
Abstract
Background
The exploration of genetic diversity is the key source of germplasm conservation and potential to broaden its genetic base. The globally growing demand for chickpea suggests superior/climate-resilient varieties, which in turn necessitates the germplasm characterization to unravel underlying genetic variation.
Methodology and results
A chickpea core collection comprising of diverse 192 accessions which include cultivated Cicer arietinum, and wild C. reticulatum, C. echinospermum, and C. microphyllum species were investigated to analyze their genetic diversity and relationship, by assaying 33 unlinked simple sequence repeat (SSR) markers. The results amplified a total of 323 alleles (Na), ranging from 2 to 8 with an average of 4.25 alleles per locus. Expected heterozygosity (He) differed from 0.46 to 0.86 with an average of 0.68. Polymorphic information content (PIC) ranged from 0.73 to 0.98 with an average of 0.89. Analysis of molecular variance (AMOVA) showed that most of the variation was among individuals (87%). Cluster analysis resulted in the formation of four distinct clusters. Cluster I represented all cultivated and clusters II, III, and IV comprised a heterogeneous group of cultivated and wild chickpea accessions.
Conclusion
We report considerable diversity and greater resolving power of SSR markers for assessing variability and interrelationship among the chickpea accessions. The chickpea core is expected to be an efficient resource for breeders for broadening the chickpea genetic base and could be useful for selective breeding of desirable traits and in the identification of target genes for genomics-assisted breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chickpea (Cicer arietinum L.) remains as the second most important cool season legume crop in the world [1] offering a cheap source of dietary protein (23% in dry seed), and provides nutritional, and income security in countries like India, Pakistan, and Bangladesh among many other developing countries [2]. It contributes 14.78 million tons to global food production from 14.56 million hectares area across the world with average productivity of 0.96 tons per hectare [3]. In India, chickpea represents national pulse acreage, and production of 35 and 46%, respectively. India serves as the largest chickpea producer (69%), consumer, and importer of chickpea globally [4]. The genome sequence of both Kabuli [5], and Desi type [6], whose advanced version was later published by [49] are available thus making chickpea a candidate crop for legume genetic, and genomic research.
The genetic variability for the traits of economic importance, and the relationship between the cultivated, and its wild relatives are of paramount importance for advanced chickpea programs. Domestication, modern plant breeding, and agricultural systems have narrowed the genetic base of cultivated chickpea. This has promoted the need for new sources of variation arising within the germplasm that might be useful in plant breeding programs [5]. However, the large size of germplasm collections, and its inadequate data/information, have resulted in low use (< 1%) of germplasm further narrowing the genetic base in many crops [7]. India is known for its largest diversity in cultivated chickpea [24]. Despite the availability of a diverse, and large number of chickpea germplasm collection at the Gene banks like the National Bureau of Plant Genetic Resources (NBPGR), and International Crop Research Institute of Semi-arid Tropics (ICRISAT), a very small proportion has been used in crop improvement/breeding programs mainly because the resources available for evaluation of such large collection is quite limited. The core approach which represents the mini crop collections capturing most of the available genetic diversity in the whole germplasm collection, is an efficacious methodology to advance crop improvement programs. Such core sets are cost-effective, easy to maintain by individual breeders, and help in the appropriate assessment of genetic diversity, population structure, association analysis, and targeted gene mining. Many national programs around the world are actively involved in the utilization of mini cores of crops like chickpea, pigeonpea, groundnut, foxtail millet sorghum, pearl millet, and finger millet, which have led to new and diverse sources of variation for, qualitative, and quantitative (biotic/abiotic stress resistance) traits in various crops.
The molecular characterization of the core collection will further enhance its use in genomics for crop improvement programs. Molecular markers can explore the genetic variation in plants, and are useful in detecting the duplicates among the core collection, and thereby help in designing efficient strategies for optimal sampling from the existing variation. The utility of DNA-based markers for unbiased estimation of molecular diversity and establishing precise phylogenetic relationships among species as compared to morphological, cytological and biochemical markers have been well understood. In chickpea, diverse molecular markers including AFLPs [8], STMS [9], RAPDs [11], ISSRs [12], and SSRs [13,14,15,16] have been already used to unravel the genetic diversity available in chickpea. The desirable genetic attributes (co-dominant inheritance, reproducibility, bi-/multi-allelic nature, and abundant genomic distribution) of sequence-based robust microsatellite/SSR markers have encouraged their use for many applications of chickpea genetics, genomics, and breeding including cultivar identification, allele mining, genetic mapping, association studies, genetic diversity analysis, population structure studies, and establishing phylogenetic relationships [32, 50]. The draft genome sequence of chickpea has led to the identification of over 48,000 SSRs for use as genetic markers [5].
Our recently published information of intensive screening and genetic evaluation of the composite set of 384 chickpea germplasm has led to the identification of chickpea core collection, which would enhance the utilization of chickpea genetic resources in crop improvement programs [10]. The present investigation was aimed to further evaluate the level of genetic diversity and allelic richness in the above selected chickpea core set of 192 accessions, using a larger number of thirty-three genome-wide SSR markers. The results derived from this study could be used for more efficient utilization of the entire collection. The microsatellite markers used in the present study were also tested for their ability to detect the extent of genetic diversity by using polymorphism information content values.
Methods
Germplasm collection
Experimental material for the study comprised of a core set of 192 chickpea accessions, consisting of landraces, elite cultivars, advanced breeding lines, and wild species from the primary, secondary, and tertiary gene pools. These lines include annual cultivated 178 Cicer arietinum lines from the three seed types (kabuli, desi, and intermediate pea-shaped) and the fourteen (14) wild relatives belonging to Cicer reticulatum Ladiz. (8), Cicer echinospermum P. H. Davis (5), and a perennial Cicer microphyllum Benth (1) species. The seed material of accessions from primary and secondary gene pools has been collected from different hot spots in Turkey under USAID project ‘‘Chickpea Innovation Lab’’, and the same has been procured by us from ICRISAT, Hyderabad, India. And tertiary gene pool has been collected from its wild natural habitat in Leh/Ladakh region of state Jammu and Kashmir, India. The seed material of cultivated chickpea has been attained from various ethnic/ global institutes. (Supplementary Table EMS_1). Details of some of the accessions like their cold tolerance/ nutrient density are available elsewhere [17, 18]. The passport data of the species is presented in Table 1.
DNA isolation
Young leaf tissues of each accession were lyophilized to extract the total genomic DNA following CTAB-DNA extraction protocol [19] with some minor modifications. The extracted DNA was purified by treating with RNase to remove the RNA contamination. The purified DNA was quantified, and its quality was assessed by 0.8% agarose gel electrophoresis). Based on the intensity of the bands, DNA was diluted for marker genotyping.
SSR markers
A total of 33 SSR markers spanning the entire chickpea genome, one marker at least covering each arm of the 8 linkage groups, were selected to characterize the set of 192 core chickpea accessions (Fig. 1). These SSR primer pairs were selected from the composite collections of the chickpea database (CicArMiSatDB: the chickpea microsatellite database (https:// cegresources.icrisat.org/CicArMiSatDB/) developed by ICRISAT. The database contains SSRs which are reported by [5]. The details of SSR primer-pairs are provided in Supplementary Table EMS_2.

The distribution of 33 SSR markers on different chickpea chromosomes used during the present study
Polymerase chain reaction (PCR)
To study polymorphism in the chickpea core collection, PCR amplification was carried out in a Universal Gradient Thermal Cycler with 96 wells using a 20-μL reaction mixture: 2 μL of template DNA (25 ng/μL), 10 × PCR buffer (1.8 mM, MgCl2, 10 mM Tris–HCl, 50 mM KCl), 2.5 mM dNTPs (Molecular Biology for Life Science, Fermentas, Lithuania, USA), 5 μM each of forward and reverse primer and 5U of Taq DNA polymerase (Sigma Aldrich USA. The thermal cycler (Peqlab) was programmed as follows: initial denaturation for 5 min at 94 °C followed by 40 cycles of denaturation for 30 s (s) at 94 °C, annealing at temperature specific for each primer pair for 30 s and extension at 72 °C for 30 s. The final extension was allowed for 10 min at 72 °C and storage at 4 °C until further use. The resulting PCR products were run on 10% Poly-acrylamide Gel Electrophoresis (PAGE) using a Dual Gel Vertical Electrophoresis System (Peqlab) and silver stained for manual visualization of bands on gel documentation system.
Data analysis
To calculate genetic diversity parameters GenALex software version 6.5b3 [20] was used. The overall diversity, number of different alleles (Na), the effective number of alleles (Ne), number of private alleles, and gene diversity measured as expected heterozygosity (He), was measured along with other parameters. Total genetic variation was partitioned among populations, among individuals, and within individuals, by performing an analysis of molecular variance (AMOVA). AllelIic frequency was calculated using Polymorphism Information Content (PIC) values which indicates the ability of markers to differentiate the genotypes was calculated as 1-Σ (fij)2 where fij is the frequency of the jth allele for ith locus summed across all alleles for the locus [21]. For the calculation of the pairwise genetic distances, DARwin version 5.0 was used [22]. The dissimilarity matrix obtained was subjected to cluster analysis, using the Unweighted Pair Group Method with Arithmetic Average (UPGMA) based hierarchical clustering for the construction of dendrograms.
Results
A set of 33 primer pairs physically mapped across eight chickpea chromosomes showing reproducible amplification, and 100% polymorphism were genotyped across the core collection of 192 cultivated and wild Cicer accessions using the polyacrylamide gel-based assay. The SSR markers used in this study were found informative enough to explain genetic variation among core chickpea accessions.
Allelic and gene diversity
A total of 323 alleles (Na) were identified at 76 marker loci generated by 33 SSR markers giving an average of 4.25 alleles per marker locus. The microsatellite marker locus CaGM06707A (mapped on LG2) was the most variable with maximum (eight) 8 alleles and microsatellite marker locus CaGM06221B was least variable with only (two) 2 alleles. The total number of effective alleles (Ne) was 263.96 with an average of 3.47 alleles/locus. The number of effective alleles was lower than the total number of alleles, showing that only a few alleles contributed to the variation. The average number of private alleles i.e., the number of alleles unique to single population was 4.25.
Gene diversity (expected heterozygosity; He) was variable across loci reflecting the different number and frequencies of the alleles found. The average gene diversity of the core collection was found moderate (0.68), showing that two randomly chosen genes will carry different alleles roughly half of the time. The lowest He (0.46) was recorded for SSR marker locus CaGM12686E and the highest He (0.86) for SSR marker locus CaGM06707A. For each population, the observed heterozygosities (Ho) were less than the expected heterozygosities (He) (Table 2, Fig. 2).

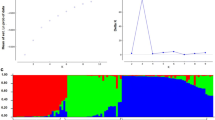
Comparison of allelic and gene diversity detected in the whole chickpea population and two sub-populations (cultivated vs wild). The figure shows comparison for number of alleles detected (Na), Number of alleles with frequency more than or equal to 5%, Number of effective alleles (Ne) and Number of private alleles, etc.
Allelic/gene diversity across sub-populations (cultivated and wild)
The allelic/genetic patterns studied across the populations are presented in Table 2. The allelic diversity, when studied within two sub-populations (cultivated v/s wild), revealed a total number of 345 alleles with an average of 4.25 alleles/locus observed in the cultivated sub-population, as against a total number of 201 alleles with an average of 3.28 alleles/locus in wild sub-population. The average number of effective alleles (Ne) was observed to be 3.46 in cultivated chickpea, and that of 2.60 in wild chickpea. The number of alleles with frequency ≥ 5 percent (Na Freq. ≥ 5%), (i.e., measurement taken to alleviate the sampling error associated with the sampling of distinct alleles) was for cultivated and wild sub-populations was 4.02 and 3.26, respectively. The average number of private alleles (alleles unique to a single population) was 0.96 in the cultivated sub-population. However, no private allele was found among the wild sub-population. Since the sample size between the two sub-populations was not equal, the resampling technique was therefore used, to make valid comparisons of diversity, wherein a small sample size i.e., 14 for wilds was drawn randomly 3000 times from the 178 cultivated group and average mean alleles were recorded. The mean alleles/locus for the cultivated group based on a sample size of 14 genotypes was found to be 3.65. The more diversity apparent in cultivated sub-population may be attributed to the presence of private alleles in the group (mean private alleles = 0.96), while no private alleles were present in the wild sub-population. Similarly, gene diversity (expected heterozygosity; He) was observed to be higher in cultivated sub-population and varied from 0.14 to 0.80 with an average of 0.68, and from 0.09 to 0.75 with an average of 0.57 in wilds (Fig. 2).
Polymorphic information content (PIC)
The PIC value provides an idea about the polymorphism detected by the marker system and its suitability for the study of genetic diversity. The higher the PIC value of a locus, the higher the number of alleles detected. During the present study, the PIC value of all the 76 polymorphic SSR marker loci for all 192 chickpea core set varied from 0.73 to 0.98 with an average of 0.89. Interestingly, all the marker loci were highly informative with the PIC value of > 0.50. Markers CaGM06707A, CaGM12686E, and CaGM19859B were most informative with high PIC value of 0.98, 0.98, and 0.97, respectively. SSR marker CaGM12686E and CaGM06221B showed the least PIC value of 0.72 and 0.73, respectively. (Table 3). The correlation worked out between the PIC value and the number of alleles was significantly positive (r = 0.67, p < 0.01).
Analysis of molecular variance (AMOVA)
AMOVA was carried out among and within the two sub-populations (cultivated and wild). The results of the analysis showed that the observed variance was mostly partitioned among individuals (87%), whereas 7 percent of the variance was partitioned within individual accessions and the rest 6 percent was among populations (Table 4). The low level of genetic variation among populations is indicative of gene flow between populations. Since the maximum number (8) of the wilds in the present study are from C. reticulatum which is the progenitor of cultivated species C. arienitum leading to smooth introgression of genes. There is a moderate amount of genetic differentiation between the groups (FST = 0.10), indicating that the groups are less genetically distinct. The FIS and FIT values are 0.92 and 0.82, respectively indicating that the chickpea accessions making up the individual groups are inbred lines. Furthermore, the gene flow (Nm) between the groups was observed to be 4.26, suggesting a high level of gene flow.
Cluster analysis
Cluster analysis based on the dissimilarity index values delineated 192 chickpea accessions into 4 main clusters by employing all the 76 marker loci, the results of which are presented in Fig. 3. The cluster-I contained 25 accessions which were all cultivated species. Cluster-II possessed 29 accessions, which include wilds from the three gene pools (C. reticulatum (3), C. echinospermum (5), and C. microplyllum (1)) and the remaining 20 in the cluster were cultivated C. arietinum. A slight speciation pattern of structure was detected especially in cluster-II, where the wild accessions belonging to three different gene pools were grouped in close vicinity. Cluster-III had a total of 53 accessions wherein all cultivated except 1 wild species (C. reticulatum) were included. Cluster-IV was the largest cluster with 85 accessions which included four C. reticulatum species and 81 cultivated species. These results infer that wild C. reticulatum belonging to primary gene pool was present with C. arietinum (cultivated) in three out of four clusters, owing to the origin of cultivated C. arietinum from the wild progenitor. Also, among all the four clusters, the cluster II was found to be the most heterogeneous.

Hierarchical clustering based on simple matching dissimilarity matrix genotyped across 192 core chickpea accessions using 33 SSR marker; Black lines indicate cultivated, Red ones are C. reticulatum, blue ones are C. echinospermum and green is C. microphyllum
Discussion
An extensive characterization of plant genetic resources, and an understanding of the genetic relationships in the germplasm collection is essential for effective conservation, management, and exploitation of genetic resources in varietal improvement programs. Also, the use of wild species provides a wider genetic base in otherwise, less diverse cultivated chickpea and is known as a potential source of resistance genes for various biotic and abiotic stresses [23]. Thus, the investigation of the nature and magnitude of genetic diversity and relatedness within and among the cultivated chickpea and its wild relatives is an obvious necessity to identify new sources of variation in the existing germplasm. A core set of chickpea acts as a working collection having an optimal and convenient size that can be evaluated for all important traits with minimum error rates. Intensive screening and evaluation of the core collections have led to the identification of diverse accessions leading to the advancement of plant breeding and thus paving the way towards booming crop improvement programs. To cite an example, the utility of the chickpea core set (300 accessions) capturing 78.1% alleles of the composite collection, developed at ICRISAT [25] has been extensively used in breeding as well as germplasm management and improvement. The molecular characterization of the core collection provides information related to unique/rare alleles from cultivated and wild species which could be used to select distinct genotypes for allele mining. Among the variety of molecular markers already developed and used in breeding programs, SSRs are considered markers of choice [26, 27]. Universal distribution and high density in a multitude of genomes along with other merits associated with SSR markers enhance their desirability, especially for germplasm characterization and have been extensively used for identification of variation in chickpea germplasm. Therefore, efforts were made during the present study to carefully select random SSR markers covering all the eight linkage groups of chickpea genome.
In this study, every SSR primer pair successfully amplified the target DNA in a core set of 192 chickpea accessions. The 33 random markers generated 76 polymorphic loci and were useful for detection of a total of 323 alleles (Na) at all marker loci. The average number of alleles/locus (4.25) found in this study are within the ranges reported by many earlier studies. A relatively higher diversity was observed in our core collection when compared to our previous study on genetic diversity in a composite set of 384 genotypes using 8 SSR markers which generated a total of 63 (Na) alleles with an average of 3.7/locus, indicating that the chickpea core maximized the diversity available in the entire composite germplasm set. Our observations are fairly comparable with earlier genetic diversity studies on the cultivated and wild Cicer accessions using random microsatellite markers with some deviations. For instance, the use of 478 microsatellites identified a total of 3703 alleles with an average of 4.26 alleles/marker locus in 94 cultivated and wild chickpea accession [28]. Similarly, 38 accessions of Cicer arietinum using 100 SSR markers generated an average of 4.8 amplicons per locus [17]. Likewise, a total of 59 alleles with an average of 4.2 alleles/locus in 60 chickpea accessions were reported using14 SSR markers [15]. Similar results were also observed by [16] where he reported an average of 3.88 alleles/locus in 66 Cicer arietinum accessions. However, the level of diversity detected in the present study was less while comparing it with average number of alleles/locus (35, 17, 10.3, 7.4, 8.61, 7.75, 10.5) detected by [13, 30,31,32,33,34,35]. There are also reports where even less diversity than in our case has been reported. For example, 33 microsatellites were used to study genetic diversity in 155 chickpea accessions which resulted in a total of 111 bands with a range of 2–5 alleles/locus and on average 3.364 bands per marker [14]. In another study, using 27 SSR markers a total of 81 alleles with an average of 3.0 alleles per locus were detected in 50 chickpea genotypes [36]. The use of more cultivated chickpea accessions which possess narrow genetic base as reported by various researchers [37,38,39,40] may have led to observance of less diversity in our study. Likewise, higher polymorphic potential among the species/accessions originating from Fertile Crescent (79.5% polymorphism) in contrast to those from Central Asia- India (35.8%) was also reported by [28]. Since most of the germplasm used in the present study is indigenous which may have attributed to the presence of less diversity in our chickpea core collection. The inclusion of more chickpea wild species could produce a higher number of allele count/locus as suggested by [15, 30]. The overall genetic diversity (He) showed relatively moderate value of expected heterozygosity (He = 0.68) which are comparable to previous studies [15, 33, 41, 42]. Differences in results for estimated genetic diversity between studies may be attributed to the different number of accessions, different number of loci examined, nature of markers, and perhaps the geographical origin of the accessions used in each study. The inter and intra- specific polymorphism, when studied within two sub-populations (cultivated vs wild), did not differ much from the single population of 192 chickpea accessions, where the average number of alleles per locus for cultivated sub-population was 4.25, while that for wild was found to be 3.28. These results indicate that the number of accessions could still be reduced to study genetic diversity. The mean private alleles in cultivated was (0.96) and no private alleles in wilds were detected within the two sub-populations. Similar results were shown by [30], where no rare alleles were found in wild accessions. Among the two sub-populations, cultivated chickpea showed more diversity (possessing more number of private alleles and an average number of alleles/locus than wild population, which could be due to very less number of wild accessions involved in our study as compared to the large number of cultivated accessions used. The inclusion of more wild accessions particularly from secondary and tertiary gene pools could result in a higher level of diversity since most of the desirable gene complexes are present in them. For instance, a higher degree of polymorphism was detected among the accessions belonging to the secondary gene pool as compared to primary gene pool [28].
The results of PIC values indicate that all the markers used in the present study were informative with a PIC value greater than 0.50. The average PIC value for the 76 marker loci was 0.89 which suggests that the majority of markers enabled a high level of polymorphism and could be effective to determine the genetic differences among the chickpea accessions studied. Similar to our results the PIC value of 0.854 has been reported by [30] and PIC values of 0.77, 0.75, 0.75, 0.72, were observed by [13, 29, 35, 43], respectively. Comparatively, [17, 36, 44, 45] revealed an average PIC value of 0.53, 0.60, 0.68, and 0.43, respectively which is relatively less than that of the present study.
AMOVA is a method to study population differentiation utilizing molecular markers [46]. To evaluate the extent of population differentiation among/between the sub-populations (cultivated and wild) in the present study, the molecular variance based on 99 permutations was analyzed using 33 random SSR markers. The analysis of the two sub-populations showed that most of the variation was among individuals (87%), and 7% of the variation was partitioned within individuals. The relatively higher variation among individuals could be attributed to the differences among genotypes within sub-populations. In agreement with our results, [47] reported higher variance (61%) among the populations compared to within (39%) populations. Similarly, 71% of the allelic variation was documented among the domesticated and the wild gene pools and the remaining 29% of the allelic variation was observed within them [48].
To decipher the phylogenetic relationships between accessions and closest and distant genotypes from each other, UPGMA based Hierarchical clustering approach was found best in the present study for interpretation of results. The 33 random SSR markers could cluster chickpea core collection of chickpea accessions into 4 main clusters, and several sub-clusters and sub-sub clusters, whereby the different members within a cluster are assumed to be more closely related to each other than with those members in different clusters. From the present investigation, it was found that accessions of different origins/species were grouped together in different clusters. The C. reticulatum accessions fell in 3 clusters out of 4 main clusters. All accessions belonging to C. echinospermum were clustered in a single cluster (cluster II). Another feature of the tree is the relative position of the single C. microphyllum accession (belonging to tertiary gene pool) in close vicinity of C. echinospermum accessions (secondary gene pool). This may suggest that both species are closely related to each other (Fig. 3). Results from the present study support the observations of several workers [10, 51] the grouping followed a definite pattern with all the cultivated accessions falling into all distinct clusters while the wild species grouped into distinct subclusters and in closer vicinity to each other. Similarly, [8] used Nei’s pairwise distance calculations and identified clustering of cultivated species Cicer arietinum, Cicer reticulatum and, Cicer echinospermum in the same group. It can be inferred from this study that SSR markers were effective in placing chickpea accessions in appropriate clusters based on relationships at an evolutionary level. The allelic/genetic diversity, PIC values, molecular variation, phylogenetic relationship information generated in this study for the set of 192 chickpea core collection, and with the availability of current genomic tools, could have implications for many genotyping applications including allele mining, gene tagging association analysis and QTL mapping targeting different qualitative and quantitative traits of agricultural importance in chickpea.
Conclusion
The present study shows the potential use of selected polymorphic microsatellite markers in discriminate among the core set chickpea accessions. Our results revealed considerable genetic diversity with most of the variation found among the individuals and provided information on phylogenetic relationships among the members of different gene pools. The core collection is regarded as the gateway to unravel the diversity estimates. The results obtained in the present investigation revealed that the core collection exhibited equal diversity when compared with the entire composite chickpea germplasm from which it was derived. Thus, shall provide greater confidence to the breeding community for assessment of distinctiveness and relationships among the various accessions. The grouping of wild species with cultivated C. arietinum indicates that they could possibly serve as good sources of broadening chickpea genetic base, and for effective pre-breeding for breaking yield barriers. The clustering also indicated different genetic basis of different gene pools. The diverse genotypes could be used as genetic resources for developing mapping populations and in transcriptomics studies. The chickpea core collection is expected to ensure effective core deployment, especially for future chickpea varietal identification, conservation, and management, and the development of broad-based/ climate resilient cultivars that should meet the immediate and future breeding challenges.
Data availability
The data analyzed and generated during the current study are available within the article [and/or] its supplementary materials.
References
Mallikarjuna BP, Samineni S, Thudi M, Sajja SB, Khan AW et al (2017) Molecular mapping of flowering time major genes and QTLs in chickpea (Cicer arietinum L.). Front Plant Sci 8:1140
Archak S, Tyagi RK, Harer PN, Mahase LB, Singh N, Dahiya OP et al (2016) Characterization of chickpea germplasm conserved in the Indian national genebank and development of a core set using qualitative and quantitative trait data. Crop J 4(5):417–424
FAOSTAT, Food and Agriculture Organization of the United Nation Statistics for (2017) Online database at https://www.fao.org/faostat/en/#data
Annual Report DPD. 2017. Directorate of Pulses Development, Ministry of Agriculture and Farmers Welfare, Government of India. DPD/Pub./TR/29/2017–18
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG et al (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31(3):240
Jain M, Misra G, Patel RK, Priya P, Jhanwar S, Khan AW (2013) A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J 74:715–729
Upadhyaya HD, Yadav D, Dronavalli N, Gowda CLL, Singh S (2010) Mini core germplasm collections for infusing genetic diversity in plant breeding programs. Electron J Plant Breed 1(4):1294–1309
Nguyen TT, Taylor PWJ, Redden RJ, Ford R (2004) Genetic diversity estimates in Cicer using AFLP analysis. Plant Breed 123(2):173–179
Sethy NK, Shokeen B, Edwards KJ, Bhatia S (2006) Development of microsatellite markers and analysis of intra specific genetic variability in chickpea (Cicer arietinum L.). Theor Appl Genet 1(12):1416–1428
Fayaz H, Mir AH, Tyagi S, Wani AA, Jan N, Yasin M, Mir JI, Mondal B, Khan MA, Mir RR (2021) Assessment of molecular genetic diversity of 384 chickpea genotypes and development of core set of 192 genotypes for chickpea improvement programs. Genet Resour Crop Evol. https://doi.org/10.1007/s10722-021-01296-0
Talebi REZA, Fayaz F, Mardi M, Pirsyedi SM, Naji AM (2008) Genetic relationships among chickpea (Cicer arietinum) elite lines based on RAPD and agronomic markers. Int J Agric Biol 10(3):301–305
Jida Z, Alemu A, Mullualem D (2018) Genetic diversity among elite chickpea (Cicer arietinum L.) varieties of ethiopia based on inter simple sequence repeat markers. Afr J Biotechnol 17(34):1067–1075
Sefera T, Abebie B, Gaur PM, Assefa K, Varshney RK (2011) Characterisation and genetic diversity analysis of selected chickpea cultivars of nine countries using simple sequence repeat (SSR) markers. Crop Pasture Sci 62(2):177–187
Keneni G, Bekele E, Imtiaz M, Dagne K, Getu E, Assefa F (2012) Genetic diversity and population structure of ethiopian chickpea (Cicer arietinum L.) germplasm accessions from different geographical origins as revealed by microsatellite markers. Plant Mol Biol Rep 30(3):654–665
Ghaffari P, Talebi R, Keshavarzi F (2014) Genetic diversity and geographical differentiation of Iranian landrace, cultivars, and exotic chickpea lines as revealed by morphological and microsatellite markers. Physiol Mol Biol Plants 20(2):225–233
Aggarwal H, Choudhary SP, Rana MK, Choudhary R (2018) Assessment or evaluation of genetic diversity among 66 cultivars of chickpea (Cicer arietinum L.) of Indian origin Using SSR MARKERS. Int J Curr Microbiol Appl Sci 7(2):523–533
Choudhary P, Khanna SM, Jain PK (2012) Genetic structure and diversity analysis of the primary gene pool of chickpea using SSR markers. Genet Mol Res 11(2):891–905
Mir AH, Bhat MA, Dar SA, Sofi PA, Bhat NA, Mir RR (2021) Assessment of cold tolerance in chickpea (Cicer spp.) grown under cold/freezing weather conditions of North Western Himalayas of Jammu and Kashmir, India. Physiol Mol Biol Plants. https://doi.org/10.1007/s12298-021-00997-1
Fayaz H, Rather IA, Wani AA, Tyagi S, Pandey R, Mir RR (2019) Characterization of chickpea gene pools for nutrient concentrations under agro-climatic conditions of North- Western Himalayas. Plant Genet Resour 17:464–467
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer length polymorphism in barley: mendelian inheritance, chromosomal location and population dynamic. Proc Natl Acad Sci USA 81:8014–8019
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci, USA 70:3321–3323
Perrier X, Jacquemoud-Collet JP (2006) Darwin software. http://darwin.cirad.fr/darwin.
Singh R, Sharma P, Varshney RK, Sharma SK (2008) Chickpea improvement: role of wild species and genetic markers. Biotechnol Genet Eng 25:267–313
Maesen LJG (1972) Cicer L, A Monograph of the Genus, with Special Reference to the Chickpea (Cicer arietinum L). Its Ecology and Distribution (Doctoral thesis) Wageningen University
Upadhyaya HD, Oritz R (2001) A mini core subset for capturing diversity and promoting utilization of chickpea genetic resources in crop improvement. Theor Appl Genet 102:1292–1298
Mir RR, Varshney RK (2013) Future prospects of molecular markers in plants. In: Henry RJ (ed) Molecular markers in plants. Blackwell Publishing Ltd, Oxford, pp 170–190
Mir RR, Hiremath PJ, Riera-Lizarazu O, Varshney RK (2013) Evolving molecular marker technologies in plants: From RFLP to GBS. In: Lubberstedt T, Varshney RK (eds) Diagnostics in plant breeding. Springer, Dordrecht, pp 229–250
Saxena MS, Bajaj D, Kujur A, Das S, Badoni S, Kumar V et al (2014) Natural allelic diversity, genetic structure and linkage disequilibrium pattern in wild chickpea. PLoS ONE 9(9):e107484
Upadhyaya HD, Dwivedi SL, Baum M, Varshney RK, Udupa SM, Gowda CLL (2008) Genetic structure, diversity, and allelic richness in composite collection and reference set in chickpea (Cicer arietinum L.). BMC Plant Biol 8:106
Imtiaz M, Materne M, Hobson K, Van Ginkel M, Malhotra RS (2008) Molecular genetic diversity and linked resistance to ascochyta blight in Australian chickpea breeding materials and their wild relatives. Aust J Agric Res 59(6):554–560
Castro P, Millan T, Gil J, Merida J, Garcia ML, Rubio J, Fernadez-Romero MD (2011) Identification of chickpea cultivars by microsatellite markers. J Agric Sci 149(4):451–460
Khan R, Khan H, Harada F, Harada K (2010) Evaluation of microsatellite markers to discriminate induced mutation lines, hybrid lines and cultigens in chickpea (’Cicer arietinum L.). Aust J Crop Sci 4(5):301
Shukla N, Babbar A, Prakash V, Tripathi N, Saini N (2011) Molecular diversity analysis in some promising lines of chickpea using SSR markers. J Food Legumes 24(4):320–323
Samyuktha SM, Kannan JR, Geethanjali S (2018) Molecular genetic diversity and population structure analysis in chickpea (Cicer arietinum L.) germplasm using SSR markers. Int J Curr Microbiol Appl Sci 7(2):639–651
Mohan S, Kalaimagal T (2020) Genetic diversity studies in chickpea (Cicer arietinum L.) genotypes using SSR markers. J Food Legumes 33(1):1–5
Udupa SM, Sharma A, Sharma RP, Pai RA (1993) Narrow genetic variability in Cicer arietinum L. as revealed by RFLP analysis. J Plant Biochem Biotechnol 2(2):83–86
Sonnante G, Marangi A, Venora G, Pignone D (1997) Using RAPD markers to investigate genetic variation in chickpea. J Genet Breed 51:303–307
Sant VJ, Patankar AG, Sarode ND (1999) Potential of DNA markers in detecting divergence and in analyzing heterosis in Indian elite chickpea cultivars. Theor Appl Genet 98:1217–1225
Singh R, Prasad CD, Singhal V, Randhawa GJ (2003) Assessment of genetic diversity in chickpea cultivars using RAPD, AFLP, and STMS markers. J Breed Genet 57:165–174
Hajibarat Z, Saidi A, Hajibarat Z, Talebi R (2014) Genetic diversity and population structure analysis of landrace and improved chickpea (Cicer arietinum) genotypes using morphological and microsatellite markers. Environ Exp Biol 12:161–166
Saeed A, Hovsepyan H, Darvishzadeh R, Imtiaz M, Panguluri SK, Nazaryan R (2011) Genetic diversity of Iranian accessions, improved lines of chickpea (Cicer arietinum L.) and their wild relatives by using simple sequence repeats. Plant Mol Biol Rep 29(4):848–858
Khamassi K, Bettaiëb Ben Kaab L, Khoufi S, Chaabane R et al (2012) Morphological and molecular diversity of Tunisian chickpea. Eur J Hortic Sci 77(1):31
Bharadwaj C, Chauhan SK, Yadav S, Tara Satyavathi C, Singh R, Kumar J, Srivastava R, Rajguru G (2011) Molecular marker-based linkage map of chickpea (Cicer arietinum) developed from desi × kabuli cross. Int J Agric Sci 81(2):116–118
Sachdeva S, Bharadwaj C, Sharma V, Patil BS, Soren KR, Roorkiwal M, Varshney RK, Bhat KV (2018) Molecular and phenotypic diversity among chickpea (Cicer arietinum L.) genotypes as a function of drought tolerance. Crop Pasture Sci 69(2):142–153
Excoffier L, Smouse PE, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131(2):479–491
Roorkiwal M, Von Wettberg EJ, Upadhyaya HD, Warschefsky E, Rathore A, Varshney RK (2014) Exploring germplasm diversity to understand the domestication process in Cicer spp using SNP and DArT markers. PLoS ONE 9(7):e102016
Van Oss R, Abbo S, Eshed R, Sherman A, Coyne CJ et al (2015) Genetic relationship in Cicer sp expose evidence for geneflow between the cultigen and its wild progenitor. PLoS ONE 10(10):e0139789
Parween S, Nawaz K, Roy R, Pole AK, Venkata S, Misra G, Jain M et al (2015) An advanced draft genome assembly of a desi type chickpea (C. arietinum L.). Sci Rep 5:12806
Thudi M, Upadhyaya HD, Rathore A, Gaur PM et al (2014) Genetic dissection of drought and heat tolerance in chickpea through genome-wide and candidate gene-based association mapping approaches. PLoS ONE 9(5):e96758
Bharadwaj C, Srivastava R, Chauhan SK, Satyavathi CT, Kumar J, Faruqui A et al (2011) Molecular diversity and phylogeny in geographical collection of chickpea (Cicer sp.) accessions. J Genet 1:7
Acknowledgements
The authors are thankful to ICRISAT (Hyderabad), IIPR (Kanpur), NBPGR (New Delhi) for providing the germplasm for the present study.
Funding
This study was not funded by any external funding agency.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies/ research with human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mir, A.H., Bhat, M.A., Fayaz, H. et al. SSR markers in revealing extent of genetic diversity and phylogenetic relationships among chickpea core collection accessions for Western Himalayas. Mol Biol Rep 49, 11469–11479 (2022). https://doi.org/10.1007/s11033-022-07858-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07858-4