
Abstract
Background
Genetic variability is the most important parameter in plant breeding based on selection. There is a need for morpho-agronomic and molecular characterization of Passiflora species, to exploit their genetic resources more efficiently. No study has yet been carried out to compare half-sib and full-sib families in relation to the magnitude of the genetic variability obtained in them, and then to elucidate the advantages or disadvantages of each one.
Methods and results
In the present study, SSR markers were used to evaluate the genetic structure and diversity of half-sib and full-sib progenies of sour passion fruit. Two full-sib progenies (PSA and PSB), and a half-sib progeny (PHS), together with their parents, were genotyped with a set of eight pairs of SSR markers. Discriminant Analysis of Principal Components (DAPC) and Structure software were used to study the genetic structure of the progenies. The results indicate that the half-sib progeny has lower genetic variability, although it has higher allele richness. By the AMOVA most of the genetic variability was found within the progenies. Three groups were clearly observed in the DAPC analysis, while two hypothetical groups (k = 2) were observed in the Bayesian approach. The PSB progeny showed a high genetic mixture between the PSA and PHS progenies.
Conclusion
Lower genetic variability is found in half-sib progenies. The results obtained here allow us to suppose that the selection within full-sib progenies will possibly provide better estimates of genetic variance in sour passion fruit breeding programs, since they provide greater genetic diversity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sour passion fruit (Passiflora edulis Sims) stands out as the most important among the several species of the Passiflora genus [1, 2]. Brazil is considered the largest producer and consumer of this fruit in the world, and most orchards are planted with P. edulis [3, 4]. Other species, such as P. alata Curtis, P. quadrangularis L., P. nitida Kunth, P. cincinnata Mast and P. setacea DC, are cultivated but have less importance and economic significance [5]. Brazilian production was approximately 593 thousand tons in 2019, in an area of 41 thousand hectares, with average yield around 13 t.ha− 1.year− 1 in Brazilian orchards [4] and cultivation for usually two cycles. This yield is considered low when compared to the potential of improved cultivars, over 30 t.ha− 1 [6, 7].
Passiflora edulis is an allogamous plant, diploid with 2n = 2x = 18 [8]. The flowers are hermaphrodite type and solitary, located in the leaf axils. Fertilization in sour passion fruit occurs exclusively by cross-fertilization due to the occurrence of genetic self-incompatibility [9, 10]. Self-incompatibility plays an important role in the breeding of passion fruit and in other species in which it occurs, since it influences the obtaining of lines for hybrid production, and in the direction of cross between parents of interest. In addition, the cultivars must exhibit enough variability for the alleles of self-incompatibility to ensure satisfactory fruiting [11].
Genetic breeding has great importance for advances in the crop in Brazil, with the availability of new cultivars. Over the last few years, the objective of sour passion fruit breeding programs has been to develop genotypes with higher yield and fruit quality [12, 13], resistance/tolerance to abiotic stress [14] and, mainly, to biotic factors, since diseases like fusariosis [15] and viral diseases [16, 17], or pests like Tetranychus mexicanus [18], stand out as the main constraints in sour passion fruit production in Brazil. However, some breeding programs of sour passion fruit have also sought to develop genotypes for other purposes, such as ornamentation and extraction of chemical compounds, mainly of pharmaceutical interest, exploring other species of Passiflora [7, 19, 20].
Knowledge of genetic structure and diversity and their behavior in populations of any species is essential not only for breeding studies, but also for conservation, maintenance, and effective use of available genetic resources [21, 22]. Studying genetic diversity in a population allows exploring heterotic effects to obtain superior hybrids. In addition to other important studies, such as genomic selection, genome-wide association, and molecular marker-assisted selection [23]. Genetic diversity studies have been considered important for the breeder, as they make it possible to understand the variations in the population and outline selection strategies, to maximize genetic gains in characteristics of interest [24].
Genetic divergence between genotypes, especially for morphoagronomic traits, allows selection or genetic recombination to generate improved cultivars [25]. In passion fruit, the use of half-sib progenies is common, mainly due to the ease of obtaining [26,27,28]. In general, the capitalization of additive genetic variance within half-sib progenies is less than the capitalization of additive genetic variance within full-sib progenies, since in full-sib progenies both parents are known [29, 30]. In addition, no study has yet been carried out to compare half-sib families with full-sib families in relation to the magnitude of the genetic variability obtained in each type of progeny, and then elucidate the advantages or disadvantages of each one. Half-sib progenies of apples showed higher estimates of heritability than full-sib progenies for a range of morphological traits [31]. In Foeniculum vulgare, full-sib progenies exhibited higher estimates of genotypic and phenotypic coefficients of variation and recorded maximum genetic gain for most of the traits evaluated [32]. Differences in heritability and genetic variation coefficients between full-sib and half-sib progenies were also reported for eucalypt species [33].
There is a need for morpho-agronomic and molecular characterization of Passiflora species, in terms of pre-breeding, to more efficiently exploit their genetic resources, as reported in some studies [34, 35]. Molecular markers consist of an important and efficient tool in the genetic characterization of plant accessions and populations, including sour passion fruit [36, 37]. Among the various molecular markers used in the characterization of plants, microsatellites (SSR – Simple Sequence Repeat) stand out due to their high reproducibility and resolution, as well as the polymorphism, codominance and multiallelic nature [38, 39]. A range of SSR markers are available for passion fruit and allow important studies to be conducted on molecular characterization, in terms of conservation and improvement [20, 40, 41].
In the present study, we used SSR markers to characterize the genetic structure and diversity of full-sib and half-sib progenies of sour passion fruit, compare these two progenies in relation to the magnitude of the genetic variability obtained within each progeny, and then infer about their possible influences on the improvement of the crop.
Materials and methods
Genetic material
The study was conducted at the Laboratory of Biotechnology and Plant Breeding, located in the Department of Agronomy of the Federal University of Viçosa (UFV), Viçosa-MG, Brazil (20°45’14”S, 42°52’54”W, and 648.74 above mean sea level). Two full-sib progenies, obtained from controlled cross between contrasting elite parents, and one half-sib progeny obtained by open-pollination, belonging to the UFV Sour Passion Fruit Breeding Program, were genotyped. The parents used to obtain each progeny are shown in Table 1. A kinship relationship is observed between the female parents (P1) of PSA and PSB progenies (P3).
To obtain the two full-sib progenies, controlled crosses were carried out between their respective parents. For the half-sib progeny (PHS), a plant in the experimental area was marked to constitute the female parent (P5), and eleven plants in the surroundings were also marked to constitute the possible male parents (P6 to P16 - Table 1). A kinship relationship is also observed between the male parent of the PSB (P4) progeny and the female parent of PHS progeny (P5). The fruits were harvested about 65 days after crossing when they were fully ripe. After harvesting, the fruits were processed to collect the seeds, which were then sown to give rise to the progenies. Thirty-six genotypes from each full-sib progeny and 45 genotypes from the half-sib progeny were genotyped (Table 1). The seedling stage was conducted in a greenhouse.
Genomic DNA extraction
For DNA extraction, fully expanded young leaves were collected from each plant (parental and progeny genotypes). DNA extraction was done from 200 mg of macerated plant tissue, following the CTAB protocol [42]. The quality and concentration of the extracted DNA were checked by reading the absorbance in a microplate spectrophotometer (MultiskanTM GO), at wavelengths of 230, 260, and 280 nm.
Microsatellite marker genotyping
The polymorphic primers between the parental genotypes were chosen from an initial set of 27 random SSR markers developed by Oliveira [40] and Cerqueira-Silva et al. [20] for P. edulis (Table 2). These primers were selected based on their quality of amplification, polymorphism, and PIC observed in previous works conducted in the Sour Passion Fruit Breeding program at UFV by Cordeiro et al. [43] and Araújo (2018) (not published).
The amplification was carried out using a reaction mix of 20 µL containing 40 ng of DNA, 1X buffer (500 mM of KCl; 100 mM of Tris-HCl pH 8.4; 1% Triton X-100), 1.5 mM of MgCl2, 0.25 µM of each dNTP, 0.3 µM of each primer, and 0.6 U of Taq DNA polymerase (Phoneutria). The PCR reaction was performed in a Veriti® thermal cycler (Applied Biosystems), under the following condition: denaturation at 94 ºC for 4 min, followed by 10 touchdown cycles with denaturation at 94 ºC for 40s, annealing temperature at 61 ºC for 40s, with 0.5 ºC decrease per cycle, and extension at 72 ºC for 50s. Then, 28 additional cycles were performed with denaturation at 94 ºC for 1 min, annealing at 58 ºC for 1 min and extension at 72 ºC for 1 min, followed by final extension at 72 ºC for 7 min. The amplified fragments were separated by 6% denaturing polyacrylamide gel electrophoresis at 60 watts, 1750 volts and 60 mA for 2 h. The amplification products were stained with 2% silver nitrate.
Statistical analysis
For each progeny, the allele frequencies for all loci, private alleles, observed (Ho) and expected (He) heterozygosity, and polymorphic information content (PIC) observed and estimated for each marker were calculated. Then, the genetic structure of the progenies was analyzed through a discriminant analysis of principal components (DAPC) and a Bayesian approach. DAPC was performed using the Adegenet package [44]. Additionally, the results of the Principal Component Analysis (PCA) were plotted separately using the Factoextra package [45]. Both analyses were performed in the R software [46]. For the Bayesian approach, the Structure software [47] was used to define a value of K groups that best represented the three progenies. The analysis was based on a mixture model, considering the allelic frequency of the progeny correlated, and with a priori information of the actual number of groups. The number of groups tested was 1 to 6, with 250.000 burn-in runs, followed by 750.000 MCMC (Monte Carlo Markov Chain) repetitions. For each group, 20 iterations were performed. The selection of the best value of K was carried out using the criterion of Δk proposed by Evanno et al. [48], calculated by the Structure Harvester software [49]. The validation of the genetic variation between and within the progenies was performed by the hierarchical analysis of molecular variance (AMOVA) of Excoffier [50], in the Genes software [51], and the significance of the variance components (ØST) was tested by 1000 permutations.
The genetic diversity of progeny was studied from a multivariate distance matrix between individuals, generated considering the complement of the Nei similarity coefficient, using the Poppr package [52] of the R software [46]. From the distance matrix, the hierarchical cluster analysis of the progenies was performed, using the unweighted pair-group method with arithmetic mean (UPGMA) to study the behavior of genetic diversity. The cut-off points were determined according to the criterion of Mojena [53], considering the constant k = 1.25 as the stop criteria to define the best number of clusters as suggested by Milligan and Cooper [54]. The results were plotted using the Factoextra package [45] of the R software [46].
Results
Polymorphism and heterozygosity of the progenies
Of the 27 SSR markers tested, eight showed polymorphisms in the parent genotypes, allowing the genetic characterization of the progenies. The number of alleles per locus varied from 2 to 3, and a total of 18 different alleles were found, resulting in an average of 2.25 alleles per locus (Table 3). In the PSA and PSB progenies, a maximum of two alleles per locus was observed. In the PHS progeny, greater allele diversity was detected, where the primers PE08 and PE24 showed up to three alleles per locus. In addition, for the primers PE08, PE24, and PE27, private alleles were identified in the PHS progeny.
Six of the eight polymorphic markers selected showed loci in heterozygosis in PSA and PSB progenies. In the PHS progeny, all markers used were heterozygous, differing from PSA and PSB, which showed some homozygous loci (Table 3). However, in PHS loci with heterozygous frequency lower than 0.1 was obtained (PE12, PE27, and PE42 loci), which was not observed in PSA and PSB, in which all polymorphic markers showed heterozygous frequency higher than 0.3. The maximum values of heterozygosis observed were 0.53, 0.63, and 0.63 in PSA, PSB, and PHS progenies, respectively. The mean heterozygosity of the full-sib progenies was higher than that of the half-sib progeny (0.33, 0.40, and 0.26 in PSA, PSB, and PHS, respectively) (Table 3). The gene diversity (He) was in a range of 0.30–0.5 in PSA, 0.36–0.5 in PSB, and 0.04-56 in PHS progenies. For some markers, like PE08 in PSB, and PE02, PE07, and PE27 in PHS progeny, the expected heterozygosity was higher than the observed heterozygosity (Ho). However, the expected heterozygosity average was lower than the observed heterozygosity average for all the progenies (0.29, 0.32, and 0.25, in PSA, PSB, and PHS, respectively). Although the greatest allelic diversity is observed in PHS, this progeny has the lowest expected and observed heterozygosity (0.25 and 0.26, respectively).
The PIC values for most markers were maximum or close to the maximum in the three progenies, with values obtained in the heterozygous locus varying from 0.26 to 0.37 in the PSA progeny, 0.30 to 0.37 in the PSB progeny, and 0.05 to 0.42 in the PHS progeny, being considered moderately informative in all progenies [55]. The loci PE24 and PE27 in PSA and the loci PE18 and PE27 in PSB were monomorphic.
Structure and genetic differentiation of progenies
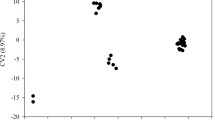
A clear genetic structure between the progenies is observed using the DAPC, where the PHS progeny is genetically closer to the PSB progeny than to PSA (Fig. 1a). By the PCA analysis, a low genetic variation can be observed within the PHS progeny (Fig. 1b). In this analysis, it is also possible to observe very close individuals between the PSA and PSB progenies.

Genetic structure of the 133 sour passion fruit genotypes belonging to the three progenies studied by discriminant analysis of principal components (DAPC) (a), and dispersion of the genotypes in the first two components of principal component analysis (PCA), delimited by ellipses following a t distribution, at a 90% confidence level (b)
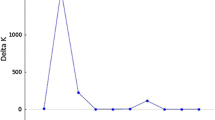
In the Bayesian approach, using the criterion of Evanno et al. [48], the highest value of ΔK is visualized in K = 2, with a subsequent reduction of ΔK as the values of K increase (Fig. 2a). In this study, when the progenies’ structures were analyzed considering the number of progenies K = 3 (Fig. 2c), a weak structure and a high percentage of mixture were observed, mainly between the PSB and PHS progenies (progenies 2 and 3, in Fig. 2b, respectively). When considering K = 2 (Fig. 2c), a strong structure can be observed. This analysis also showed that PSA (progeny 2, Fig. 2b) and PHS (progeny 3, Fig. 2b) maintain a more characteristic genetic profile. On the other hand, the genotypes of the PSB progeny maintain higher percentage of mixture in their genetic profile and can be considered as intermediate between the two hypothetical genetic structures considered.

Genetic structure of the 133 genotypes of sour passion fruit. a. Estimation of the probabilities of K, from the method of Evanno et al. (2005). b and c. Barplots showing the genetic structure of the three progenies studied, using the Structure program, based on the Bayesian model of the population structure, with K = 3 (b) and K = 2 (c). Each individual of the progeny is divided into the number of hypothetical populations, proportionally to their genetic profile, expressed by the scale ranging from 0 to 1, shown on the left in the figures. In this figure, the progenies PSA, PSB, and PHS are represented by numbers 1, 2, and 3, respectively
By the AMOVA, approximately 70% of the variation is found within, which indicates a high degree of variation among the individuals that constitute these progenies (Table 4). Despite the lower percentage of variation among the progenies, it is observed that the degree of genetic differentiation is moderate to low (0.31).
Inter and intra-progeny diversity
There was a considerable difference of diversity within the full-sib and half-sib progenies. High proximity was observed between the genotypes of PHS progeny. Besides the formation of eight groups, approximately, corroborating the results observed in the PCA analysis. The male parental genotypes P6, P7, P8, P10 and P15 were grouped separately, evidencing the non-participation of these genotypes as pollen donors for the formation of the PHS progeny. The PHS31 and PHS42 segregating genotypes also clustered and curiously exhibited alleles exclusive to the P8 and P10 male parental genotypes. Considering that the genotypes P6, P7, P8, P10 and P15 and the genotypes PHS31 and PHS42 may be influencing the genetic divergence between the other segregating genotypes, these genotypes were removed from the dataset and a new analysis was performed. It was observed that the remaining individuals were more distributed in the new clustering (Fig. 1 – Online Resource 1). Thus, subsequent analyses were carried out without these genotypes. It is important to note that, despite the redistribution, low genetic diversity continued to be observed within the PHS progeny, in which the longest distance observed within the PHS progeny was 0.47, and the mean distance between the genotypes was 0.2.

Clustering of PHS progeny accessions obtained by the UPGMA cluster method, with numbers of groups defined by the Mojena (1977) method, using k = 1.25. P5: female PHS parent; P6 to P16: possible male PHS parent; PHS1 to PHS45: segregating PHS progeny genotypes
On the other hand, in the full-sib progenies, the individuals were well distributed in the clustering (Figs. 2 and 3 – Online Resource 1). In both progenies, the greatest distance between the genotypes within the progenies was 0.57, and the mean distance was 0.26. As expected, the parents of both progenies were clustered in different groups, and subgroups composed only of hybrid individuals were observed (Figs. 2 and 3 – Online Resource 1).
In the clustering with the three progenies simultaneously (Fig. 4), it is possible to visualize the greater diversity of hybrid progenies. From the 15 groups formed, only six had genotypes belonging to the PHS progeny. More than 50% of the PHS progeny genotypes clustered in just one group, together with parents P5, P9, P12 and P13. 10 other genotypes were grouped with their possible parents P11. No pattern of genotypes’ distribution was observed for the hybrid progenies, as observed for PHS. The mean distance was 0.35 between the PSA and PSB progenies, 0.64 between PSA and PHS, and 0.38 between PSB and PHS.

Cluster analysis of the 133 genotypes belonging to the progenies studied, obtained by UPGMA, with numbers of groups defined from the method of Mojena (1977), using k = 1.25. PSA1 to PSA36: segregating genotypes of the PSA progeny; P1 and P2: Female and male parents of the PSA progeny, respectively; PSB1 to PSB36: segregating genotypes of the PSB progeny; P3 and P4: Female and male parents of the PSB progeny, respectively; PHS1 to PHS45: segregating genotypes of the PHS progeny; P5: female parent of PHS; P9 to P16: possible male parents
Discussion
The PIC values obtained were moderately informative according to Botstein et al. [55]. This can be attributed to the low number of alleles per locus found, since for most markers only two allelic forms were identified, which attributes a bi-allelic nature to the markers, restricting the PIC values to a maximum of 0.5 [23]. Higher PIC values in the PHS progeny are due to higher allelic diversity in this progeny.
The results show a satisfactory level of genetic diversity in the progenies studied. The progenies PSB and PHS show deficit of heterozygosity for some loci, with most of them being in the PHS progeny (PE02, PE07, and PE27 in PHS, and PE08 in PSB), where He was higher than Ho. Besides that, the expected heterozygosity average was lower than the observed heterozygosity average in all the progenies. The excess of heterozygosity was also reported by Cordeiro et al. [43] in progenies of sour passion fruit, with similar results of He and Ho for some loci used. The expected heterozygosity (He) gives a very important information about the genetic diversity of a population [56].
The number of alleles per locus found (2.25 alleles) can be considered low for the species but is corroborated by results found in the literature for P. edulis [41, 57]. The low number of alleles per locus can be attributed to the degree of selection of the accessions, due to the genetic narrowing in the population breeding. Lower allelic means have been observed in accessions of sour passion fruit with higher degrees of selection, such as commercial cultivars [20]. In contrast, higher allelic diversity has been reported in populations with lower levels of selection. Cerqueira-Silva et al. [6] reported an allele mean of 5.52 alleles per locus when studying sour passion fruit accessions from the several Brazilian States and some foreign accessions from Portugal and Venezuela. Likewise, Ocampo et al. [37] detected 6 to 18 alleles per locus in Colombian accessions of P. edulis. However, these same authors reported 1.61 and 2.8 alleles per locus considering the accessions of specific regions, values close to those found in the present study.
Studies show that the genetic variability of cultivated germplasm in Brazil is low, and this is a consequence of low exploration and lack of collections of wild accessions that have not been taken into account in breeding programs. This is possibly due to the loss of alleles in the sour passion fruit selection processes, so crosses with divergent accessions are needed to recovery these alleles, which would avoid the reduction of genetic variability in these programs [58].
Genetic structure and progeny relationships
The reduced degree of progeny differentiation is due the high degree of selection for specific traits in breeding programs, which reduces the genetic basis of breeding populations [23]. In addition to these factors, sour passion fruit is an allogamous plant, and therefore a greater gene flow is expected, which tends to provide a lower degree of structuring, as demonstrated by Ocampo et al. [37]. Another cause of reduced degree of differentiation in the present study might be the relationship between the parents, especially the PSB progeny, which has a kinship relationship with both PSA and PHS progenies (data not shown), which may have increased the frequency of common alleles between the progenies. The results obtained by the Bayesian approach are especially important since, in addition to the genetic structure, they made it possible to identify kinship relationships between the progenies. Despite kinship, in sour passion fruit, the lowest degree of differentiation in breeding accessions has been attributed to the high percentage of common alleles [58]. Less genetic variation between progenies, like the results obtained here, has also been observed in wheat [23].
Genetic diversity and implications for breeding
The low genetic diversity observed within the PHS progeny and the restricted crosses with some male parents allow us to suppose the occurrence of correlated crosses in the formation of open-pollination progenies in sour passion fruit. Crosses between related individuals and a low number of male parents associated with the process of pollination can be the causes of correlated cross [59]. In the present study, we observed a negative inbreeding coefficient in the PHS progeny (f = -0.04, data not shown), allowing us to infer that crosses between relatives would not be the cause of the occurrence of correlated crosses, and that possibly the low participation of the parents would be the most likely reason. Another factor that may alter mating patterns in sour passion fruit is flower pollination, which is entomophilous type done by wasps [60]. The behavior of the wasps in the field, associated with the number of pollinators in the area, can increase mating between close plants [61].
Correlated crosses lead to a higher proportion of siblings in a population, and consequently lead to an increase in the coancestry coefficient within the progeny [62]. Cordeiro et al. [43] demonstrated that half-sib progeny obtained from one fruit is not sufficient to genetically represent the original population, so larger samples are required. In this study, alleles unique to P8 and P10 male parents were observed only in PHS31 and PHS42 genotypes from PHS progeny, which shows the low representation of these alleles in the progeny, which would possibly be better represented in larger samples.
In breeding programs, the evaluation of progeny with high genetic diversity, as well as the presence of favorable alleles, is fundamental to obtain greater gains [63]. Half-sib progenies have advantages such as easy obtaining, lower costs, and are often used to select genotypes in sour passion fruit breeding [28, 64]. Besides that, they can provide greater allelic diversity in the breeding populations, as observed in the results presented. On the other hand, half-sib progenies show very low index of genetic diversity, which is a problem for the breeding program, especially in breeding of quantitative traits. The absence of enough genetic diversity is a limiting factor for the breeding of quantitative traits since it is highly influenced by environmental conditions [65]. Furthermore, the genetic variance is essential to the estimation of genetic parameters, through the components of variance that enable the selection for these traits and enhance the breeding strategies.
Conclusion
This study assessed the pattern and the extent of genetic diversity of different progenies of sour passion fruit obtained by controlled (full-siblings) and open-pollinated (half-siblings) crosses. Most of the genetic diversity was found within the progenies, with the coefficient of differentiation between the progenies being moderate to low, indicating that the progenies share higher percentage of alleles, possibly due the direct selection for specific traits that breeding populations have undergone. Considerable differences of genetic diversity were observed between full-sib and half-sib progenies. Half-sib progenies show more allele richness; however, the highest levels of genetic diversity were found in the full-sib progenies, highlighted by the higher amount of expected and observed heterozygosity. Genetic variability in any breeding program is a priming factor to practice selection of superior genotypes, so the choice of suitable types of progenies that exhibited higher genetic diversity becomes important for the success of the program. For passion fruit breeding, the results obtained indicated that the use of full-sib progenies instead of half-sib progenies in the selection process may be more appropriate to obtain greater genetic gains.
Data Availability
The authors declare that the data used to prepare this manuscript is available under request.
References
Bernacci LC, Soares-scott MD, Junqueira VTN et al (2008) PASSIFLORA edulis SIMS: THE CORRECT TAXONOMIC WAY TO CITE THE YELLOW PASSION FRUIT (AND OF OTHERS COLORS). Rev Bras Frutic 30:566–576
He X, Luan F, Yang Y et al (2020) Passiflora edulis: an insight into current researches on phytochemistry and pharmacology. Front Pharmacol 11:617
Altendorf S (2018) Minor tropical fruits. In: Food Outlook - FAO. Accessed 7 Oct 2022
IBGE (2019) IBGE. In: IBGE - Instituto Brasileiro de Geografia e Estatísticas. Produção agrícola municipal. https://sidra.ibge.gov.br/tabela/5457#resultado. Accessed 9 Oct 2019
Cerqueira-silva CBM, Jesus ON, Santos ESL et al (2014) Genetic breeding and diversity of the Genus Passiflora: Progress and Perspectives in Molecular and Genetic Studies. Int J Mol Sci 15:14122–14152. https://doi.org/10.3390/ijms150814122
Cerqueira-Silva CBM, Jesus ON, Oliveira EJ et al (2015) Characterization and selection of passion fruit (yellow and purple) accessions based on molecular markers and disease reactions for use in breeding programs. Euphytica 202:345–359. https://doi.org/10.1007/s10681-014-1235-9
Gioppato HA, da Silva MB, Carrara S et al (2019) Genomic and transcriptomic approaches to understand Passiflora physiology and to contribute to passionfruit breeding. Theor Exp Plant Physiol 31:173–181. https://doi.org/10.1007/s40626-018-0134-1
Snow N, MacDougal JM (1993) New chromosome reports in Passiflora (Passifloraceae).Syst Bot261–273
Ocampo J, Arias JC, Urrea R (2016) Interspecific hybridization between cultivated and wild species of genus Passiflora L. Euphytica 209:395–408
Madureira HC, Pereira TNS, da Cunha M et al (2014) Self-incompatibility in passion fruit: cellular responses in incompatible pollinations. Biol (Bratisl) 69:574–584
Bruckner CH, Suassuna T, de MF, Rêgo MM do, Nunes ES (2005) Auto-incompatibilidade no maracujá - Implicações no melhoramento genético. In: Faleiro FG, Junqueira NTV, Braga MF (eds) Maracujá: germoplasma e melhoramento genético. Embrapa Cerrados, Planaltina, DF, p 670
Rodrigues DL, Viana AP, Vieira HD et al (2017) Contribution of production and seed variables to the genetic divergence in passion fruit under different nutrient availabilities. Pesqui Agropecu Bras 52:607–614. https://doi.org/10.1590/S0100-204X2017000800006
de Jesus CAS, de Carvalho EV, Girardi EA et al (2018) Fruit quality and production of yellow and sweet passion fruits in northern state of São Paulo. Rev Bras Frutic 40:e–968
Cordeiro MHM, Rosado RDS, de Souto AG L, et al (2019) Sour passion fruit hybrids with a low photoperiod and temperature requirement for genetic improvement in higher-latitude regions. Sci Hortic 249:86–92. https://doi.org/10.1016/j.scienta.2019.01.032
Teixeira L, Coelho L, Tebaldi ND (2017) Characterization of Fusarium oxysporum isolates and resistance of passion fruit genotypes to fusariosis. Rev Bras Frutic 39:e–415
Freitas JC, de O, Pio Viana A, Santos EA et al (2016) Sour passion fruit breeding: Strategy applied to individual selection in segregating population of Passiflora resistant to Cowpea aphid-born mosaic virus (CABMV). Sci Hortic 211:241–247. https://doi.org/10.1016/j.scienta.2016.09.002
Fontenele RS, Abreu RA, Lamas NS et al (2018) Passion fruit chlorotic mottle virus: molecular characterization of a new divergent geminivirus in Brazil. Viruses 10:169
Silva PR, dos Santos CEM, da Silva RS et al (2020) Assessing the resistance of passion fruit genotypes as control method to Tetranychus mexicanus. Crop Prot 128:15–19. https://doi.org/10.1016/j.cropro.2019.104990
Abreu PP, Souza MM, Santos EA et al (2009) Passion flower hybrids and their use in the ornamental plant market: perspectives for sustainable development with emphasis on Brazil. Euphytica 166:307–315. https://doi.org/10.1007/s10681-008-9835-x
Cerqueira-Silva CBM, Santos ESL, Vieira JGP et al (2014) New microsatellite markers for wild and commercial species of Passiflora (Passifloraceae) and cross-amplification. Appl Plant Sci 2:1300061. https://doi.org/10.3732/apps.1300061
Silva FHL, Muñoz PR, Vincent CI, Viana AP (2016) Generating relevant information for breeding Passiflora edulis: genetic parameters and population structure. Euphytica 208:609–619. https://doi.org/10.1007/s10681-015-1616-8
Geethanjali S, Anitha Rukmani J, Rajakumar D et al (2018) Genetic diversity, population structure and association analysis in coconut (Cocos nucifera L.) germplasm using SSR markers. Plant Genet Resour 16:156–168. https://doi.org/10.1017/S1479262117000119
Eltaher S, Sallam A, Belamkar V et al (2018) Genetic diversity and population structure of F3:6 Nebraska Winter wheat genotypes using genotyping-by-sequencing. Front Genet 9:1–9. https://doi.org/10.3389/fgene.2018.00076
Govindaraj M, Vetriventhan M, Srinivasan M (2015) Importance of genetic diversity assessment in crop plants and its recent advances: An overview of its analytical perspectives. Genet Res Int 2015:14. https://doi.org/10.1155/2015/431487
Pandey P, Anurag PJ, Tiwari DK et al (2009) Genetic variability, diversity and association of quantitative traits with grain yield in rice (Oryza sativa L.). J Biosci (Rajshari) 17:77–82
Oliveira EJ, da Silva Santos V, Souza de Lima D et al (2008) Seleção em progênies de maracujazeiro-amarelo com base em índices multivariados. Pesqui Agropecu Bras 43:1543–1549. https://doi.org/10.1590/S0100-204X2008001100013
Rosado RDS, Rosado LDS, Borges LL et al (2019) Genetic diversity of sour passion fruit revealed by predicted genetic values. Agron J 111:165–174. https://doi.org/10.2134/agronj2018.05.0310
Chagas K, Alexandre RS, Schmildt ER et al (2016) Divergência genética em genótipos de maracujazeiro azedo, com base em características físicas e químicas dos frutos1. Revista Ciencia Agronomica 47:524–531. https://doi.org/10.5935/1806-6690.20160063
Casler MD, Brummer EC (2008) Theoretical expected genetic gains for among-and‐within‐family selection methods in perennial forage crops. Crop Sci 48:890–902
Cruz CD, Regazzi AJ, Carneiro PCS (2004) Modelos Biométricos Aplicados ao Melhoramento Genético, 2nd edn. Imprensa Universitária, Viçosa
Hajnajari H, Chashnidel B, Vahdati K et al (2012) Heritability of morphological traits in apple early-ripening full-sib and half-sib offspring and its potential use for assisted selection. HortScience 47:328–333
Singh V, Sastry EVD, Singhania DL (2004) Comparison of full sib, half sib and S1 progenies in fennel (Foeniculum vulgare Mill.). J Spices Aromatic Crops 13:129–131
Hodge GR, Volker PW, Potts BM, Owen J (1996) A comparison of genetic information from open-pollinated and control-pollinated progeny tests in two eucalypt species. Theor Appl Genet 92:53–63
Cerqueira-Silva CBM, Faleiro FG, de Jesus ON et al (2018) Passion fruit (Passiflora spp.) breeding. In: Al-Khayri JM, Jain SM, Johnson D (eds) Advances in plant breeding strategies: fruits. Springer, Itapetinga, pp 929–951
Ocampo J, Marín V, Urrea R (2021) Agro-morphological characterization of yellow passion fruit (Passiflora edulis f. flavicarpa degener) reveals elite genotypes for a breeding program in Colombia. Agron Colomb 39:156–176
Castro JA, Oliveira EJ, Jesus ON et al (2016) Molecular markers for conservation genetic resources of four Passiflora species. Sci Hortic 212:251–261. https://doi.org/10.1016/j.scienta.2016.10.005
Ocampo J, Acosta-Barón N, Hernández-Fernández J (2017) Variability and genetic structure of yellow passion fruit (Passiflora edulis f. flavicarpa degener) in Colombia using microsatellite DNA markers. Agron Colomb 35:135–149. https://doi.org/10.15446/agron.colomb.v35n2.59973
Buschiazzo E, Gemmell NJ (2010) Conservation of human microsatellites across 450 million years of evolution. Genome Biol Evol 2:153–165. https://doi.org/10.1093/gbe/evq007
Caixeta ET, Ferrão LFv, Maciel-Zambolim E (2013) Marcadores moleculares. In: BORÉM A, FRITSCHE-NETO R (eds) Biotecnologia aplicada ao melhoramento de plantas. Suprema, Visconde do Rio Branco, pp 9–30
Oliveira EJ (2006) Desenvolvimento e uso de marcadores microssatélites para construção e integração de mapas genéticos de maracujá-amarelo (Passiflora edulis Sims f. flavicarpa Deg.)
Araya S, Martins AM, Junqueira NTv et al (2017) Microsatellite marker development by partial sequencing of the sour passion fruit genome (Passiflora edulis Sims). BMC Genomics 18. https://doi.org/10.1186/s12864-017-3881-5
Doyle JL, Doyle JJ (1990) Isolation of plant DNA from fresh tissue. Focus (Madison) 12:13–15
Cordeiro MHM, Bruckner CH, Rosado RDS et al (2020) Contribution of male genitors in open-pollination progenies of sour passion fruit. Euphytica 216:1–16. https://doi.org/10.1007/s10681-020-02681-z
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Kassambara A, Mundt F (2017) Factoextra: extract and visualize the results of multivariate data analyses. R package version 1:337–354
R core team (2019) R: A language and environment for statistical computing. R Foundation for Statistical Computinghttps://www.R-project.org Acessed 27 January 2023
Pritchard JK, Stephens M, Donnelly P (2000) Inference of Population Structure using Multilocus Genotype Data. 155:945–959
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Earl DA, VonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of Molecular Variance Inferred from Metric distances among DNA haplotypes: application to human mitochondrial DNA Restriction Data. Genetica 131:479–491. https://doi.org/10.3354/meps198283
Cruz CD (2013) GENES - Software para análise de dados em estatística experimental e em genética quantitativa. Acta Sci Agron 35:271–276. https://doi.org/10.4025/actasciagron.v35i3.21251
Kamvar ZN, Tabima JF, Gr̈unwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:1–14. https://doi.org/10.7717/peerj.281
Mojena R (1977) Hierarchical grouping methods and stopping rules: an evaluation. Comput J 20:359–363
Milligan GW, Cooper MC (1985) An examination of procedures for determining the number of clusters in a data set. Psychometrika 50:159–179
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in Man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Yu J, Zhao H, Zhu T et al (2013) Transferability of rice SSR markers to Miscanthus sinensis, a potential biofuel crop. Euphytica 191:455–468
Araujo GL, Reis EF, Moraes WB et al (2011) Influência do déficit hídrico no desenvolvimento inicial de duas cultivares de café Conilon. Irriga 16:115–124. https://doi.org/10.15809/irriga.2011v16n2p115
Costa JL, Jesus ON, Oliveira GAF, de Oliveira EJ (2012) Effect of selection on genetic variability in yellow passion fruit. Crop Breed Appl Biotechnol 12:253–260. https://doi.org/10.1590/s1984-70332012000400004
Mori ES, Sebbenn AM, Tambarussi EV, Guries RP (2013) Sistema de reprodução em populações naturais de Peltophorum dubium. Scientia Forestalis/Forest Sciences 41:307–317
Silva SR, Almeida NM, Siqueira KMM et al (2019) Isolation from natural habitat reduces yield and quality of passion fruit. Plant Biol 21:142–149. https://doi.org/10.1111/plb.12910
Wagenius S, Hangelbroek HH, Ridley CE, Shaw RG (2010) Biparental inbreeding and interremnant mating in a perennial prairie plant: fitness consequences for progeny in their first eight years. Evol (N Y) 64:761–771. https://doi.org/10.1111/j.1558-5646.2009.00860.x
Mori NT, Mori ES, Tambarussi EV et al (2015) Sistema de cruzamento em populações de Handroanthus heptaphyllus (Vell.) Mattos e suas implicações para a coleta de sementes para fins de conservação e melhoramento genético. Scientia Forestalis/Forest Sciences 43:675–681
Viegas MP, Silva CLSP, Moreira JP et al (2015) Diversidade genética e tamanho efetivo de duas populações de myracrodruon urundeuva Fr. All., sob conservação ex situ. Revista Árvore 35:769–779. https://doi.org/10.1590/s0100-67622011000500002
Ferreira RT, Viana AP, Silva FHDL et al (2016) Seleção Recorrente Intrapopulacional em maracujazeiro-azedo via modelos mistos. Rev Bras Frutic 38:158–166
Chavarría-Perez LM, Giordani W, Dias KOG et al (2020) Improving yield and fruit quality traits in sweet passion fruit: evidence for genotype by environment interaction and selection of promising genotypes. PLoS ONE 15:e0232818
Funding
Coordination for the Improvement of Higher Education Personnel (CAPES) (grant number 001). This study was conducted using the physical structure and personnel of the Federal University of Viçosa.
Author information
Authors and Affiliations
Contributions
Carlos Eduardo Magalhães dos Santos and Eveline Teixeira Caixeta contributed to hypothesis elaboration and supervision of manuscript writing. Ana Claudia Costa and Luciana Domiciano Silva Rosado contributed to the experimental analysis and data acquisition. Edilson Marques Junior contributed to experimental analysis and data acquisition, statistical analysis, and wrote the manuscript.
Corresponding author
Ethics declarations
Ethical aproval
This article does not contain any studies with human participants or animals performed by any of the authors.
Competing Interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Junior, E.M., Rosado, L.D.S., Costa, A.C. et al. Full-sib progenies show greater genetic diversity than half-sib progenies in sour passion fruit: an approach by ssr markers. Mol Biol Rep 50, 4133–4144 (2023). https://doi.org/10.1007/s11033-023-08340-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-023-08340-5