
Abstract
Background
The aim of this study was to evaluate the phenotypic features and the clinical significance of the R202Q mutation of the MEFV gene.
Methods
We retrospectively reviewed the medical records of Familial Mediterranean Fever patients with M694V/- and M694V/R202Q mutations. We compared the patients regarding disease severity, symptoms, age at the onset of symptoms, gender, consanguinity, and family history.
Results
Twenty-one patients (9 males, 12 females) had compound heterozygote mutation (M694V/R202Q), and 37 patients (23 males, 14 females) had M694V/- mutation. The mean age of the patients at the time of diagnosis was 7.3 ± 4.3 and 9.2 ± 3.7 years. The rate of arthritis was significantly higher in patients with M694V/R202Q heterozygote mutation than those with M694V/- heterozygote mutation (76.2% vs 32.4%; p = < 0.001). The mean severity score was higher in M694V/R202Q heterozygote group although it did not reach statistical significance (8.43 ± 1.69 vs 7.49 ± 1.50; p = 0.082). However, the rate of having a high severity score was significantly higher in the M694V/R202Q mutation group than in the other group (47.6% vs 21.6%, respectively; p = 0.039). The rate of arthritis was significantly higher in patients with M694V/R202Q heterozygote mutation than those with M694V/- heterozygote mutation (76.2% vs 32.4%; p = < 0.001).
Conclusion
Our finding supports the possibility that R202Q may be pathogenic rather than a variation. We found that the R202Q mutation is associated with the inflammatory phenotype of FMF; hence, the typical clinical findings of FMF especially arthritis can be observed in patients with compound mutation including R202Q.
Key Points • We found that the R202Q mutation is associated with the inflammatory phenotype of FMF • The patients with the R202Q mutation had a greater rate of arthritis symptoms |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Familial Mediterranean Fever (FMF) is a hereditary inflammatory disorder transmitted in autosomal recessive trait. The FMF gene, MEFV, is located on chromosome 16p13.3 [1]. It encodes a 781-amino-acid protein which is called pyrin [2, 3]. This protein appears to play an important role in the regulation of inflammation since it causes IL-1ß secretion in FMF [4]. The majority of the FMF-associated mutations are located on exon 2, 3, 5, and 10. The most frequent ones are M694V, M680I, V726A, and M694I in exon 10 and E148Q in exon 2. Most FMF patients have homozygote or compound heterozygote FMF mutations. The R202Q (c.605G>A) mutation was described as a frequent polymorphism, where a G>A transition at nucleotide 605 results in glutamine (Q) substituting for arginine (R). The INFEVER genetic database reported that the G allele of the mutation was in linkage disequilibrium with M694V. Linkage disequilibrium is a way of genetic diversity so that certain alleles of each gene are inherited together more often than that would be expected by chance [5]. Although R202Q mutation that has been commonly linked to this mutation has been regarded as a polymorphism, recent studies have shown that it may be a disease-causing mutation [6,7,8]. The aim of this study was to evaluate the phenotypic features and the clinical significance of the R202Q mutation of the MEFV gene.
Material and methods
We retrospectively reviewed the medical records of patients with Familial Mediterranean Fever having M694V/- and M694V/R202Q mutations, who were under the follow-up of our nephrology-rheumatology center between 2015 and 2021. The diagnosis of FMF was based on the criteria described by Livneh et al. [9]. We compared the patients regarding disease severity, symptoms, age of symptom onset, gender, and family history. We used the Pras criteria to grade the disease severity [10], which has six elements, including age of symptom onset, dose of colchicine, number of attacks per month, presence of arthritis, erysipelas-like erythema, and amyloidosis. The severity score is the sum of each parameter. According to Pras, 3–5 points indicate mild (M) disease, 6–8 points indicate intermediate (I) disease, and greater than 9 points are indicative of severe (S) disease.
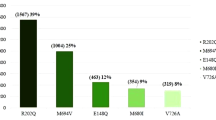
MEFV exons were PCR-amplified from genomic DNA that detects the 12 most frequent MEFV mutations (E148Q, R202Q, P369S, F479L, M680I (G/C), M680I (G/A), M694V, M694I, K695R, V726A, A744S, R761H). Patients with homozygous M694V mutation were excluded due to their severity profile [11]. Hence, other homozygous mutations were included. Patients having chronic disease other than FMF were excluded.
Statistical analysis
The study data were analyzed with SPSS (Statistical Package for Social Science) 16.0 software package. Categorical values were expressed as percentage, frequency, while non-categorical variables were expressed as mean ± standard deviation (SD). Male rate, early onset of symptoms, rate of symptoms (fever, abdominal pain, arthritis, chest pain), and family history of FMF were compared using Chi-square test. The mean severity score and age at symptom onset were compared with Mann–Whitney U and Student’s t tests. The level of significance was set at p < 0.05. Kırıkkale University Clinical Research Ethics Committee approved the study (Date: 30.06.2021/Decision no.: 2021.06.12). Informed consent was signed by all patients prior to the genetic testing.
Results
In order to evaluate the phenotype-genotype correlation, we reviewed the records of 58 patients (M/F=32/26). Twenty-one patients (9 males, 12 females) had compound heterozygote mutation (M694V/R202Q), and 37 patients (23 males, 14 females) had M694V/- mutation. The mean age of the patients at the time of diagnosis was 7.3±4.3 and 9.2±3.7 years, in respective groups. The demographic and clinical characteristics of mutation groups were shown in (Table 1).
The comparison of the demographic and clinical findings of the groups showed no significant difference regarding gender distribution, the mean age of diagnosis, early symptom onset, and the rates of fever, abdominal pain, and chest pain. However, the rate of arthritis was significantly higher in patients with M694V/R202Q heterozygote mutation than those with M694V/- heterozygote mutation (76.2% vs 32.4%; p=<0.001). The mean severity score based on the Pras criteria [10] was higher in the M694V/R202Q heterozygote group although the difference did not reach statistical significance (8.43±1.69 vs 7.49±1.50; p=0.082). However, the rate of having a high severity score (≥9) was significantly higher in the M694V/R202Q mutation group than in the other group (47.6% vs 21.6%, respectively; p=0.039) (Table 1). The rate of a positive family history was higher in the M694V/R202Q heterozygote group (71.4% vs 43.2%; p=0.038). None of the patients had erysipelas or amyloidosis.
Discussion
Recent molecular genetic studies have revealed disease-causing mutations in FMF [11]. There is a clinical heterogeneity of FMF; therefore, genetic analysis may provide a definitive diagnosis. We herein reported the clinical manifestations and genotype-phenotype correlations among a selected group of Turkish children with and without R202Q mutation. Today, there are extremely scarce literature data in relation to this mutation.
The R202Q alteration has been previously reported as a common polymorphism [12]. Although several studies [13] on Turkish patients have suggested that R202Q is associated with the FMF phenotype, there is currently limited data about the significance of R202Q alteration. Ritis et al. [8] showed R202Q homozygosity in 4 of 26 Greek patients with FMF compared to none in 60 healthy controls, suggesting that R202Q gene alteration may be a mutation more than a polymorphism. In another prior study, Arpacı et al. [14] reported that the clinical findings of patients with R202Q were similar to the diagnostic clinical findings of FMF reported in the literature. A recent study also showed that R202Q mutation is associated with the inflammatory phenotype of FMF and its presence contributes to disease expression [15]. Another recent study [16] showed that seven patients (23.3%) with R202Q alterations had typical episodes of FMF. Similarly, our findings may suggest that R202Q alteration is associated with an inflammatory phenotype, and it has clinical significance for FMF.
We found no significant difference between the study groups with respect to the rates of fever, abdominal pain, or chest pain. The rate of arthritis, on the other hand, was higher in the R202Q compound group. The rate of having a high severity score was significantly higher in that group, too. Ozturk et al. found that R202Q had no effect in its heterozygous state; however, when combined with another disease-causing mutation, the clinical spectrum becomes apparent [17]. Our comparison of patients with M694V/R202Q and heterozygous M694V mutations supports this finding. Hence, we revealed that the R202Q alteration of the MEFV gene may lead to an increase in symptom severity, especially among patients with pathogenic mutations (M694V, V726A, M680I).
We found that the patients with the R202Q mutation had a greater rate of arthritis. This finding may suggest that when all etiologies were excluded in a patient who has arthritis, it should be kept in mind that these patients may have a R202Q mutation. Treatment can be arranged in this direction as well. The tendency of arthritis in patients with R202Q was also shown by the study of Ayar et al. They showed that 9 (18.75%) patients out of 48 patients with arthropathy (spondyloarthropathy) had R202Q mutation in the compound form [18]. R202Q alteration may be associated with a different phenotype while it showed phenotypic differences other than the common MEFV mutations. R202Q alteration may affect a patient’s phenotype under some still undefined genetic and epigenetic conditions. Although it is known that the clinical presentation is more severe in compound heterozygotes than in heterozygotes, our finding supports the fact that R202Q may be pathogenic rather than variation.
Our study has some limitations. It has the disadvantages of its retrospective design, and the patient records may not have been standard; thus, some data could be missing in some patients. To compensate for this limitation, we used a scoring system. Additionally, we compared the M694V mutation with R202Q and without R202Q in a small number of patients. We retrospectively reviewed the medical records of patients with Familial Mediterranean Fever who were under follow-up at our pediatric nephrology-rheumatology center between 2015 and 2021. Among these, the total number of pediatric patients with FMF was 154. Out of these patients, 21 had M694V/R202Q. Since our study was a single-center study that was conducted in a relatively short time, the sample size was small. We think that our results provide grounds for future studies.
Conclusion
Our finding supports the possibility that R202Q may be pathogenic rather than a variation. We found that the R202Q mutation is associated with the inflammatory phenotype of FMF; hence, the typical clinical findings of FMF especially arthritis can be observed in patients with compound mutation including R202Q. Comprehensive prospective studies are needed to further investigate the relationship between R202Q mutations and clinical findings.
References
Centola M, Wood G, Frucht DM et al (2000) The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood 95:3223–3231
Moradian MM, Babikyan D, Banoian D et al (2017) Comprehensive analysis of mutations in the MEFV gene reveal that the location and not the substitution type determines symptom severity in FMF. Mol Genet Genomic Med 5:742–750
Touitou I (2001) The spectrum of Familial Mediterranean Fever (FMF) mutations. Eur J Hum Gen 9:473
Berkun Y, Ben-Chetrit E (2007) Pyrin and cryopyrin–similar domain sequence but opposite inflammatory consequence. Clin Exp Rheumatol 45(Suppl):6–8
Slatkin M (2008) Linkage disequilibrium—understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9:477–485
Giaglis S, Papadopoulos V, Kambas K et al (2007) MEFV alterations and population genetics analysis in a large cohort of Greek patients with familial Mediterranean fever. Clin Genet 71:458–467
Yigit S, Karakus N, Tasliyurt T et al (2012) Significance of MEFV gene R202Q polymorphism in Turkish familial Mediterranean fever patients. Gene 506:43–45
Ritis K, Giaglis S, Spathari N et al (2004) Non-isotopic RNase cleavage assay for mutation detection in MEFV, the gene responsible for familial Mediterranean fever, in a cohort of Greek patients. Ann Rheum Dis 63:438–443
Livneh A, Langevitz P, Zemer D, Kees S, Lidav T (1997) Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 40:1879–1885
Pras E, Livneh A, Balow JE Jr et al (1998) Clinical differences between North African and Iraqi Jews with familial Mediterranean fever. Am J Med Genet 75:216–219
de Sarrauste MC, Terriere S, Pugnere D et al (2003) INFEVERS: the Registry for FMF and he-reditary inflammatory disorders mutations. Nucleic Acids Res 31:282–285
Touitou I, Lesage S, McDermott M et al (2004) Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat 24:194–198
Yildirim ME, Kurtulgan HK, Ozdemir O et al (2019) Prevalence of MEFV gene mutations in a large cohort of patients with suspected familial Mediterranean fever in Central Anatolia. Ann Saudi Med 39:382–387
Arpacı A, Doğan S, Erdoğan HF, El Ç, Cura SE (2021) Presentation of a new mutation in FMF and evaluating the frequency of distribution of the MEFV gene mutation in our region with clinical findings. Mol Biol Rep 48:2025–2033
Sgouropoulou V, Farmaki E, Papadopoulos T, Tzimouli V, Pratsidou-Gertsi J, Trachana M (2021) Sequence analysis in familial mediterranean fever patients with no confirmatory genotype. Rheumatol Int. https://doi.org/10.1007/s00296-021-04913-4.
Comak E, Akman S, Koyun M et al (2014) Clinical evaluation of R202Q alteration of MEFV genes in Turkish children. Clin Rheumatol 33:1765–1771
Öztürk A, Özçakar B, Ekim M, Akar N (2008) Is MEFV gene Arg202Gln (605 G>A) a disease-causing mutation? Turk J Med Sci 38:205–208
Ayar K, Ermurat S, Toka D, Öztürk EK (2021) Comparison of clinical findings and pathogenic mutations among axial spondyloarthritis subgroups in Familial Mediterranean Fever disease. Uludağ Üniversitesi Tıp Fakültesi Dergisi 47:35–42
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Standards
All human studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki. All persons gave their informed consent prior to their inclusion in the study. Kırıkkale University Clinical Research Ethics Committee approved the study (Date: 30.06.2021 / Decision no: 2021.06.12).
Disclosures
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kandur, Y., Kocakap, D.B.S., Alpcan, A. et al. Clinical significance of MEFV gene variation R202Q. Clin Rheumatol 41, 271–274 (2022). https://doi.org/10.1007/s10067-021-05906-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-021-05906-1