
Abstract
In guava (Psidium guajava), the impact of self-pollination on the quantitative traits of the fruits is not fully understood, necessitating further investigation. This study aimed to estimate the effects of selfing on fruit traits in S1 and S2 inbred families of guava and to explore potential impacts on genetic diversity. Eighteen S1 families were generated through selfing of progenies from biparental crosses, and ten S2 families were produced by selfing superior genotypes from S1 families. The experiment was conducted at the Experimental Station of Ilha Barra do Pomba, in the municipality of Itaocara-Rio de Janeiro, Brazil. It utilized a randomized complete block design, with three replications and ten plants per plot. Evaluated traits included fruit weight, length, diameter, length-diameter ratio, endocarp thickness, mesocarp thickness, pulp weight, and soluble solids content. The data underwent individual analysis of variance, yielding predicted mean trait values for S1 and S2 generations, alongside correlation and homozygosity estimates. Genetic diversity was assessed using Mahalanobis distance and UPGMA cluster analysis, and comparative box plots between inbred populations were created for the evaluated traits. Box plot analysis revealed symmetry in most evaluated traits, suggesting uniformity in the data due to the selfing strategy. Analysis of variance indicated statistically significant differences in all traits, highlighting variability between populations S1 and S2. Fruit and pulp weights exhibited high homozygosity levels, with values of 90.86 and 102.59 respectively, linked to increased fruit traits in the S2 population, indicating their importance in the fixation of favorable alleles. Fruit weight, length, and diameter, endocarp thickness, and mesocarp thickness showed strong correlations, exceeding 0.70. Genetic diversity assessment via Mahalanobis distance indicated a decrease in genetic variability, evidenced by fewer groups in S2 compared to the S1 population. However, this reduction did not noticeably affect the average performance of the S2 population. The results indicate that the two generations of self-pollination did not negatively affect the phenotypic values of the evaluated traits.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In Brazil, the cultivation of guava (Psidium guajava L.) covers an extensive area of 22,137 thousand hectares, leading to a production of 552,393 thousand tons of fruit (IBGE 2023). Several cultivars are utilized by farmers, with Pedro Sato, Cortibel, Kumagai, Paluma, Rica, Sassaoka, and Século XXI being particularly prominent (Santos et al. 2011). The Paluma cultivar dominates, covering approximately 70% of Brazil's guava cultivation area (Pereira and Kavati 2011). This dominance underlines the vulnerability of the crop due to limited exploitation of the genetic diversity of the species. Accordingly, there is a pressing need to develop new cultivars that more effectively cater to the specific demands of the market and growers. Such development would facilitate the diversification of guava cultivars in the field with different alleles, particularly focusing on varied fruit types and resistance to pests and diseases (Krause et al. 2017).
Guava possesses a mixed reproductive system that includes both allogamy and autogamy. Although self-pollination is prevalent, accounting for 25.7% to 41.3% of cases (with an average of 36.6%), cross-pollination remains significantly high (Singh and Sehgal 1968). The high rate of cross-pollination, combined with sexual propagation, contributes to the establishment of heterogeneous orchards, resulting in non-uniformity among the cultivated genotypes. This considerable variability poses significant challenges to guava production, disrupting the uniformity of cultivation practices and the marketing system due to a lack of standardization in key aspects such as yield, plant growth, and fruit quality (Santos et al. 1998; Manica et al. 2000; Oliveira et al. 2009).
Self-pollination, or selfing, results in increased homozygosity and reduced heterozygosity in progeny, potentially leading to inbreeding depression. This condition manifests as a decline in the average values of quantitative traits related to the reproductive capability or physiological efficiency of the plant, largely due to the homozygosity of deleterious alleles (Falconer and Mackay 1996). In guava, the specific consequences of selfing are not extensively studied, primarily because of the challenges associated with the long reproductive cycle of perennial species. This gap in research is intriguing, given the economic significance of guava and the complexity of its reproductive system.
Scientific research has demonstrated that inbreeding depression varies across different species. An example of this can be observed in corn (Zea mays L.), where selfing can have severe consequences, including the potential extinction of a variety (Paterniani 1978). Conversely, studies on perennial species like eucalyptus (Eucalyptus spp.) suggest that inbreeding does not significantly affect the germination, survival, or growth of self-pollinated plants (Estopa et al. 2007). Regarding guava, Maitan (2022) investigated the effects of inbreeding on the physiological quality of seeds from self-pollinated guava plants over two generations, finding no adverse impacts on seed characteristics and physiological quality. A more comprehensive study by Ambrósio et al. (2022) focused on 18 S1 inbred families of guava to select superior genotypes for future generations of selfing based on morpho-agronomic traits. The research demonstrated genetic gains, emphasizing the potential for selecting superior genotypes in subsequent generations of selfing.
Although inbreeding is employed in certain breeding programs to fix favorable traits, its use in guava breeding research is limited. In this context, the State University of Northern Rio de Janeiro (UENF) has been conducting a guava breeding program for the past 15 years, yielding promising outcomes (Pessanha et al. 2011; Campos et al. 2013, 2016; Oliveira et al. 2014; Quintal et al. 2017; Gomes et al. 2017; Silva et al. 2020, 2021, 2023; Ambrósio et al. 2021, 2022). These studies demonstrate that the development of inbred guava families through selfing is a feasible approach for generating superior populations.
In the establishment of a breeding program, it is essential for breeders to ensure the presence of variability within the population not only at the initiation phase but also throughout the program's duration. It is essential to monitor the preservation of this variability to avoid hindering future genetic advancements and, consequently, the sustainability of the program. Thus, the target population must exhibit genetic diversity and agronomic traits that are of interest to both producers and consumers. The synergy of genetic variation and superior mean values is crucial for yielding productive cultivars in the breeding program (Krause et al. 2017).
The study of genetic diversity via multivariate analysis plays a significant role in breeding programs, as the variance among genotypes is essential for achieving selection gains by crossing genetically distinct groups that display desirable traits (Cruz et al. 2011).
Among the various metrics developed to assess genetic diversity among genotypes, the generalized Mahalanobis distance is particularly noteworthy. It is extensively employed in studies with replications due to its ability to account for correlations between evaluated traits (Cruz et al. 2012; Simon et al. 2012). When integrated with clustering techniques such as the hierarchical UPGMA (Unweighted Pair Group Method with Arithmetic Mean), it provides a systematic approach to interpreting genetic diversity. In UPGMA, genotypes are clustered through a recursive process until a dendrogram with highly informative value is produced (Cruz 2005). Both techniques are widely utilized in the analysis of genetic diversity in fruit crops (Bohry et al. 2019; Mendes et al. 2020; Radaelli et al. 2020; Torres et al. 2019; Santos et al. 2022).
Accordingly, this study offers a valuable contribution to guava breeding research by addressing specific gaps in the scientific literature. By exploring the development of guava lines and the potential implications of inbreeding, it addresses a significant void in targeted studies on these topics. Moreover, by further elucidating the guava reproductive system, this research aids in formulating effective breeding strategies.
Therefore, the primary goal of this research is to evaluate the effects of self-pollination on the fruit characteristics of S1 and S2 inbred guava families, in addition to examining its potential impacts on the genetic diversity of the species.
Material and methods
Study area
The research was carried out at the experimental station situated on Barra do Pomba Island, within the municipality of Itaocara, northwest region of the state of Rio de Janeiro, Brazil (21°40' S and 42°04' W, 76 m asl). The soil of the experimental site is classified as Argisol, as per the categorization by the Soil Survey Staff (2015). The area is characterized by an Aw climate type according to the Köppen classification, with an average annual temperature around 22 °C and annual precipitation averaging 1,297 mm, as documented by Alvares et al. (2013).
Origin of the studied populations
Initially, 18 S1 guava families were established through the selfing of superior individuals from controlled bi-parental crosses, as conducted by Quintal et al. (2017) during September and October 2008 in Bom Jesus do Itabapoana, also in the northwest of Rio de Janeiro. These families were later assessed by Ambrósio et al. (2021), who selected the most promising genotypes for further selfing, resulting in ten S2 families (Table 1).
Experimental design
Plants from the 18 S1 population were installed in July 2014, while the ten families from the S2 population were established on January 24, 2020. A randomized block experimental design was utilized for evaluating these populations, featuring three replications and ten plants per plot.
Cultivation treatments
Throughout the experiments, pruning and thinning practices were implemented as required. Additionally, liming and fertilization procedures were conducted according to the guidelines provided by Costa and Costa (2003). These agronomic practices aimed to promote optimal plant growth, adhering to the standards set forth in specialized literature.
Evaluated traits
A total of 400 genotypes from the S1 families and 150 genotypes from the S2 families were evaluated. For each genotype, five fruits underwent assessment based on eight specific descriptors for P. guajava L., as stipulated by the Ministry of Agriculture (MAPA 2018). The employed descriptors included: Fruit Weight (FW)—measured in five fruits from each genotype, using a semi-analytical scale, with the results presented in grams; Fruit Transverse Diameter (FD)—measured in five fruit samples was with a caliper, with values reported in millimeters; Fruit Length (FL)—determined in five selected fruits per genotype using a caliper, with measurements in millimeters; Fruit Length/Diameter Ratio (L/D)—computed by dividing the fruit's length by its diameter, presented as a dimensionless value; Mesocarp Thickness (MT)—gauged at two points on each fruit's median region with a caliper, with measurements in millimeters; Endocarp Thickness (ET)—measured at the seed-concentrated area, specifically the placental region, using a digital caliper, with the thickness expressed in millimeters; Pulp Weight (PW)—calculated by deducting the weight of the placenta from the total fruit weight, representing the weight of the mesocarp area; Soluble Solids Content (SSC)—ascertained by analyzing the juice extracted from the fruit pulp using a manual press. The readings were obtained with an ATAGO N1 portable refractometer, with the soluble solids content expressed in degrees Brix, ranging from 0 to 32º Brix.
Genetic-statistical analysis
Initially, descriptive statistics were conducted to provide an overview of the data. Subsequently, linear mixed models were implemented in the R programming language using the lme4::lme4 function from the nlme package (Pinheiro and Bates 2000). We employed restricted maximum likelihood (REML) as an estimator and the best unbiased linear predictor (BLUP) for individual trait response variables. The model used is as follows:
where \({{\varvec{Y}}}_{({\varvec{i}}{\varvec{j}}{\varvec{k}})}\) is the vector of phenotypic values of each trait in genotype i of family j in block k; \({{\varvec{\beta}}}_{0}\) is a parameter associated with the model (intercept); \({{\varvec{g}}}_{[{\varvec{i}}=1,\dots ?]}\) is the parametric vector of the random effects of genotype i, associated with vector Y by the known incidence matrix \({{\varvec{\beta}}}_{1}\), assuming that \({\varvec{g}}\sim {\varvec{N}}({{\varvec{\mu}}}_{{\varvec{g}}},{{\varvec{\sigma}}}_{{\varvec{g}}}^{2})\), where \({{\varvec{\sigma}}}_{{\varvec{g}}}^{2}\) is the genetic variance matrix; \({{\varvec{s}}}_{[{\varvec{j}}=1,...,2]}\) is the parametric vector of the fixed effects of generation j, associated with vector Y by the known incidence matrix \({{\varvec{\beta}}}_{2}\), assuming \({\varvec{s}}\sim {\varvec{N}}({{\varvec{\mu}}}_{{\varvec{s}}},{{\varvec{\sigma}}}_{{\varvec{s}}}^{2})\), where \({{\varvec{\sigma}}}_{{\varvec{s}}}^{2}\) is the generation variance; b/f is the parametric vector of the random effects of family j within block k, influencing both the intercept and the slope, associated with the vector Y by the known incidence matrix \({{\varvec{\beta}}}_{3}\); and \({\varvec{\epsilon}}\) is the vector of random residual effects, assuming \({\varvec{\epsilon}}\sim {\varvec{N}}(0,{{\varvec{\sigma}}}_{{\varvec{\epsilon}}}^{2})\), where \({{\varvec{\sigma}}}_{{\varvec{\epsilon}}}^{2}\) is the residual variance.
After evaluating the significant effect of generation, an individual analysis of variance, following the same model mentioned above and filtering data for each generation, was conducted for each trait as a response variable. From the individual model, we obtained the predicted values of trait means for both generations, Pearson’s correlations between generations for each variable, the sum of squares of the generation effect, and its significance.
Homozygosity estimates were also calculated based on the model:
where: \(Ho\) represents the homozygosity of the S2 generation relative to the S1 generation; \(\overline{M{S }_{2}}\) is the estimated mean of the S2 generation in the joint model, disregarding all effects of sources of variation; and \(\overline{M{S }_{1}}\) is the estimated mean of generation S1 in the joint model, disregarding all effects of sources of variation. This model was adapted from the description provided by Gardner and Eberhart (1966). The significance of the estimated homozygosity was assessed using the following formula:
where \(t\): t-statistics to evaluate the significance of homozygosity of one generation relative to another; \(MSresidual\) is the residual mean square of the joint model; and r is the number of replicates.
Subsequently, Mahalanobis' generalized distance (D2) (Mahalanobis 1936) was estimated as a measure of dissimilarity among families, considering the phenotypic means for each generation separately. The utilization of Mahalanobis distance aimed to assess the genetic diversity levels in two consecutive generations of selfing in guava. Then, the UPGMA hierarchical method was employed to visually represent the distances between families. To determine the optimal number of groups in the UPGMA method, the criterion proposed by Mojena (1977) was applied. All analyses were conducted using Genes software (Cruz 2016).
Additionally, box plots were generated using the following formula to provide graphical representations of the data, with the goal of distinguishing the performance of population S1 in comparison to S2. Box plots are a data exploration tool that relies on separatrix estimates, including the median (md) and quartiles (Q1 and Q3). They present information from a dataset in a format that allows for easy visualization and comprehension (Iemma 1992). The median divides the data into two equal halves (50%) of the distribution and can be estimated using the following equation:
where \({1}_{md}\) is the lower limit of the median class; \({F}_{prc}\) is the cumulative frequency of the class preceding the median class (cumulative value); \({f}_{md}\) is the absolute frequency of the median class (number of observed data); and \(a\_md\) is the range of the median class (difference between the maximum and minimum value of the class).
Quartiles represent specific percentages of the data. The first quartile (Q1) encompasses 25% of the data, and the third quartile (Q3) encompasses 75%. They can be calculated using the following equations:
where: \({1}_{Q1}\) and \({1}_{Q3}\): lower limits of the classes containing the first (Q1) and the third (Q3) quartiles, respectively; \({F}_{prc}\): cumulative frequency of the class preceding the class containing the quartile being estimated; \({f}_{Q1}\) and \({f}_{Q3}\): absolute frequencies of the classes that contain the quartiles; and a: range of the class of the quartile being estimated.
The box plots were created using R Studio software (R Core Team 2021) with the assistance of the biplot2 package (Wickham 2016).
Results and discussion
Upon analyzing the box plots, we identified symmetry in the traits of FW, PW, FL, FD, L/D, MT, and ET for both populations under study. Symmetry is evident through the alignment of the line representing the second quartile (median) with the central point in the box plot (Fig. 1). This alignment indicates a balanced distribution relative to the mean values associated with these traits, implying uniformity in the arrangement of the data. The uniform distribution signifies reduced intrapopulation variation concerning the evaluated trait. In simpler terms, most individuals exhibit similar values for these specific attributes, leading to a decrease in differences among population members. This uniformity can be attributed to the crossing system employed in this study. Despite the reduction in genetic variability, S2 families have seen quantitative improvements in most analyzed traits. This highlights the effectiveness of this approach in enhancing fruit characteristics in plants from the S2 population.

Box plot for eight agronomic traits of P. guajava fruits in 18 S1 and 10 S2 families, evaluated in Rio de Janeiro state
In the analysis of the SSC trait, negative skewness was observed in the S1 population. Specifically, it was noticed that the gap between the first quartile and the median was more pronounced compared to the difference between the median and the third quartile. This observation suggests a wider dispersion of smaller values on the left side of the distribution, along with a lower mean compared to the median. In contrast, the S2 population exhibited a more uniform and symmetrical distribution around the median. This indicates that the data are evenly spread around the median without any directional bias.
Table 2 provides a summary of analysis of variance for populations S1 and S2 concerning the eight fruit traits under evaluation. The individual mean squares for the generations displayed significant effects (p < 0.05) on all traits, indicating variation between the populations. Consequently, an in-depth examination of the significant interaction between guava inbred populations S1 and S2 becomes crucial as it can yield valuable insights for the development of effective breeding strategies.
Notably, the S2 generations outperformed the S1 generations in all evaluated traits, with particular emphasis on the most agronomically important traits in the crop, namely FW and PW. These results indicate that the observed superiority can be attributed to the fixation of favorable alleles for the traits being improved (Table 3).
Furthermore, the FW and PW traits exhibited high levels of significant homozygosity, with values of 90.86 and 102.59, respectively. These findings are closely associated with a remarkable increase in fruit-related traits within the S2 population. The discovery of high homozygosity, coupled with the enhanced average fruit weight in the S2 population, suggests that homozygosity plays a substantial role in fixing favorable alleles. Within the context of guava breeding, the improvement of fruit traits is considered satisfactory, especially given the scarcity of studies exploring the development of lines through inbred families. This condition allows for the selection of homozygous individuals with desired traits, thereby increasing the likelihood of transmitting these traits to future generations.
Correlation estimates between populations S1 and S2 varied from 0.65 to 0.87, with the traits of FW, FL, FD, L/D, ET, and MT exhibiting correlations exceeding 0.70, which is considered strong according to Dancey and Reidy (2018). In the context of endogamous populations where reproduction primarily occurs through selfing of the most productive genotypes, this robust association suggests that the studied traits are being consistently passed on to subsequent generations.
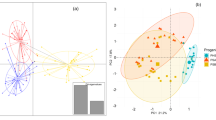
To further explore the relationship between the two populations under examination, scatterplots were generated for both populations and the eight traits, as depicted in Fig. 2.

Scatterplot of the S1 and S2 inbred populations of P. guajava for eight agronomic fruit traits, evaluated in Rio de Janeiro state
The analysis revealed a positive correlation between the variables and a narrow dispersion of the data, evident from the linear upward trend of the traits in the graph. This linear relationship signifies that an increase in one variable corresponds to an increase in the other. These findings align with the breeding strategy employed in this study, indicating that guava exhibits a low susceptibility to inbreeding depression and benefits positively from selfing. This is underscored by the rise in trait means in the S2 generation compared to the S1 generation, suggesting that the fixation of favorable alleles may explain this increase.
While relatively few studies have focused on investigating inbreeding depression in P. guajava, Maitan (2022) evaluated the effect of inbreeding on the physiological quality of seeds from the two populations under examination. The results did not indicate any negative impact resulting from inbreeding on seed traits and quality. In contrast, Ambrósio et al. (2022) conducted a more extensive investigation, evaluating 18 S1 inbred families of guava to select superior genotypes for future generations of selfing. Utilizing morpho-agronomic descriptors, their results were positive, demonstrating genetic gain. As a result, they selected superior genotypes for the subsequent generations of selfing.
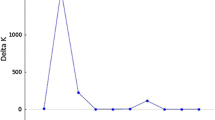
Using the UPGMA method for average group linkage, cutoff points were determined within the dendrogram (Fig. 3) at distances of 17.5 for the S1 population and 36.5 for the S2 population. These cutoff points were defined based on the criteria outlined by Mojena (1977) and correspond to 76.05% and 99.47% of the maximum distance observed in the fusion levels of the analyzed populations, respectively.

Representative dendrogram showcasing genetic dissimilarity between 18 S1 and 10 S2 families of P. guajava, considering the phenotypic means in each generation, obtained using the UPGMA method with the Mahalanobis distance
Upon applying the cutoff points, four groups were established for the S1 population, while the S2 population was divided into two groups. It is noteworthy that a greater number of families were assessed in the S1 population (18 families) compared to the S2 population (10 families), leading to a more significant prevalence of clusters in the former. Furthermore, these results hint at a potential reduction in genetic variability within the S2 population due to the selfing process. Nevertheless, despite the decrease in the number of groups and consequently reduced genetic diversity in the S2 population, no adverse impact on the average fruit traits of these families was observed as a result of the selfing process. On the contrary, it was found that, despite having a mixed reproductive system, guava exhibits positive outcomes from increased homozygosity induced by selfing.
Studies focusing on genetic diversity, particularly concerning agronomic traits, play a fundamental role in characterizing and comprehending the genetic variability within a population. In the case of guava, such studies have been extensively employed to identify more divergent genotypes, as documented in research by Santos et al. (2011), Krause et al. (2017), and Ambrósio et al. (2022). However, it is essential to note that there is limited research investigating genetic diversity at the level of inbred families.
The genetic variability observed within the S1 families can be attributed to two main factors. Firstly, it can be ascribed to the substantial genetic diversity identified in the initial population (S0) as reported by Pessanha et al. (2011). This initial diversity likely contributed to increased genetic differentiation within the S1 generation. Additionally, the species under investigation exhibits a preference for cross-pollination (Alves and Freitas 2007; Pessanha et al. 2011). This preference results in a higher likelihood of genetic exchange between different individuals, ultimately leading to greater genetic diversity within the population. Secondly, the use of seeds from heterozygous parents in seedling production, as emphasized by Alves and Freitas (2007), also plays a significant role in enhancing genetic diversity. Thus, the combination of high initial genetic variability, cross-pollination preference, and the use of heterozygous parents in seedling production collectively explains the greater number of clusters observed in the S1 population in comparison to the S2 families.
The findings of this study regarding phenotypic traits align with the conclusions drawn by Reis (2023), who employed SSR molecular markers to characterize both populations examined. The author identified genetic variability in both populations S1 and S2 and emphasized the necessity of multiple rounds of selfing to mitigate genetic variability between genotypes within the same family.
The outcomes of this study yield valuable knowledge for the fixation of favorable alleles in genotypes of interest, leading to the production of high-quality and uniform guava materials for producers. Therefore, it is essential to continue assessments with the S3 population, closely monitoring the levels of inbreeding at each selfing generation and exploring the potential for developing superior lines through seed propagation.
Conclusion
-
1.
The two generations of selfing did not result in a detrimental reduction in guava genetic diversity.
-
2.
The guava exhibits a low susceptibility to inbreeding depression for the traits studied in this work, which is advantageous when conducting selfing.
-
3.
These results provide robust support for the continuation of the breeding program using inbred families and suggest progressing to S3 families as a promising strategy.
Data availability
Data availability Inquiries about data availability should be directed to the authors.
References
Alvares CA, Stape JL, Senteljhas OS, Moraes JLM, Sparovek G (2013) Koppen’s climate classification map for Brazil. Meteorol Z 22:711–722. https://doi.org/10.1127/0941-2948/2013/0507
Alves JE, Freitas BM (2007) Requerimento de polinização da goiabeira. Ciênc Rural, Santa Maria 37:1281–1286. https://doi.org/10.1590/S010384782007000500010
Ambrósio M, Viana AP, Ribeiro RM, Preisigke SC, Cavalcante NR, Silva FAD, Sousa CMBD (2021) Genotypic superiority of Psidium guajava S1 families using mixed modeling for truncated and simultaneous selection. Sci Agric 78:e20190179. https://doi.org/10.1590/1678-992X-2019-0179
Ambrósio M, Viana AP, Pureza Da Cruz D, Da Costa PS, Ramos Cavalcante N, Herênio Gonçalves Júnior D, Pequeno De Souza Y (2022) Categories of variables in analysis of genetic diversity in S1 progenies of Psidium guajava. Sci Rep 12:22367. https://doi.org/10.1038/s41598-022-26950-0
Bohry D, Berilli APCG, Berilli SS, Almeida RF, Zooca AAF (2019) Characterization and genetic divergence of araçá-boi based on physicochemical and colorimetric traits of fruits. Rev de Ciênc agrár 62:1–8
Campos BM, Viana AP, Quintal SSR, Gonçalves LSA, Pessanha PGDO (2013) Quantification of the genetic divergence among guava accessions using Ward-MLM strategy. Rev Bras Frutic 35:571–578. https://doi.org/10.1590/S0100-29452013000200028
Campos B, Viana AP, Quintal SSR, Barbosa CD, Daher RFH (2016) Heterotic group formation in Psidium guajava L. by artificial neural network and discriminant analysis. Rev Bras Frutic 38:151–157. https://doi.org/10.1590/0100-2945-258/14
Costa AFS, Costa NA (2003) Tecnologias para produção de goiaba. Vitória.
Cruz CD (2016) Genes Software - extended and integrated with the R, Matlab and Selegen. Acta Sci Agron 38:547–552. https://doi.org/10.4025/actasciagron.v38i4.32629
Cruz CD, Ferreira FM, Pessoni LA (2011) Biometria aplicada ao estudo da diversidade genética. Visconde do Rio Branco: Suprema.
Cruz CD, Regazzi AJ, Carneiro PCS (2012). Modelos biométricos aplicados ao melhoramento genético. Viçosa.
Cruz CD (2005). Princípios de genética quantitativa. Viçosa.
Dancey C, Reidy J. Estatística Sem Matemática para Psicologia 7. 3. ed. Porto Alegre: Artmed., 2018. 530 p.
Estopa RA, Ramalho MAP, Rezende GDSP, Abad JIM, Gonçalves FMA (2007) Desempenho dos descendentes de clones de Eucalyptus spp. Autofecundados e Cruzados Cerne 13:264–270
Falconer, DS, Mackay TFC (1996). Mackay. Introduction to Quantita essentially defined the subject, Longmans.
Gardner CO, Eberhart AS (1966) Analysis and interpretation of the variety cross diallel and related populations. Biometrics, Tucson 22:439–452
Gomes VM, Ribeiro RM, Viana AP, De Souza RM, Santos EA, Rodrigues DL, De Almeida OF (2017) Inheritance of resistance to Meloidogyne enterolobii and individual selection in segregating populations of Psidium spp. Eur J Plant Pathol 148:699–708. https://doi.org/10.1007/s10658-016-1128-y
IBGE - Instituto Brasileiro de Geografia e Estatística. Produção agrícola municipal. Disponível em: https://sidra.ibge.gov.br/tabela/1613#resultado. Acessado em 12 de agosto de 2023.
Iemma AF. 1992. Parâmetros que descrevem a posição ou a tendência central In: Iemma AF (ed). Estatística descritiva. Piracicaba QOP Publicações, 4ª ed. Piracicaba, pp87–107.
Krause W, Cavalcante NR, Viana AP, Ambrósio M, Santos EA, Vieira HD (2017) Genetic variability in inbred guava families mediated by agronomic traits. Funct Plant Breed J 3(2):61–71
Mahalanobis PC (1936) On the generalized distance in statistics. Proc Natl Inst Sci India 2:49–55. https://doi.org/10.1007/s13171-019-00164-5
Maitan MQ (2022) Estimativa da depressão endogâmica em Psidium guajava via atributos de sementes utilizando famílias S0, S1 e S2. Tese de Doutorado. Universidade Estadual do Norte Fluminense Darcy Ribeiro, Campos dos Goytacazes
Manica I. 2000. Fruticultura Tropical. Porto Alegre
MAPA - Ministério da Agricultura Pecuária e Abastecimento. Tabela de descritores de goiabeira (Psidium guajava L.). Disponível em: <http://www.in.gov.br/materia//asset _publisher /Kujrw0TZC2Mb/content/id/26173654/do1–2018–06–18-ato-n-5- 754 de-14-de-junho-de-201826173582 > Acesso em: 10 de agosto de 2023.
Mendes GC, Gusmão MTA, Martins TGV, Rosado RDS, Sobrinho RSAS, Nunes ACP, Ribeiro WS, Zanuncio JC (2020) Genetic divergence of native palms of oenocarpus distichus considering biometric fruit variables. Sci Rep 9:4943. https://doi.org/10.1038/s41598-019-41507-4
Mojena R (1977) Hierarchical grouping methods and stopping rules: an evaluation. Comput J 20:359–363. https://doi.org/10.1093/comjnl/20.4.359
Oliveira MG, Oliveira JGD, Gomes Filho A, Pereira MG, Viana AP, Souza Filho GAD, Lopes GEM (2009) Diversidade genética de aceroleiras (malphigia emarginata DC), utilizando marcadores moleculares RAPD e características morfoagronômicas. Rev Bras Frutic 31:162–170. https://doi.org/10.1590/S0100-29452009000100023
Oliveira NNS, Viana AP, Quintal SSR, Paiva CL, Marinho CS (2014) Analysis of genetic distance between access of the gender Psidium by route markers ISSR. Rev Bras Frutic 36:917–923. https://doi.org/10.1590/0100-2945-413/13
Paterniani E (1978) Melhoramento e produção de milho no Brasil. São Paulo.
Pereira FM, Kavati R (2011) Contribution of Brazilian scientific research in developing some of subtropical fruit. Rev Bras Frutic 33:92–108. https://doi.org/10.1590/S0100-29452011000500013
Pessanha PGDO, Viana AP, Amaral Júnior ATD, Souza RMD, Teixeira MC, Pereira MG (2011) Assessment of genetic diversity in access to psidum spp. via RAPD markers. Rev Bras Frutic 33:129–136. https://doi.org/10.1590/S0100-29452011000100018
Pinheiro JC, Bates DM (2000) Modelos de efeitos mistos em S e S-PLUS. Springer, Nova York. https://doi.org/10.1007/b98882
Quintal SSR, Viana AP, Campos BM, Vivas M, Amaral Júnior AT (2017) Analysis of structures of covariance and repeatability in guava segreganting population. Rev Caatinga 30(4):885–891. https://doi.org/10.1590/1983-21252017v30n408rc
R Core Team. 2021. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing, Vienna, Austria. https://www.r-project.org/
Radaelli JC, Oliveira LS, Fabiane KC, Domingues LS, Júnior AW (2020) Phenotypic divergence of Plinia spp. genotypes based on plant growth behavior. Colloquium Agrariae 16:77–86
Reis NV (2023) Caracterização molecular de famílias S1 e S2 de goiabeira (Psidium guajava L.). Dissertação, Universidade Estadual do Norte Fluminense Darcy Ribeiro, Campos dos Goytacazes.
Santos RR, Martins FP, Ribeiro IJA, Nascimento LM, Igue T (1998) Avaliação de variedades de goiabeira em monte alegre do Sul (SP). Bragantia 57:117–126. https://doi.org/10.1590/S0006-87051998000100014
Santos CAF, Corrêa LC, Costa SR (2011) Genetic divergence among psidium accessions based on biochemical and agronomic variables. Crop Breed Appl Biotechnol 11:149–156. https://doi.org/10.1590/S1984-70332011000200007
Santos TG, Pedroza Neto JL, Chaves SFS, Alves RM, Silva ABM, Jose ARM (2022) Characterization of the genetic structure of a cupuassu tree population collected in primary forest. Rev Bras De Ciênc Agrár 17:e1592. https://doi.org/10.5039/agraria.v17i4a1592
Silva FA, Viana AP, Corrêa CCG, Carvalho BM, De Sousa CMB, Amaral BD, Glória LS (2020) Impact of bayesian inference on the selection of Psidium guajava. Sci Rep 10:1999. https://doi.org/10.1038/s41598-020-58850-6
Silva FA, Viana AP, Correa CCG, Santos EA, De Oliveira JAVS, Andrade JDG, Glória LS (2021) Bayesian ridge regression shows the best fit for SSR markers in psidium guajava among bayesian models. Sci Rep 11:1–11. https://doi.org/10.1038/s41598-021-93120-z
Silva FAD, Viana AP, Corrêa CCG, Leal LSDS, Glória LS (2023) Repeatability estimates in longitudinal data on guava trees. Sci Agric 80:1–7. https://doi.org/10.1590/1678-992X-2022-0065
Simon GA, Takeshi K, Moiteiro M (2012) Divergência genética no cultivo de milho na primeira e na segunda safra. Semina Ciênc Agrár 33(2):449–457
Singh R, Sehgal OP (1968) Studies on the blossom biology of Psidium guajava L. (guava); 2, Pollen studies stigmatal receptivity pollination and fruit set. Indian J Hortic 25:52–59
Soil Survey Staff (2015) Illustrated guide to soil taxonomy. Version 2.0. Lincoln: USDA, NRCS.
Torres GX, Viana AP, Vieira HD, Rodrigues DL, Santos VO (2019) Contribution of seed traits to the genetic diversity of a segregating population of Passiflora spp. Chil J Agric Res 79:288–295. https://doi.org/10.4067/S0718-58392019000200288
Wickham H (2016) Ggplot2: Gráficos elegantes para análise de dados. Springer-Verlag. Nova Iorque. https://ggplot2.tidyverse.org. Acessado em 28 de setembro de 2023.
Acknowledgements
To the National Development Council Scientific and Technological Development (CNPq), the Rio de Janeiro State Research Support Foundation (FAPERJ). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.
Author information
Authors and Affiliations
Contributions
JAL: Investigation, methodology, formal analysis, writing – original draft, writing—review and editing. APV: Supervision, conceptualization, methodology, visualization, validation, project administration, funding acquisition. CCGC: Research and data curation. DSM: Investigation, data curation. EAS: Conceptualization, methodology, visualization, elaboration, validation, writing – review and editing. FAS: methodology, visualization, validation. LSA: Investigation, data curation. LCLC: Investigation, data curation. MZM: Investigation, data curation. NVR: Investigation, data curation, NRC: methodology, visualization, data curation, validation. RFD: Conceptualization, methodology, visualization, elaboration, validation, writing – review and editing. TCC: Investigation, data curation.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lima, J.A., Viana, A.P., Correa, C.C.G. et al. Impact of self-pollination on the genetic diversity of inbred families of Psidium guajava L. Euphytica 220, 132 (2024). https://doi.org/10.1007/s10681-024-03389-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-024-03389-0