
Abstract
Objective
Obstructive sleep apnea is closely related to oxidative stress. 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (Tempol) can scavenge reactive oxygen species (ROS) and ameliorate oxidative damage in the body. The mechanism by which Tempol alleviates chronic intermittent hypoxia-induced lung injury has rarely been reported. This study aimed to confirm the molecular mechanism by which Tempol alleviates lung injury.
Methods
The levels of miR-212-5p and Sirtuin 6 (SIRT6) in injured lungs were analyzed using bioinformatics. In vitro, intermittent hypoxia (IH) treatment induced hypoxia in BEAS-2B cells and we established a model of chronic intermittent hypoxia (CIH) in mouse using a programmed hypoxia chamber. We used HE staining to observe the morphology of lung tissue, and the changes in lung fibers were observed by Masson staining. The levels of inflammatory factors in mouse serum were detected by ELISA, and the levels of the oxidative stress indicators GSH, MDA, SOD and ROS were detected using commercially available kits. Moreover, a real-time qPCR assay was used to detect miR-212-5p expression, and Western blotting was used to detect the levels of SIRT6, HIF-1α and apoptosis-related proteins. CCK-8 was used to detect cell proliferation. Subsequently, we used flow cytometry to detect cell apoptosis. Dual-luciferase gene reporters determine the on-target binding relationship of miR-212-5p and SIRT6.
Results
SIRT6 was highly expressed in CIH-induced lung injury, as shown by bioinformatics analysis; however, miR-212-5p expression was decreased. Tempol promoted miR-212-5p expression, and the levels of SIRT6 and HIF-1α were inhibited. In BEAS-2B cells, Tempol also increased proliferation, inhibited apoptosis and inhibited oxidative stress in BEAS-2B cells under IH conditions. In BEAS-2B cells, these effects of Tempol were reversed after transfection with an miR-212-5p inhibitor. miR-212-5p targeted and negatively regulated the level of SIRT6 and overexpression of SIRT6 effectively reversed the enhanced influence of the miR-212-5p mimic on Tempol’s antioxidant activity. Tempol effectively ameliorated lung injury in CIH mice and inhibited collagen deposition and inflammatory cell infiltration. Likewise, the therapeutic effect of Tempol could be effectively reversed by interference with the miR-212-5p inhibitor.
Conclusion
Inhibition of the SIRT6-HIF-1α signaling pathway could promote the effect of Tempol by upregulating the level of miR-212-5p, thereby alleviating the occurrence of lung injury and providing a new underlying target for the treatment of lung injury.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Through repeated episodes of hypoxemia, obstructive sleep apnea (OSA) is characterized by hypercapnia and sleep disorders during nighttime sleep, resulting in ischemia and hypoxia of tissues and organs, which in turn lead to functional limitations of multiple organ systems [1, 2]. The pathophysiological mechanism of OSA is quite complicated and its pathogenesis is not completely clear. Studies have shown that chronic intermittent hypoxia (CIH) and its underlying related oxidative stress are important pathogenic mechanisms of OSA. Recent studies have shown that sleep apnea is an oxidative stress disease, and oxidative stress is an important mechanism of target organ damage in sleep apnea [3, 4]. Repeated sleep apnea and related hypoxemia and reoxygenation after apnea can induce tissue ischemia‒reperfusion injury (IRI), which can lead to oxidative stress, endothelial cell injury, cardiovascular and cerebrovascular diseases and lipid metabolism disorders [4,5,6]. OSA is a risk factor for lung injury [7]. Therefore, exploring the molecular mechanism of OSA induced lung injury may provide insights for the treatment of OSA.
Reactive oxygen species (ROS) have an important influence in regulating signal transduction and cell function. Produced ROS during normal cellular oxygen metabolism can be eliminated by enzymatic and nonenzymatic antioxidant systems [8]. When a large amount of ROS is produced in cells and antioxidants cannot quickly remove them, cells and tissues will be damaged, and autophagy will increase excessively, which can cause excessive cell death and promote necrosis and apoptosis under oxidative stress [9]. In addition, ROS are an important cause of systemic inflammation, which can initiate the inflammatory cascade reaction, lead to the overexpression of inflammatory cytokines, form a large amount of extracellular matrix and regenerative node deposits, and lead to fibrosis at the injured site [10]. Studies have shown that CIH is related to the production of ROS. Racanelli et al. [11] reported that CIH is accompanied by pulmonary hypertension, which not only promotes an increase in ROS but also promotes a high level of inflammatory factors. An increasing number of studies have confirmed that the core factor of multisystem damage in OSA patients is continuous intermittent hypoxia (IH), which frequently alternates with reoxygenation. Its stress pathway produces excessive endogenous ROS, which damage DNA, protein, carbohydrates, lipids and cell functions and weaken the body’s antioxidant defense system [12, 13]. Therefore, oxidative stress is considered to have an important influence on the development of OSA. This study will further explore the specific mechanism of oxidative stress in OSA.
The Sirtuin 6(SIRT6) gene contains 8 exons, encodes 355 amino acids in length and is located on chromosome 19p13.3. As a member of the Sirtuin family, SIRT6 is an NAD + -dependent protein deacetylase that participates in resistance and metabolic homeostasis [14]. Studies have shown that SIRT6 is expressed at a high level in a mouse model of chronic IH with sleep apnea [15]. In contrast, as a downstream factor of SIRT6, hypoxia-inducible factor-1α (HIF-1α) is stable and accumulates under hypoxic conditions and dimerizes with HIF-1β, inducing the transcription of more than 100 downstream genes and resulting in hypoxia-induced reactions [16]. Chronic upregulation of HIF-1α in the serum of patients with OSA has been observed. Therefore, SIRT6/HIF-1α signaling molecules have an important influence on the development of OSA, but there is no research to confirm the specific mechanism of action; thus, it is necessary to study this aspect thoroughly.
In recent years, less than 2% of the human genome has been indicated to encode proteins, and the transcripts of genes that do not encode protein are collectively called noncoding RNA. Among them, microRNA (miRNA) is a class of noncoding small RNA with a length of about 20–24 nucleotides, which can complement and bind to the untranslated region (UTR) at the 3' end of the target mRNA, and miRNAs could induce mRNA degradation via the RNA induced silencing complex (RISC), translational repression, or total inhibition of mRNA translation [17]. miRNA has an important influence on the development of IH. For example, the level of miR-3574 could be inhibited by IH and the influence of IH on viability and apoptosis could be improved by the overexpression of miR-3574 in H9c2 cells [18]. There is miR-223 expression in adult Sprague‒Dawley rats when they were exposed to CIH or normoxia (N), while the downregulation of miR-223 could be reversed by 2-Me in vivo and in vitro, thus reversing the pulmonary hypertension induced by CIH [19]. MiR-212-5p, as a type of miRNA, has not been reported in IH; thus, this study aimed to investigate the mechanism of miR-212-5p in IH. In addition, a targeted relationship has been found between miR-212-5p and SIRT6 [20] and miR-212-5p is involved in the inflammatory reaction of apoptosis induced by oxidative stress [21]. For example, a low level of NEAT1 can inhibit the MPP + -induced apoptosis and inflammation of SK-N-SH cells by regulating the axis of miR-212-5P/RAB3IP [22]. MiR-212-5p has a neuroprotective influence on cognitive dysfunction induced by isoflurane by inhibiting neuroinflammation [23]. Therefore, we hypothesized that the level of ROS could be inhibited by miR-212-5p in cells by regulating the SIRT6/HIF-1α axis, thus alleviating the oxidative stress of cells and reducing the damage caused by oxidative stress.
4-hydroxy-2, 2, 6, 6-tetramethylpiperidine-1-oxyl (Tempol) is a superoxide dismutase mimetic with low molecular weight, high stability and good cell membrane permeability [24]. This compound can scavenge ROS and inhibit the production of OH, thus inhibiting oxidative damage and reducing inflammation. Tempol has many biological activities, such as antioxidant, anti-inflammatory, anti-lipid metabolism, cardiovascular and neuroprotective activities [25,26,27,28]. Xiong et al. confirmed in an in vitro study of traumatic spinal cord injury that Tempol protects mitochondrial respiratory function, thus improving mitochondrial oxidative dysfunction [29, 30]. The role of Tempol in IH has also been confirmed. For example, Tempol can reduce lung injury in rats with chronic IH by inhibiting inflammation and oxidative stress [31]. However, the specific mechanism of Tempol in IH is still unclear.
Based on the above background, this study mainly is focused on defining the undelying mechanism of Tempol in relieving IH to obtain a richer theoretical basis to treat OSA.
Materials and methods
Bioinformatics analysis
The GEO database collects a large amount of high-throughput sequencing results. In this study, the GEO database was used to search for cases of lung injury caused by intermittent oxygenation and the sample sources and treatments were screened to minimize the differences between datasets. The samples in GSE189958 were all from C57BL/6 J mice and were treated with uniform IH, including four intermittent hypoxic mice and four untreated controls. To explore the upstream regulatory mechanisms, the GSE181067 dataset was selected for differential miRNA analysis, which was based on the GPL21572 Affymetrix Multispecies miRNA-4 Array [ProbeSet ID version] platform and contained 6 IH samples and 6 control samples. These mRNA data were uniformly background-checked and normalized by the Limma package in R language, and further differential analysis was carried out with the differential expression judgment threshold set at |log2FC|≥ 1 and P < 0.05. The differential miRNAs were then filtered according to the statistical significance threshold according to the actual conditions.
Animals and cell culture
C57BL/6 female mice were provided by Animal Experimental Center of Kunming Medical University. The experimental mice were provided food and water normally and were placed in a 12 h/12 h light–dark cycle. The mice were randomly divided into the IH and normoxic groups and placed in experimental environments with IH or normoxic conditions, respectively. All experimental animals were given humane care according to the 3R principles, and the protocol was performed according to the regulations of the Chinese Technical Committee for Animal Science Standardization.
Human lung epithelial BEAS-2B cells were obtained from the cell culture unit of the Shanghai Academy of Sciences, China, and cultured in DMEM containing fetal bovine serum (10%) at 37 °C. The culture conditions were CO2 (5%) and O2 (21%). The Lipofectamine 2000 kit (Invitrogen, USA) was used to transfect NC-mimic, miR-212-5p mimic, NC-inhibitor, miR-212-5p inhibitor, NC-OE and OE-SIRT6 RNA oligonucleotides, which were purchased from GenePharma (Shanghai, China).
IH model and groupings
CIH is an important pathogenesis of OSA. CIH model has good controllability, wide operating range, less harm to animals and can better simulate the pathological process of IH, so it has become the most common model to simulate OSA in various disciplines. In order to clarify the pathogenesis of OSA syndrome in animals and suggesting potential treatment for OSA syndrome, we established an IH mouse model. In this process, 30 8-week-old C57BL/6 female mice were selected and randomly divided into the following four groups (n = 5/group): the normal oxygen treatment group (named control group); the IH model group; the IH group treated with Tempol (named IH + Tempol); and the IH group treated with Tempol + miR-212-5p inhibitor (named IH + Tempol + miR-212-5p inhibitor). The four groups were housed in four transparent methacrylate boxes (26 cm × 18 cm × 6 cm), and air was cyclically varied to 5% oxygen (15 s), except for the control group. The room air inlet (40 s) was gradually shifted to the reservoir containing hypoxic air (20 s) to simulate 60 apneas per hour. Mice in the treatment and IH + Tempol + miR-212-5p inhibitor groups were injected intraperitoneally with Tempol (200 μM/24 h). Finally, the mice in the IH + Tempol + miR-212-5p inhibitor group were injected intraperitoneally with human epithelial BEAS-2B cells containing the miR-212-5p inhibitor once daily for 5 days.
Human epithelial BEAS-2B cells were divided into the following groups: the NC group; the IH group; the IH + Tempol group; the IH + Tempol + NC-inhibitor group; the IH + Tempol + miR-212-5p mimic group; the NC-mimic group; the NC-inhibitor group; the IH + Tempol + miR-212-5p inhibitor group; the IH + Tempol + miR-212-5p mimic + NC-OE group; the IH + Tempol + miR-212-5p mimic + OE-SIRT6 group; the miR-212-5p mimic group; and the miR-212-5p inhibitor group. The cells were first treated for 12 h with serum-free medium and then incubated in preparation for in vitro IH treatment. As a control, the cells under normoxic conditions (21% O2 with 5% CO2) were used in the same medium. In the IH group, the medium was placed in a hypoxic chamber and injected with 5% CO2 for 15 s and 1% O2 for 25 s in a cycle chamber for a total of 60 cycles. After hypoxia treatment, maintenance medium was used to culture human epithelial BEAS-2B cells in a normal incubator, and the cells were allowed to recover for 60 min. After transfection, the cells were treated with 200 μM Tempol every 24 h.
Real-time qPCR
A total RNA kit was used to obtain total RNA, which was treated with DNase I (Takara, Japan). One microliter of RNA sample was used to detect RNA integrity on 1% agarose gel electrophoresis, and 1 μl of RNA sample was taken after dilution to measure the OD value through the ratio of OD260/OD280 to identify total RNA purity. The RNA sample was subsequently stored at − 80 °C. For miRNA quantification, the internal reference gene was U6, and the level was detected on an ABI7500 real-time PCR system. The real-time qPCR conditions were as follows: 95 °C for 30 s; 3 s at 95 °C; followed by annealing at 60 °C for 30 s for 40 cycles. All primers (Table 1) used in this study were designed using Premier 5.0 and we used the 2−ΔΔCt method to calculate and analyze the detection results.
Western blot analysis
The proteins were extracted utilizing RIPA lysis buffer (Sangon Biotech, Shanghai), and a lysate containing benzoyl fluoride (PMSF) was added. A BCA assay was used to determine the protein content. SDS-PAGE gel electrophoresis (12%) was performed for the target protein with an initial voltage of 90 V and an adjustment voltage of 120 V. Then, skim milk (5%) was used to block the film for 3 h. The isolated proteins were transferred to PVDF membranes and the PVDF membranes were incubated with the following Abcam antibodies (UK): anti-SIRT6 (1:1000); anti-HIF-1α (1:10,000); anti-cleaved-caspase3 (1:1000); anti-Bax (1:500); anti-Bcl-2 (1:500); anti-β-actin (1:1000); and goat anti-rabbit IgG (UK, 1:300). ECL was used for color development, a gel imaging system was used for imaging analysis and ImageJ was used to analyze the bands.
Enzyme-linked immunosorbent assay (ELISA)
In this study, ELISA kits (ThermoFisher, USA) were used to detect the levels of tumor necrosis factor TNF-α, cytosolic interleukin IL-1β and IL-6 in the BAL solution and cell supernatant. Briefly, to obtain bronchoalveolar lavage fluid (BAL), we inserted a 23-G catheter into the trachea. Then, we washed the lungs with 0.8 mL of PBS and centrifuged the collected BAL solution for 5 min at 4 °C at 1500 rpm. The total cells were counted with a blood cell counter. Cell needles on microscope slides were used for differential cell counting, and cells were stained by Diff-Quick staining (Sysmax, Japan). To identify different populations of inflammatory cells, we counted 400 cells in randomly selected sections. Finally, glutathione (GSH), peroxidase (SOD) and malondialdehyde (MDA) levels in samples lysates were determined using a commercial ELISA kit.
Tissue collection and staining
The mice were euthanized, and their lungs were collected immediately after euthanasia. Formalin (10%) was used to fix the left lung tissues. The tissues were sectioned to a thickness of 4 µm and stained after being embedded in paraffin. Hematoxylin–eosin (HE) staining and Masson staining were used to evaluate fibrosis. HE staining was performed with hematoxylin staining for 10 min and then rinsed with tap water. Subsequently, the sections were stained with eosin for 30 min and rinsed with double-distilled water to remove excess stain. Then, the sections were dehydrated with ethanol, cleared with xylene and sealed with neutral glue. Masson staining was performed with picric acid Sirius red staining and hematoxylin restaining. Then, muscle morphology and myofiber changes were examined under a light microscope and photographed. The remaining tissue was used for protein and supernatant extraction.
Flow cytometry
We used flow cytometry to measure apoptosis and ROS levels. After BEAS-2B cells were incubated for 24 h (1 × 105 cells/culture dish), the cells were trypsin-digested with EDTA-free (0.25%) from each treatment group, washed three times with PBS, and resuspended using 100 μL of buffer. At 25 °C, the cells were co-incubated with 5 μL of Annexin V-PE and 5 µL of PI (BD Biosciences) for 10 min. Finally, after adding termination buffer, the apoptosis rate was determined by flow cytometry. In the dark, the cells were resuspended in HBSS containing 10 M H2DCFDA. After that, the cell suspensions were incubated for 15 min at 37 °C in the dark and washed 3 times with HBSS. To remove unbound dye, the cells were washed with PBS buffer. Ultimately, flow cytometry was used to observe the ROS levels.
Cell counting kit 8 (CCK-8)
A CCK-8 kit (Solarbio, Beijing) was used to determine cell viability. Cells were plated in 96-well dishes and incubated in an incubator environment (37 °C, 5% CO2) for preculture. In a 37 °C incubator, 100 μL of medium was added to every plate and incubated for 24 h. After transfection or dosing, ten microliters of CCK-8 reagent was added and incubated for 2 h. Finally, an enzyme marker (ELX800, BioTeK, USA) was used to measure the absorbance at 450 nm.
Dual luciferase reporter
The 293 T cells were inoculated into the 24-well plate, and the experiment was carried out when the cell confluence reached 60–70%. pmirGLO-SIRT6-WT (wild type) and pmirGLO-SIRT6-MUT (mutant) reporter plasmids were constructed in advanced. 50 nmol/L NC-mimic or miR-212-5p mimic was co-transfected into cells with 0.1 µg pmirGLO-SIRT6-WT or pmirGLO-SIRT6-MUT vector using Lipofectamine 2000(Invitrogen, USA). After 48 h, firefly and Renilla luciferase activity was detected using a dual luciferase reporter assay system (Beyotime, Shanghai, China). Firefly luciferase activity was normalized by Renilla luciferase activity.
Statistical analysis
All experiments in this paper were repeated 3 times and GraphPad Prism 8.0 was used for all statistical analyses. The statistical significance value was p < 0.05. The mean value and standard deviation (SD) are shown. One-way analysis and Student's t test were used to analyze the data.
Results
Analysis of differentially expressed genes and miRNAs in lung injury induced by IH
In this study, 1663 genes with significant changes were identified in the GSE189958 dataset, including 716 downregulated genes and 947 upregulated genes. The 100 most significantly expressed genes among the upregulated genes were used to construct the volcano plot, and these genes among the downregulated genes showed that the gene expression patterns between the control and IH treatment groups were significantly different.
Differential analysis of the GSE181067 dataset by the Limma package yielded 89 differentially expressed miRNAs, including 40 downregulated miRNAs and 49 upregulated miRNAs. miR-212, which is upstream of the SIRT6 gene, was identified (Fig. 1A, B).

GEO GeneChip volcano map. A GEO database GSE189958 gene volcano map. SIRT6 is highly expressed in CIH-induced lung injury. B GEO database GSE181067 chip data volcano map. miR-212-5p is low expressed in CIH-induced lung injury
Downregulation of miR-212-5p in CIH mice is associated with the inflammatory response and oxidative stress
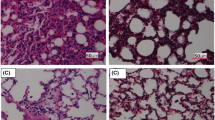
Through bioinformatics analysis, the aforementioned studies found that the expression of many miRNAs was differentially expressed in CHI-induced lung injury diseases, among which miR-212-5p expression was downregulated, indicating that miR-212-5p may be involved in the regulation of CIH. To confirm the physiological influence of miR-212-5p regulation in CIH-induced lung injury, we constructed a mouse model induced by CIH (5% O2) and detected the changes in miR-212-5p and STRT6 levels. The changes in IL-1β, TNF-α, IL-6 and oxidative stress indexes (i.e., MDA, ROS, GSH and SOD) were detected by ELISA and flow cytometry . The results showed that miR-212-5p expression was downregulated significantly, and SIRT6 expression was upregulated in CIH mice compared with normal mice (Fig. 2A, B). The levels of inflammatory factor IL-1β, TNF-α and IL-6 were upregulated (Fig. 2C) and there were upregulated levels of MDA and ROS, while GSH and SOD showed downregulated levels in IH group (Fig. 2D). Through HE staining, in the lung tissues of the control group, there were no inflammatory cells, and the alveolar septum was normal. In the lung tissues of the IH group, there were inflammatory cells, and the alveolar septum was thickened (Fig. 2E). In the control group, Masson staining showed that lung fibers were aligned, while in the IH group, collagen accumulation and interstitial cell proliferation were observed (Fig. 2F). miR-212-5p is downregulated in CIH-induced lung injury mice and may participate in oxidative stress and inflammation.

CIH induced lung injury in mice. A Real-time qPCR analysis of miR-212-5p expression in the different models, and CIH treatment reduced the level of miR-212-5p; B Western blot analysis of SIRT6 expression, and the protein level of SIRT6 was increased after CIH treatment; C ELISA analysis of the expression of inflammatory factors (IL-1β, TNF-α, and IL-6); D The expression levels of oxidative stress indicators (MDA, ROS, GSH and SOD) were detected by kits; E HE staining to observe lung tissue morphology. Scale bar, 100 μm; F Masson staining to observe lung fiber changes, collagen deposition (blue). Scale bar, 100 μm.*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus control
Tempol can affect CIH-induced expression of inflammatory and oxidative factors and reduce cell apoptosis in BEAS-2B human lung epithelial cells
In this study, to confirm the mechanism of Tempol in CIH-induced lung injury, we constructed a CIH-induced BEAS-2B human lung epithelial cell injury model and treated the cells with Tempol. The results showed that compared with the NC group, BEAS-2B human lung epithelial cells in the IH group showed significantly decreased cell viability, an increased apoptosis rate, and the levels of inflammatory factors IL-1β, TNF-α and IL-6 were upregulated, while the cell activity, apoptosis rate and levels of inflammatory factors were reversed after Tempol treatment (Fig. 3A–C). The expression of apoptosis-related proteins (cleaved-caspase3, Bax, Bcl-2) was detected. Compared with the NC group, the levels of Caspase3 and Bax were upregulated, and the levels of Bcl-2 was downregulated in the IH group, while the expression trend of these proteins was reversed after Tempol treatment (Fig. 3D). And the levels of oxidative stress indicators (MDA, ROS, GSH and SOD) were detected. There were upregulated levels of MDA and ROS and downregulated levels of GSH and SOD in the IH group, while Tempol reversed the changes in the above indexes (Fig. 3E). These results indicate that Tempol can alleviate CIH-induced oxidative stress and inflammatory reactions and reduce BEAS-2B human lung epithelial cell apoptosis.

Tempol alleviates CIH-induced BEAS-2B injury. A CCK-8 analysis of BEAS-2B proliferation, and Tempol promoted cell proliferation; B Flow cytometry analysis of apoptosis in BEAS-2B cells, and Tempol inhibited apoptosis; C ELISA analysis of the expression of inflammatory factors (IL-1β, TNF-α, and IL-6); D Western blot analysis of the expression of apoptosis-related proteins (cleaved-caspase3, Bax, Bcl-2); E Expression levels of oxidative stress indicators (MDA, ROS, GSH and SOD). **P < 0.01, ****P < 0.0001 versus NC; +P < 0.05, ++P < 0.01,+++P<0.001, ++++P < 0.0001 versus IH
Tempol can inhibit apoptosis, oxidative stress and inflammatory reactions in BEAS-2B human lung epithelial cells by regulating miR-212-5p expression
In previous studies, miR-212-5p expression was low in a CIH-induced mouse lung injury model and was related to the inflammatory response and oxidative stress. In addition, we found that Tempol could affect the expression of inflammatory factors and oxidation factors in BEAS-2B human lung epithelial cells under CIH and reduced cell apoptosis. It appears that miR-212-5p and Tempol are both related to injury, inflammatory reactions and oxidative stress induced by CIH. However, it is still unknown whether there is an interaction between Tempol and miR-212-5p; thus, we tested this possible mechanism in BEAS-2B human lung epithelial cells under CIH induction. The changes of miR-212-5p at the cellular level were detected. miR-212-5p expression decreased in BEAS-2B human lung epithelial cells induced by CIH. Then, between Tempol and miR-212-5p, we added inhibitors of miR-212-5p on the basis of CHI induction and Tempol treatment to observe the interaction. The cell viability, apoptosis, inflammatory factors and oxidative stress factors were detected. Compared with the IH + Tempol + NC-inhibitor group, the level of miR-212-5p was downregulated, the activity of BEAS-2B human lung epithelial cells was decreased, apoptosis was increased, the levels of IL-1β, TNF-α, IL-6, cleaved-caspase3 and Bax were increased, and the level of Bcl-2 was downregulated. The levels of MDA and ROS were increased, while the levels of GSH and SOD were decreased (Fig. 4). In summary, miR-212-5p has an important influence on BEAS-2B human lung epithelial cells induced by CIH, and Tempol inhibits the apoptosis, oxidative stress and inflammatory reaction of BEAS-2B human lung epithelial cells by regulating the level of miR-212-5p.

Tempol regulates miR-212-5p and inhibits oxidative stress in lung epithelial cells. A Real-time qPCR analysis of miR-212-5p expression; B CCK-8 analysis of BEAS-2B cell proliferation, and miR-212-5p inhibitor inhibited cell proliferation; C Flow cytometry analysis of apoptosis in BEAS-2B cells, and miR-212-5p inhibitor promoted apoptosis; D Western blot analysis of apoptosis-related protein (cleaved-caspase3,Bax,Bcl-2) expression levels; E ELISA analysis of inflammatory factor expression(IL-1β, TNF-α, and IL-6); F Expression levels of oxidative stress indicators (MDA, ROS, GSH and SOD). **P < 0.01, ***P < 0.001, ****P < 0.0001 versus NC; +P < 0.05, ++P < 0.01, +++P < 0.001, ++++P < 0.0001 versus IH; ^P < 0.05, ^^P < 0.001, ^^^P < 0.001, ^^^^P < 0.0001 versus IH + Tempol + NC-inhibitor
miR-212-5p negatively regulates SIRT6 expression
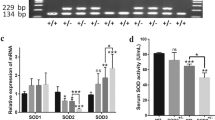
In the initial study, miR-212-5p was differentially expressed in mice induced by CIH and SIRT6 expression was upregulated. Therefore, we questioned whether there might be an interaction between the two. As shown in the mRNA expression profile GSE189958 in the GEO database, the binding of miR-212-5p and SIRT6 through the prediction of target relationships was detected by StarBase and the detection of double luciferase gene reports. miR-212-5p and SIRT6 have a targeted regulatory relationship (Fig. 5A, B). Second, we detected changes in SIRT6 protein expression by western blot to confirm the specific mechanism of miR-212-5p and SIRT6. There was a decreased level of SIRT6 in the miR-212-5p mimic group, while that in the miR-212-5p inhibitor group was increased compared with that in the NC mimic group (Fig. 5C). Therefore, SIRT6 expression could be negatively regulated by miR-212-5p.

The effect of miR-212-5p on SIRT6 expression. A StarBase prediction of the targeting relationship between miR-212-5p and SIRT6; B Dual luciferase gene reporter assay to identify the binding of miR-212-5p and SIRT6; C Western blot analysis showing that miR-212-5p targeting negatively regulates SIRT6. **P < 0.01, ***P < 0.001 versus NC-mimic; ++P < 0.01 versus NC-inhibitor
Tempol can inhibit the expression of SIRT6 and HIF-1α by promoting the expression of miR-212-5p, thus inhibiting the apoptosis, inflammatory reaction and oxidative stress reaction of BEAS-2B human lung epithelial cells induced by CIH.
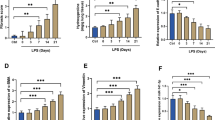
The SIRT6-HIF-1α axis is related to apoptosis and oxidative stress, and miR-212-5p can negatively regulate the expression of SIRT6. To confirm the specific molecular mechanism by which miR-212-5p and SIRT6-HIF-1α are regulated by Tempol, we detected changes in various indexes in BEAS-2B human lung epithelial cells after the overexpression of miR-212-5p and SIRT6. Apoptosis and the levels of SIRT6 and HIF-1α in the IH group were upregulated, and cell activity was obviously decreased compared with that in the NC group. The levels of cleaved-caspase3 and Bax were increased, while Bcl-2 expression was decreased. The levels of IL-1β, TNF-α, IL-6, MDA and ROS were upregulated, while GSH and SOD were downregulated. After Tempol treatment, the changes in the above indexes were opposite to those noted in the CIH-treated group, and after adding miR-212-5p mimic, the effect of Tempol was further enhanced. On the other hand, after the overexpression of SIRT6 on the basis of Tempol treatment and overexpression of miR-212-5p, it was found that the trends of the above indexes have reversed (Fig. 6). In summary, Tempol inhibited the expression of SIRT6 and HIF-1α by promoting the expression of miR-212-5p, thus inhibiting the apoptosis, inflammatory reaction and oxidative stress reaction of BEAS-2B human lung epithelial cells induced by CIH.

Tempol alleviates CIH-induced BEAS-2B cell injury by regulating the miR-212-5p-SIRT6-HIF-1α axis. A Western blot analysis of the expression of pathway-related proteins (SIRT6 and HIF-1α); B Proliferation of BEAS-2B cells was examined by CCK-8 assays, and overexpression of SIRT6 inhibited cell proliferation; C Flow cytometry analysis of apoptosis, and overexpression of SIRT6 promoted apoptosis; D Western blot analysis of apoptosis-related protein(cleaved-caspase3,Bax,Bcl-2) expression levels; E ELISA analysis of the expression of inflammatory factors(IL-1β, TNF-α and IL-6); F Expression levels of oxidative stress indicators(MDA, ROS, GSH and SOD). ***P < 0.001, ****P < 0.0001 versus NC; +P < 0.05, ++P < 0.01, +++P < 0.001, ++++P < 0.0001 versus IH group; ^P < 0.05, ^^P < 0.01,^^^P < 0.001, ^^^^P < 0.0001 versus IH + Tempol group; #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 versus IH + Tempol + miR-212-5p mimic + NC-OE
Tempol promoted the expression of miR-212-5p, inhibited the expression of SIRT6 and HIF-1α, and inhibited the inflammatory reaction and oxidative stress reaction in CIH-induced lung injury mice to alleviate lung injury.
In a CIH-induced cell model, we verified the mechanism by which miR-212-5p is regulated by Tempol. To confirm whether it has the same mechanism in the animal model, we injected miR-212-5p inhibitor-transfected cells into the IH mouse model after Tempol treatment. Changes in the levels of SIRT6, HIF-1α, inflammatory factors and oxidative stress factors were detected. Finally, Masson staining and HE staining were used to detect tissue damage and repair. The level of miR-212-5p in the CIH-induced group was significantly decreased, the levels of SIRT6, HIF-1α IL-1β, TNF-α and IL-6 were increased, the levels of the oxidative stress indexes MDA and ROS were increased, and the levels of GSH and SOD were decreased compared with those in the control group. In the IH + Tempol group, after treatment with Tempol, the effect of IH was alleviated. After addition of the miR-212-5p inhibitor, the changes in the above indexes were opposite to those in the IH + Tempol group (Fig. 7A–D). In the control group, HE staining showed no inflammatory cell infiltration and normal alveolar septum, but inflammatory cell infiltration and alveolar septum thickening was evident in the IH group. The alveolar septum of the IH + Tempol group was obviously thinner than that of the IH group, and the infiltration level of inflammatory cells decreased to be similar to that of the control group. The staining results of lung tissue of mice after miR-212-5p inhibitor injection deteriorated compared with the IH + Tempol group. Through Masson staining, the lung fibers in the normal control group were arranged neatly, while there was increased collagen accumulation and interstitial cells in the IH group. The interstitial cells of the IH + Tempol group were significantly reduced to a level similar to that of the control group compared with the IH group, and the degree of pulmonary fibrosis was reduced. The collagen accumulation in the IH + Tempol + miR-212-5p inhibitor group were increased compared with those in the IH + Tempol group (Fig. 7E, F). The evidence is sufficient to demonstrate that the levels of SIRT6 and HIF-1α could be inhibited by Tempol through promoting the level of miR-212-5p and inhibiting the inflammatory reaction and oxidative stress reaction of CIH-induced lung injury mice to alleviate lung injury.

Analysis of the mechanism of action of Tempol in CIH-induced mice lung injury. A: Real-time qPCR analysis of miR-212-5p expression in the different mice models; B: Western blot analysis of the expression of pathway-related proteins (SIRT6 and HIF-1α); C: ELISA analysis of inflammatory factors (IL-1β, TNF-α, and IL-6); D: Expression levels of oxidative stress indicators (MDA, ROS, GSH and SOD); E: HE staining to observe lung tissue morphology. Scale bar, 100 μm; F: Masson staining to observe lung fiber changes, collagen deposition (blue). Scale bar, 100 μm. ***P < 0.001, ****P < 0.0001 versus control; ++P < 0.01, +++P < 0.001, ++++P < 0.0001 versus IH group; ^P < 0.05, ^^P < 0.01, ^^^P < 0.001, ^^^^P < 0.0001 versus IH + Tempol group
Discussion
The pathogenesis of OSA is very complex. CIH and the oxidative stress response are important in the pathogenesis of OSA. Sleep apnea is a disease of oxidative stress, and oxidative stress is an important mechanism of target organ damage in sleep apnea [3]. In this study, CIH-induced mouse and BEAS-2B cells CIH models were established, and changes in oxidative stress indicators were detected. The results showed that inflammation and the levels of inflammatory cytokines (IL-1β, TNF-α and IL-6) and oxidative stress indicators (MDA, ROS, GSH and SOD) were significantly changed in animals and cells, and inflammation and oxidative stress levels increased after CIH induction, thereby verifying the key role of oxidative stress in OSA. Therefore, finding the molecular mechanism that can effectively improve OSA and regulate oxidative stress can provide new insights and influence the treatment of OSA.
Tempol is a type of superoxide dismutase simulant that can prevent several diseases caused by oxidative stress, including hypertension, obesity, insulin resistance and pancreatic injury [32]. In addition, the role of Tempol in CIH has also been confirmed. For example, the antioxidation effect of Tempol can block IH-induced liver injury in rats [33]. In this study, we found that Tempol could effectively inhibit apoptosis, oxidative stress and inflammation of BEAS-2B human lung epithelial cells induced by CIH, and alleviate lung injury induced by CIH in mice.
The expression of genes in cells after transcription can be regulated by miRNA, which affects many major cellular processes, including cell death and oxidative stress [34]. As mentioned in the previous discussion, CIH is closely involved in oxidative stress, while miRNA has an important influence on the regulation of oxidative stress. Different miRNAs play a role in CIH. In this study, we found that there was decreased miR-212-5p expression in both the CIH-induced mouse model and cell model, and the inflammatory response and oxidative stress response could be reduced by overexpression of miR-212-5p in the CIH model group. The level of miR-212-5p was increased after Tempol treatment, and CIH-induced lung injury was repaired. However, the status of CIH-induced lung injury was not improved when the miR-212-5p inhibitor was added. This finding indicates that miR-212-5p has an important influence on CIH-induced lung injury. Tempol has an important function in the treatment of CIH-induced lung injury by regulating the level of miR-212-5p and the oxidative stress process.
In addition, the SIRT6/HIF-1α signaling pathway was found to be associated with oxidative stress and inflammatory responses [20, 35]. In our study, SIRT6 expression increased in CIH-induced mouse models and cell models, and the level of HIF-1α, which is the downstream pathway of SIRT6, also increased. After Tempol treatment, the levels of SIRT6 and HIF-1α decreased and the levels of inflammatory factors and oxidative stress-related factors were reversed, thus alleviating lung injury in mice. Therefore, the SIRT6/HIF-1α signaling pathway also has an important influence in regulating CIH-induced lung injury, and Tempol can reduce inflammation and oxidative stress by regulating the SIRT6/HIF-1α signaling pathway to repair and reverse the lung injury induced by CIH.
In the transcription and translation process, miRNAs regulate downstream genes by binding to the 3′ end of target genes [36]. The binding of miR-212-5p and SIRT6 was detected through StarBase prediction of the targeting binding relationship and double luciferase gene reporter detection, and a targeting regulatory relationship between miR-212-5p and SIRT6 was found. Finally, we verified the overall molecular regulatory pathway by overexpressing miR-212-5p and SIRT6 in the CIH-induced mouse model. The results showed that overexpression of miR-212-5p could further enhance the effect of Tempol, while overexpression of SIRT6 weakened the effect of miR-212-5p mimic, thus intensifying CIH-induced lung injury.
In conclusion, Tempol can promote the expression of miR-212-5p, inhibit the SIRT6/HIF-1α signaling pathway, inhibit inflammation and the oxidative stress response and thus achieve the therapeutic effect of CIH-induced lung injury. This study enriches the understanding of the mechanism by which Tempol alleviates CIH lung injury and provides a new molecular basis for the treatment of OSA.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Huang P, Zhou J, Chen S et al (2018) The association between obstructive sleep apnea and shortened telomere length: a systematic review and meta-analysis. Sleep Med 48:107–112
Tsou YA, Huang CW, Wu TF et al (2018) The effect of tongue base suspension with uvulopalato-pharyngoplasty on sleep quality in obstructive sleep apnea. Sci Rep 8(1):8788
Eisele HJ, Markart P, Schulz R (2015) Obstructive sleep apnea, oxidative stress, and cardiovascular disease: evidence from human studies. Oxid Med Cell Longev 2015:608438
Passali D, Corallo G, Yaremchuk S et al (2015) Oxidative stress in patients with obstructive sleep apnoea syndrome. Acta Otorhinolaryngol Ital 35(6):420–425
Gupta MA, Jarosz P (2018) Obstructive sleep apnea severity is directly related to suicidal ideation in posttraumatic stress disorder. J Clin Sleep Med JCSM Off Publ Am Acad Sleep Med 14(3):427–435
Hensen HA, Krishnan AV, Eckert DJ (2017) Sleep-disordered breathing in people with multiple sclerosis: prevalence, pathophysiological mechanisms, and disease consequences. Front Neurol 8:740
Chen J, Zhu H, Chen Q et al (2022) The role of ferroptosis in chronic intermittent hypoxia-induced lung injury. BMC Pulm Med 22(1):488
Zhang D, Ren L, Chen GQ et al (2015) ROS-induced oxidative stress and apoptosis-like event directly affect the cell viability of cryopreserved embryogenic callus in Agapanthus praecox. Plant Cell Rep 34(9):1499–1513
Hua W, Huang HZ, Tan LT et al (2015) CD36 mediated fatty acid-induced podocyte apoptosis via oxidative stress. PLoS ONE 10(5):e0127507
Zhang J, Wang X, Vikash V et al (2016) ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev 2016:4350965
Racanelli AC, Kikkers SA, Choi AMK et al (2018) Autophagy and inflammation in chronic respiratory disease. Autophagy 14(2):221–232
Belaidi E, Morand J, Gras E et al (2016) Targeting the ROS-HIF-1-endothelin axis as a therapeutic approach for the treatment of obstructive sleep apnea-related cardiovascular complications. Pharmacol Ther 168:1–11
Kim SJ, Cheresh P, Williams D et al (2014) Mitochondria-targeted Ogg1 and aconitase-2 prevent oxidant-induced mitochondrial DNA damage in alveolar epithelial cells. J Biol Chem 289(9):6165–6176
Michishita E, McCord RA, Berber E et al (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452(7186):492–496
Castro-Grattoni AL, Suarez-Giron M, Benitez I et al (2021) The effect of chronic intermittent hypoxia in cardiovascular gene expression is modulated by age in a mice model of sleep apnea. Sleep. https://doi.org/10.1093/sleep/zsaa293
Hu Y, Liu J, Huang H (2013) Recent agents targeting HIF-1α for cancer therapy. J Cell Biochem 114(3):498–509
Lu TX, Rothenberg ME (2018) MicroRNA. J Allergy Clin Immunol 141(4):1202–1207
Chen Q, Lin G, Chen Y et al (2021) miR-3574 ameliorates intermittent hypoxia-induced cardiomyocyte injury through inhibiting Axin1. Aging 13(6):8068–8077
Hao S, Jiang L, Fu C et al (2019) 2-Methoxyestradiol attenuates chronic-intermittent-hypoxia-induced pulmonary hypertension through regulating microRNA-223. J Cell Physiol 234(5):6324–6335
Lu H, Lin J, Xu C et al (2021) Cyclosporine modulates neutrophil functions via the SIRT6-HIF-1α-glycolysis axis to alleviate severe ulcerative colitis. Clin Transl Med 11(2):e334
Yang Z, Yu W, Huang R et al (2019) SIRT6/HIF-1α axis promotes papillary thyroid cancer progression by inducing epithelial-mesenchymal transition. Cancer Cell Int 19:17
Gou L, Zou H, Li B (2019) Long noncoding RNA MALAT1 knockdown inhibits progression of anaplastic thyroid carcinoma by regulating miR-200a-3p/FOXA1. Cancer Biol Ther 20(11):1355–1365
Si J, Jin Y, Cui M et al (2021) Neuroprotective effect of miR-212-5p on isoflurane-induced cognitive dysfunction by inhibiting neuroinflammation. Toxicol Mech Methods 31(7):501–506
El-Sayed NS, Mahran LG, Khattab MM (2011) Tempol, a membrane-permeable radical scavenger, ameliorates lipopolysaccharide-induced acute lung injury in mice: a key role for superoxide anion. Eur J Pharmacol 663(1–3):68–73
Gonzalez EJ, Peterson A, Malley S et al (2015) The effects of tempol on cyclophosphamide-induced oxidative stress in rat micturition reflexes. Sci World J 2015:545048
Li L, Lai EY, Luo Z et al (2017) Superoxide and hydrogen peroxide counterregulate myogenic contractions in renal afferent arterioles from a mouse model of chronic kidney disease. Kidney Int 92(3):625–633
Neil S, Huh J, Baronas V et al (2017) Oral administration of the nitroxide radical TEMPOL exhibits immunomodulatory and therapeutic properties in multiple sclerosis models. Brain Behav Immun 62:332–343
Salvi A, Patki G, Khan E et al (2016) Protective effect of tempol on buthionine sulfoximine-induced mitochondrial impairment in hippocampal derived HT22 cells. Oxid Med Cell Longev 2016:5059043
Xiong Y, Hall ED (2009) Pharmacological evidence for a role of peroxynitrite in the pathophysiology of spinal cord injury. Exp Neurol 216(1):105–114
Xiong Y, Singh IN, Hall ED (2009) Tempol protection of spinal cord mitochondria from peroxynitrite-induced oxidative damage. Free Radical Res 43(6):604–612
Wang Y, Hai B, Ai L et al (2018) Tempol relieves lung injury in a rat model of chronic intermittent hypoxia via suppression of inflammation and oxidative stress. Iran J Basic Med Sci 21(12):1238–1244
Li T, Zhang T, Gao H et al (2021) Tempol ameliorates polycystic ovary syndrome through attenuating intestinal oxidative stress and modulating of gut microbiota composition-serum metabolites interaction. Redox Biol 41:101886
Zhao HY, Chen BY, Guo R et al (2013) Impact of chronic intermittent hypoxia upon rat liver lipid metabolism and interventional effect of Tempol. Zhonghua Yi Xue Za Zhi 93(6):407–410
Climent M, Viggiani G, Chen YW et al (2020) MicroRNA and ROS crosstalk in cardiac and pulmonary diseases. Int J Mol Sci 21(12):4370
Zhang M, Liu W, Liu Y et al (2023) Astragaloside IV inhibited podocyte pyroptosis in diabetic kidney disease by regulating SIRT6/HIF-1α axis. DNA Cell Biol 42(10):594–607
Ho PTB, Clark IM, Le LTT (2022) MicroRNA-based diagnosis and therapy. Int J Mol Sci 23(13):7167
Acknowledgements
Not applicable.
Funding
This research is supported by the National Natural Science Foundation of China (Project Number: 81960025).
Author information
Authors and Affiliations
Contributions
Conceptualization, LA and LR; methodology, LA, YL and YC; software, YC and ZL; validation, XN; formal analysis, RL, YC, ZL and XN; investigation, LA and YL; resources, LA and YL; data curation, RL and YC; writing—original draft preparation, LA and YL; writing—review and editing, LA and YL; visualization, LR, YC, ZL and XN; supervision, LA and YL; funding acquisition, LA and YL. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Ethical approval
All animal experiments were approved by the Experimental Animal Ethics Committee of Kunming Medical University (kmmu20211432). All authors confirm that all the methods were in accordance with the ARRIVE guidelines.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ai, L., Li, R., Cao, Y. et al. 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (Tempol) alleviates lung injury by inhibiting SIRT6-HIF-1α signaling pathway activation through the upregulation of miR-212-5p expression. Mol Biol Rep 51, 129 (2024). https://doi.org/10.1007/s11033-023-09039-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-023-09039-3