was recently reported in two Japanese patients [9]; however, the clinical presentation of patients with these MEFV variants has not been fully described. Here, we report the precise clinical features of three patients with these novel or uncommon mutations.
Patients and methods
Over 100 patients with clinically suspected FMF are referred to our laboratory annually [Department of Medicine (Neurology and Rheumatology), Shinshu University School of Medicine, Nagano, Japan] for identification of MEFV variants. Among these patients, we retrospectively analyzed three FMF patients with novel or very uncommon mutations. In all patients, clinical diagnosis of FMF was made according to Tel-Hashomer’s criteria [10], and all patients fulfilled it. Mutation analysis was performed by direct sequencing, and polymerase chain reaction (PCR) amplification was performed using forward and reverse primers for hotspot regions (exons 1, 2, 3, 5, and 10) as described previously [6].
Results
We identified one novel (N679H) and two uncommon (T681I and R410H) mutations in three different patients. Summarized clinical pictures of three Japanese FMF patients are shown in Table 1. MEFV gene analysis revealed that all three patients had compound heterozygote or complex genotypes, including novel or uncommon mutations. All patients had recurrent episodes of febrile attacks with a high fever of > 39 °C lasting 1 to 5 days. Pleuritis was observed in two patients, and arthritis was observed in one patient, with none of the patients presenting peritonitis. The response to colchicine therapy was very good in two patients.
Patient 1
A 31-year-old female had experienced periodic fever (maximum temperature: 39 °C) and left-side chest pain one to four times monthly since 18 years of age; however, the symptoms spontaneously disappeared within 24 h, and a correct diagnosis had not been made, despite visits to several different hospitals. The attacks seemed to be triggered by overwork or emotional stress, and there were no relationships between the attacks and menstrual periods. The father of the patient also experienced episodic fever and abdominal pain. Laboratory examinations 3 days after the fever showed elevated C-reactive protein (CRP) levels (0.6 mg/dL) and white blood-cell count (10,000/µL). Antinuclear antibody was negative.
We suspected that the patient was suffering from FMF, and MEFV gene analysis revealed a complex genotype comprising a novel mutation (N679H) in exon 10 (Fig. 1a) in addition to L110P, E148Q, and R202Q. Colchicine treatment was recommended, but the patient declined treatment due to fears it would influence future pregnancies. A symptomatic treatment plan using antipyretic analgesics was provided for the febrile and pain attacks.

MEFV gene analysis in three patients. a Asparagine-to-histidine substitution (p.Asn679His) as a result of an A-to-C transition at nucleotide position 2035 (c.2035A > C) in exon 10. b Threonine-to-isoleucine substitution (p.Thr681Ile) as a result of a C-to-T transition at nucleotide position 2042 (c.2042C>T) in exon 10. c Arginine-to-histidine substitution (p.Arg410His) as a result of a G-to-A transition at nucleotide position 1229 (c.1229G > A) in exon 3
Patient 2
A 50-year-old female had suffered from various inflammatory episodes, including periodic fever and left-side chest pain, for > 20 years. The symptoms had occurred several times annually, but the frequency of febrile attacks had increased to monthly ~ 3 months prior to consultation. The patient experienced nonspecific arthropathy of the hip, knee, and ankle, but apparent monoarthritis was not observed. Overwork or emotional stresses appeared to provoke inflammatory attacks, but a correct diagnosis had not been made, despite the patient visiting several different hospitals. The patient had taken anti-inflammatory drugs or antibiotics during the attacks, which provided relief for several days. The patient had no family history of similar periodic febrile episodes, but repeatedly experienced febrile attacks with a fever of 39 °C accompanied by strong chest and back pain, probably due to pleuritis. Laboratory examinations revealed elevated serum CRP levels (2.25 mg/dL), but the cause of the chest pain was not identified by chest X-ray or computer tomography.
We suspected that the patient was suffering from FMF, and genetic analysis revealed that the patient was compound heterozygous for MEFV mutations, including a rare mutation (T681I) in exon 10 (Fig. 1b) and E148Q. After administration of 1.5 mg/day colchicine, the symptoms markedly improved, although the patient continued to report chest pain. The colchicine dosage was increased to 2 mg/day, which completely inhibited the febrile attacks.
Patient 3
A 49-year-old female experienced periodic episodes of high fever since childhood and diagnosed as resulting from tonsillitis. At 45 years of age, the patient was diagnosed with infectious mononucleosis and thrombocytopenic purpura due to clinical and laboratory findings, including hepatosplenomegaly, lymphadenopathy, positive Epstein–Barr virus-antibody titers, and thrombocytopenia. The patient was administered 40 mg prednisolone daily, and the symptoms remitted temporarily; however, the patient suffered from recurrent fevers of 39 °C, erythema of the trunk and limbs, and arthritis of the hip joint once in 2 weeks and lasting for 1–3 days. The rash was an itchy, palpable erythema up to 3 cm in diameter and appeared along with fevers and gradually changed circularly. The patient was completely asymptomatic in between attacks, and there was no family history suggestive of FMF. Laboratory examinations at the peak of the attacks revealed elevated CRP levels (4.8 mg/dL), and blood cultures were negative for pathogenic microorganisms. Chronic active Epstein–Barr virus infection was rejected based on the virus-antibody titers and a skin biopsy, neither of which provided information useful for diagnosis.
We suspected that the patient was suffering from FMF and administered 0.5 mg/day colchicine, which provided complete remission of the febrile attacks. MEFV gene analysis revealed a complex genotype, including a rare mutation (R410H) in exon 3 (Fig. 1c) along with E84K and E148Q. She stopped taking colchicine daily due to side effects of severe diarrhea and instead took colchicine only for a few days while she felt the onset of inflammatory attacks, resulting in control of the symptoms.
Discussion
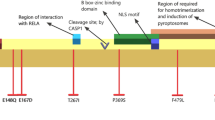
We identified one novel mutation (N679H) and two rare mutations, T681I and R410H, in the MEFV gene. Allele frequencies of these three mutations in 51 healthy individuals (102 alleles) were zero, respectively. The C-terminal B30.2 (PRYSPRY) domain of pyrin is mainly encoded by MEFV exon 10 and plays a key role in the inflammatory response. Indeed, the activation of caspase-1 and secretion of interleukin-1β are directly regulated by the C-terminal B30.2 (PRYSPRY) domain [11]. Therefore, the impact of exon 10 variants on pyrin function and the clinical phenotype of FMF might be significant. Patients with MEFV exon 10 variants tend to exhibit a “typical FMF phenotype”, including recurrent episodes of high fever lasting a few days and severe serositis symptoms. In addition, such variants tend to induce reactive amyloid A amyloidosis due to recurrent severe inflammation without appropriate treatments [12]. Therefore, it is important to detect MEFV exon 10 variants to obtain a definite diagnosis of FMF and to predict clinical severity in patients with suspected FMF. Although a large number of Mediterranean patients with FMF carry exon 10 mutations [4], the frequency of MEFV exon 10 mutations is relatively low in Japanese patients with FMF. To date, there have been few reports of MEFV exon 10 mutations, with the exception of M694I, in Japanese patients [13, 14].
In our study, two exon 10 mutations were identified in patients 1 and 2: one (N679H) is novel and another (T681I) is very uncommon and represents the first case reported in Japanese populations. Both patients revealed typical FMF symptoms similar to those observed in other exon 10 mutations. We used a bioinformatics tool (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/) [15] to predict the impact of the N679H mutation identified in patient 1 on pyrin structure and function. This mutation was predicted as most likely damaging (score: 0.986; sensitivity: 0.74; specificity: 0.96). It is possible that the N679H mutation changes the molecular structure of pyrin due to the substitution of a neutral amino acid with a basic residue, which changes the pyrin isoelectric point. The T681I mutation, identified in patient 2, was not detected among 1275 Turkish FMF patients [16] and has thus far only been reported in one female Iranian patient [8]. This patient also had the M694V mutation in another allele and presented with classical FMF complicated by amyloidosis.
Patient 3 had an R410H mutation in MEFV exon 3 and clinically presented with typical FMF symptoms. This mutation was recently described, and two patients with the R410H mutation were identified among 601 Japanese patients experiencing unexplained fever or suspected FMF [9]. Although detailed clinical characteristics of the reported patients are unavailable, one patient was classified as belonging to the “sure FMF” group similar to patient 3 in this study. In MEFV exon 3, P369S and R408Q are often detected in FMF patients and reportedly result in a highly variable phenotype and atypical clinical presentation [17]. In contrast to P369S and R408Q, the R410H mutation might be associated with a typical FMF phenotype, as seen in this study.
In this study, the three patients analyzed carried additional MEFV mutations in exon 1 or 2 (E84K, L110P, E148Q, or R202Q). These exon 1 or 2 mutations are not always considered disease-causing mutations [18,19,20]. The E148Q mutation was found in 23% of healthy Japanese [18], and its status as a pathogenic mutation or a simple polymorphism remains controversial [19]. Furthermore, the R202Q mutation reportedly causes FMF only in homozygotes [20]. In addition, patients carrying an uncertain variant, such as E148Q or R202Q, tend to develop FMF symptoms only in the presence of a clearly pathogenic mutation in the other allele [21]. These data clearly support that the three mutations identified in this study are pathogenic mutations associated with FMF.
Autoinflammatory diseases are characterized by recurrent episodes of systemic inflammation and defined as a group of disorders exhibiting abnormal innate-immunity activation [22]. Differential diagnosis is important, as many kinds of diseases have been reported recently. We diagnosed our three patients as FMF based on various findings, such as age at onset, good response to colchicine treatment, positivity of MEFV gene mutations, and disagreement with the diagnostic criteria of other diseases.
Recently, testing only for specific MEFV mutations was thought to be sufficient for gene screening, because many single-nucleotide polymorphisms were not associated with a disease phenotype. However, our data suggest that a number of as-yet unrecognized or quite rare pathogenic mutations might exist in MEFV, even in Japanese populations. Therefore, screening only limited numbers of mutations is insufficient and likely to result in misdiagnosis of individuals with rare or novel FMF-causing mutations, and careful genetic testing might be required for patients with suspected FMF without common MEFV variants.
References
Tunca M, Akar S, Onen F, Ozdogan H, Kasapcopur O, Yalcinkaya F et al (2005) Familial Mediterranean fever (FMF) in Turkey: results of a nationwide multicenter study. Medicine 84:1–11
The International FMF Consortium (1997) Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90:797–807
Sarrauste de Menthière C, Terrière S, Pugnère D, Ruiz M, Demaille J, Touitou I (2003) INFEVERS: the Registry for FMF and hereditary inflammatory disorders mutations. Nucleic Acids Res 31:282–285
Touitou I (2001) The spectrum of familial Mediterranean fever (FMF) mutations. Eur J Hum Genet 9:473–483
Kishida D, Nakamura A, Yazaki M, Tsuchiya-Suzuki A, Matsuda M, Ikeda S (2014) Genotype-phenotype correlation in Japanese patients with familial Mediterranean fever: differences in genotype and clinical features between Japanese and Mediterranean populations. Arthritis Res Ther 16:439
Tsuchiya-Suzuki A, Yazaki M, Nakamura A, Yamazaki K, Agematsu K, Matsuda M, Ikeda S (2009) Clinical and genetic features of familial Mediterranean fever in Japan. J Rheumatol 36:1671–1676
Migita K, Uehara R, Nakamura Y, Yasunami M, Tsuchiya-Suzuki A, Yazaki M et al (2012) Familial Mediterranean fever in Japan. Medicine 91:337–343
Booth DR, Gillmore JD, Booth SE, Pepys MB, Hawkins PN (1998) Pyrin/marenostrin mutations in familial Mediterranean fever. QJM 91:603–606
Migita K, Izumi Y, Jiuchi Y, Iwanaga N, Kawahara C, Agematsu K et al (2016) Familial Mediterranean fever is no longer a rare disease in Japan. Arthritis Res Ther 18:175
Pras M (1998) Familial Mediterranean fever: from the clinical syndrome to the cloning of the pyrin gene. Scand J Rheumatol 27:92–97
Chae JJ, Aksentijevich I, Kastner DL (2009) Advances in the understanding of familial Mediterranean fever and possibilities for targeted therapy. Br J Haematol 146:467–478
Drenth JP, van der Meer JW (2001) Hereditary periodic fever. N Engl J Med 345:1748–1757
Oshima K, Yamazaki K, Nakajima Y, Kobayashi A, Kato T, Ohara O, Agematsu K (2010) A case of familial Mediterranean fever associated with compound heterozygosity for the pyrin variant L110P-E148Q/M680I in Japan. Mod Rheumatol 20:193–195
Umeda M, Migita K, Ueki Y, Nonaka F, Aramaki T, Terada K, Koga T, Ichinose K, Eguchi K, Kawakami A (2017) A Japanese familial Mediterranean fever patient with a rare G632S MEFV mutation in exon 10. Mod Rheumatol 27:378–379
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249
Zamani AG, Acar A, Yildirim MS (2013) Spectrum of mutations in the familial Mediterranean fever gene (MEFV) in Turkish patients of the Central Anatolia region: a comparison of two mutation detection system. Genet Mol Res 12:5152–5159
Ryan JG, Masters SL, Booty MG, Habal N, Alexander JD, Barham BK, Remmers EF, Barron KS, Kastner DL, Aksentijevich I (2010) Clinical features and functional significance of the P369S/R408Q variant in pyrin, the familial Mediterranean fever protein. Ann Rheum Dis 69:1383–1388
Sugiura T, Kawaguchi Y, Fujikawa S, Hirano Y, Igarashi T, Kawamoto M, Takagi K, Hara M, Kamatani N (2008) Familial Mediterranean fever in three Japanese patients, and a comparison of the frequency of MEFV gene mutations in Japanese and Mediterranean populations. Mod Rheumatol 18:57–59
Ozen S, Batu ED (2015) The myths we believed in familial Mediterranean fever: what have we learned in the past years? Semin Immunopathol 37:363–369
Yigit S, Karakus N, Tasliyurt T, Kaya SU, Bozkurt N, Kisacik B (2012) Significance of MEFV gene R202Q polymorphism in Turkish familial Mediterranean fever patients. Gene 506:43–45
Ozen S, Bilginer Y (2014) A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat Rev Rheumatol 10:135–147
Jamilloux Y, Belot A, Magnotti F, Benezech S, Gerfaud-Valentin M, Bourdonnay E, Walzer T, Sève P, Henry T (2017) Geoepidemiology and immunologic features of autoinflammatory diseases: a comprehensive review. Clin Rev Allergy Immunol. doi: https://doi.org/10.1007/s12016-017-8613-8
Author information
Authors and Affiliations
Contributions
DK: conception and design, data collection and analysis, and drafting of the manuscript. MY: conception and design, help in drafting the manuscript. AN: conception and design, help in drafting the manuscript. FN, TK, TU, MI, AO, NW, RE, SK, YSH, and YSE: data collection and interpretation, critically review of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Funding
This work was supported by JSPS KAKENHI (Grant Numbers JP26860447 and JP17K15892).
Informed consent
Genetic analysis of MEFV was approved by the Institutional Review Board (No. 314; March 8, 2011) in the Shinshu University School of Medicine, Matsumoto, Japan. All patients provided informed consent prior to MEFV genetic testing and agreed to the publication of their clinical characteristics under anonymity.
Rights and permissions
About this article

Cite this article
Kishida, D., Yazaki, M., Nakamura, A. et al. One novel and two uncommon MEFV mutations in Japanese patients with familial Mediterranean fever: a clinicogenetic study. Rheumatol Int 38, 105–110 (2018). https://doi.org/10.1007/s00296-017-3886-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-017-3886-z