
Abstract
Stephania is a medicinal plants-rich genus of Menispermaceae. However, the identification of morphologically-similar species in Stephania is difficult using the currently reported methods. The indiscriminate overexploitation of Stephania plants has resulted in clinical misuse and endangerment of many species, which necessitates the development of an efficient and reliable method for species authentication. Therefore, six candidate DNA barcode sequences (ITS, ITS2, psbA-trnH, matK, rbcL, and trnL-F) were tested for their capacity to identify Stephania species. The barcodes were analyzed either as a single region or in combination by tree-based [neighbor-joining (NJ) and Bayesian inference (BI)], distance-based (PWG-distance), and sequence similarity-based (TaxonDNA) methods. Amplification and sequencing success rates were 100% for all six candidate barcodes. A comparison of six barcode regions showed that ITS exhibited the highest number of variable and informative sites (182/179), followed by psbA-trnH (173/162). DNA barcoding gap assessment showed that interspecific distances of the six barcodes were greater than intraspecific distances. The identification results showed that species discrimination rates of combination barcodes were higher than those of single-region barcodes. Based on best match and best close match methods, the ITS+psbA-trnH combination exhibited the highest discrimination power (93.93%). Further, all Stephania species could be resolved in the phylogenetic trees based on ITS+psbA-trnH (NJ, BI). This study demonstrates that DNA barcoding is an efficient method to identify Stephania species and recommends that the ITS+psbA-trnH combination is the best DNA barcode for the identification of Stephania species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Stephania Lour. (Menispermaceae) includes approximately 60 species of herbaceous or woody vines with tropical and subtropical distribution in Asia and Africa. China is the diversity center of Stephania, with 40 species distributed mainly to the south of the Yangtze River. This genus has been classified into three subgenera (subgen. Stephania, subgen. Tuberiphania, and subgen. Botryodiscia) [1, 2]. Many species of the genus Stephania are used as traditional medicines for the treatment of diseases such as dysentery, fever, tuberculosis, rheumatism, and cancer [2, 3]. Stephania plants contain a variety of alkaloids that possess various pharmacological activities, e.g., antimicrobial, analgesic, anticancer, anti-viral, anti-inflammatory, and antipsychotic activities [2,3,4]. Apart from their diverse medicinal uses, several Stephania plants exert toxic effects [3]. Thus, clinical misuse of Stephania plants may be ineffective or even detrimental to patients.
In recent decades, overexploitation of medicinal Stephania plants has resulted in 22 Stephania species being listed on the China Biodiversity Red List [5]. However, indiscriminate harvesting of Stephania plants is still prevalent, mainly due to misidentification of species with similar morphological characteristics. Despite many studies describing the taxonomic delimitation, bioactive components, and genetic diversity of Stephania plants [1,2,3,4, 6,7,8], relatively few articles have addressed identification at species level [1, 9]. Due to the morphological similarities and variations, it is very difficult to identify many Stephania species with certainty using the conventional morphological methods [1]. A UPLC-QTOF-MS/MS method developed for profiling the alkaloids of Stephania plants can differentiate between the three species being investigated [9]. However, the three species, belonging to the three different subgenera, have relatively pronounced morphological features for differentiation, and may be relatively easy to be distinguished by chemicals. The efficiency of phytochemical analysis for distinguishing between Stephania plants, especially closely related species belonging to the same subgenus, needs further study. Thus, it is difficult to efficiently identify Stephania species using the currently reported methods.
DNA barcoding has become an effective tool for species identification of medicinal plants [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30]. The psbA-trnH intergenic spacer [14, 19,20,21] and nuclear ribosomal ITS or ITS2 [22, 23] have been successfully applied to species identification. Barcode combinations such as ITS+matK [25] and ITS/ITS2+psbA-trnH [13, 15, 24, 26, 29] have achieved high identification rates at the species level. Although our group has studied the phylogeny and infragenetic classification of Stephania using ITS and trnL-F regions [7], species-level differentiation remains problematic because of insufficient information for accurate species identification and limited sample size of closely related medicinal species.
In this study, six barcoding loci, including ITS, ITS2, psbA-trnH, matK, rbcL, and trnL-F, were evaluated either as single region or in combination for species discrimination in the genus Stephania, and their species discrimination efficiencies were compared by different analytical methods. In addition, some insights into the phylogenetic relationship between Stephania species from a DNA barcode perspective were provided.
Materials and methods
Plant materials
Sixty-four accessions from 23 species were sampled (Table S1), of which 12 species were listed on the China Biodiversity Red List. Two additional samples of Cyclea hypoglauca (Schauer) Diels were used as out-groups. All species were identified by Dr. Yun Kang, Fudan University. The fresh leaf and root samples were desiccated by silica gel immediately after collection. The voucher specimens were deposited in the herbarium of the School of Pharmacy, Fudan University (SHMU).
DNA extraction, polymerase chain reaction (PCR) amplification, and sequencing
Total genomic DNA was extracted from samples using the Plant Genomic DNA Kit (Axygen, Biheng Biotechnology Co., Shanghai, China) following the manufacturer’s protocol. Six DNA barcode sequences were amplified using the primers and conditions described in Table 1. The PCR amplification was performed in a 25 µL reaction mixture with 30 ng template genomic DNA, 2.5 µL of 10 × PCR buffer (Mg2+ Plus) (TaKaRa, Biheng Biotechnology Co., Shanghai, China), 2 µL dNTP (2.5 mM each), 0.2 µL Taq DNA Polymerase (5 U/µL), and 0.5 µL of 10 µM forward and reverse primers (Sangon Biotech (Shanghai) Co., Ltd., Shanghai, China). The PCR products were detected by 1.0% agarose gel electrophoresis. Finally, the PCR products were purified using PCR purification kit (AxyPrepTM Gel Extraction Kit, Axygen Biosciences) and sequenced on an ABI 3730XL automated sequencer (Thermo Fisher Scientific Co., Suzhou, China).
Sequence alignment and data analysis
Bidirectional sequences were assembled and edited by Contig and the edited sequences were aligned using Clustal X2 [31]. The assembled sequences were submitted to the National Center for Biotechnology Information (NCBI). Intraspecific and interspecific genetic distances were calculated using the Kimura two-parameter (K2-P) model [32] by MEGA 6.0 [33]. Further, the barcoding gap histograms were estimated. Jmodeltest 2.1.10 [34] was used to calculate the most suitable evolution model for the DNA barcode sequences of Stephania species. Phylogenetic trees were constructed by MEGA 6.0 [33] and Mrbayes 3.1.2 [35]. The discriminatory power of the DNA barcodes was evaluated by TaxonDNA [36].
Results
Sequence analysis
In this study, the amplification and sequencing reactions were performed with high success (100%) for all loci of the 64 samples from 23 Stephania species. Among six DNA barcodes, ITS provided the largest number of variable and informative sites (182/179), followed by psbA-trnH (173/162), trnL-F (103/98), matK (91/82), ITS2 (76/74), and rbcL (30/28). All barcodes except rbcL contained insertions and deletions (indels). The largest length variation was detected in psbA-trnH, consisting of 18 indels. The GC content of psbA-trnH ranged from 24.8 to 27.5% and was lower than the other five barcodes. Intraspecific distances in the six barcodes ranged from 0.0000 to 0.0278, while interspecific distances ranged from 0.0000 to 0.2832 (Table 2).
DNA barcoding gap assessment
A PWG-distance method based on K2-P distance was adopted to estimate the barcoding gap among the six barcodes. When interspecific and intraspecific distances of the six barcodes were compared, it was found that interspecific distances were greater than intraspecific distances in Stephania species (Tables 2, S2–S5). ITS exhibited the shortest mean intraspecific distance (0.0005), and ITS2 showed the longest mean interspecific distance (0.1019). For an ideal barcoding gap, the minimum interspecific divergence should be greater than the maximum intraspecific variation. However, the overlap of genetic distances increases as the number of closely related species increases [14, 27]. Yet, despite this overlap, histograms of the barcoding gap showed that there existed a defined range between the intraspecific and interspecific divergence of the six barcodes (Fig. S1). Interspecific variation was greater than intraspecific variation. The results were confirmed by Wilcoxon two sample tests and a median test (Tables S4, S5).
Species discrimination
TaxonDNA was employed to analyze all sequences generated in this study. Both the “best match” and “best close match” methods achieved similar species discrimination success (Table 3). PsbA-trnH and trnL-F obtained the highest species discrimination for single regions (both 83.33%), followed by rbcL (50.00%), matK (48.48%), ITS (42.42%), and ITS2 (36.36%). With respect to the two-region combinations, ITS+psbA-trnH and ITS+trnL-F exhibited the highest discriminatory power (both 93.93%), followed by ITS2+psbA-trnH (92.92%), psbA-trnH+matK (83.83%), psbA-trnH+rbcL (83.83%), and psbA-trnH+trnL-F (83.83%). Combinations of three and four regions did not increase the discrimination rate any further (Table 3).
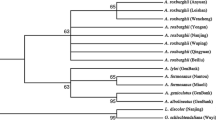
Tree-based analyses (Figs. 1, S2–S6) showed that the species resolution rates for combination barcodes were higher than those of single-region barcodes. Trees using one barcode could not provide an accurate resolution for Stephania species. The neighbor-joining (NJ) trees displayed similar clustering patterns to those of the Bayesian inference (BI) trees. The highest identification rate (100%) was achieved by the combination of nuclear ITS and chloroplast psbA-trnH using either NJ or BI method (Fig. 1, S2).

Phylogenetic tree of ITS+psbA-trnH barcode using neighbor-joining method
Discussion
The results of this study showed that the barcode candidates exhibited different ability of species discrimination in the genus Stephania and combination barcodes could achieve higher discrimination rates than single-region barcodes. The Consortium for the Barcode of Life (CBOL) Plant-Working Group proposed ITS/ITS2 as a core barcode in seed plants [23]. The psbA-trnH intergenic spacer is the most viable region of the chloroplast genome in angiosperms, and its species identification ability is generally higher than that of other chloroplast genes due to its fast evolution rate [14, 20, 21]. In this study, based on the best and best close match (Table 3), all four chloroplast DNA barcodes had higher rates of accurate identification than the two nuclear barcodes. The chloroplast psbA-trnH and trnL-F achieved the highest success rate (83.33%), while the nuclear ITS and ITS2 obtained a low success rate (about 40%) with a substantial amount of ambiguous (about 50%) and incorrect identification (about 10%). Tree-based analysis also demonstrated that the best barcode was able to authenticate most, but not all, Stephania species. Consequently, combinations of the single-region barcodes were analyzed for higher identification efficiency.
CBOL proposed matK+rbcL as the best DNA barcode to identify plants at a genetic level [28]. However, the number of matK+rbcL variable sites was insufficient to differentiate between closely related species in our study (Table 2). Further, the species discrimination rate of matK+rbcL was only 63.63% based on best match and best close match methods (Table 3). The phylogenetic trees based on either matK+rbcL or all four chloroplast genes combined (Figs. S5, S6) were also unable to authenticate all species. Previous studies showed that combining chloroplast with nuclear genes could greatly improve species identification [15, 18, 23, 25, 29], which is consistent with the satisfactory discrimination achieved by ITS+psbA-trnH and ITS+trnL-F combinations in our study. Based on the best match and best close match methods, ITS+trnL-F and ITS+psbA-trnH showed the same discrimination success rate (both 93.93%; Table 3); however, the phylogenetic tree (NJ, BI) of ITS+psbA-trnH indicated a higher discrimination rate (100%) (Figs. 1, S2). Thus, ITS+psbA-trnH was chosen as the best DNA barcode to identify Stephania species.
Phylogenetic trees reveal interrelations between different species, which can be used for species identification. Based on flower, inflorescence, leaf, and tuber traits, species of the genus Stephania in China have been classified into three subgenera, namely S. subgenus Botryodiscia, S. subgenus Stephania, and S. subgenus Tuberiphania [2]. In this study, the phylogenetic trees (NJ, BI) base on ITS+psbA-trnH contained three major clades. Those clades corresponded to the three subgenera of the genus Stephania. S. hainanensis and S. succifera belong to the same subgenus (Tuberiphania), and both are distributed in Hainan province. The samples of these two species clustered as two independent clades in the phylogenetic tree constructed based on the ITS+psbA-trnH combination. The two clades were well supported as sister groups with high bootstrap values (Figs. 1, S2), indicating that these two closely related species could be distinguished at the genetic level. The three sympatric species, S. kwangensis, S. micrantha, and S. kuinanensis—all of which are the source plants of the traditional Chinese medicine Radix Stephaniae—have similar morphology. Thus, they are usually harvested indiscriminately even though S. kuinanensis is an endangered species. The samples of the three species also formed different clades in the phylogenetic trees constructed using ITS+psbA-trnH (NJ, BI). These results show that tree-based phylogeny is a feasible method to identify these closely related species in the genus Stephania.
Further, S. kwangensis samples, collected from different populations, formed two clades in the phylogenetic trees established by ITS+psbA-trnH (NJ, BI). There might be two explanations for this pattern of S. kwangensis. One is that different evolutionary lineages of S. kwangensis exhibit similar morphology because of similar habitats, introgression, and incomplete lineage sorting. Another possible explanation is that there might be a cryptic species exists within S. kwangensis. Thus, this DNA barcoding method could provide valuable information regarding the formation of variants, hybrids and cryptic species at the genetic level.
The proposed DNA barcoding method is applicable to not only leaf, but also to root samples for species identification, which improves its practicability because roots are the medicinal parts for most Stephania plants. However, this technique has an inherent limitation which is related to the quality of genomic DNA. In this study, high-quality total DNA could be extracted from samples dried immediately with silica-gel and stored in dry conditions even for seven years. Genomic DNA from air-dried or sun-dried samples was seriously degraded, especially for moldy samples. Then the following amplification and sequencing were failed. In general, DNA may be degraded partially or even completely by various factors including extensive heat treatment, irradiation, and microbiological corrosion [30]. Consequently, this method may be not feasible for some processed crude drugs. Thus, other methods such as chemical analysis and microscopic observation are needed to be developed for identifying these samples. Herein, we recommend DNA barcoding combined with other techniques to further resolve the identification problems in Stephania plants.
Conclusions
In this study, six barcodes were analyzed either as single region or in combination for their potential to discriminate 23 Stephania species, and a combination of ITS+psbA-trnH was recommended as the best DNA barcode for the species discrimination based on tree-based, distance-based, and sequence similarity-based methods. Both ITS and psbA-trnH are relatively short regions (< 600 bp) and easy to be amplified (100%), and this barcode combination exhibits high discrimination power. The results demonstrate that DNA barcoding is an efficient and practical method for the species identification of Stephania. The application of this method will be of great help to the correct usage of Stephania plants and the protection of endangered resources.
References
Lo HS (1982) A systematic notes on the genus Stephania of China. Bull Bot Res 2(1):33–59
Editorial Committee of Flora of China (1996) Flora Reipublicae Popularis Sinicae, 1st edn., vol. 30, no. 1. Science Press, Beijing, p 40–70
Semwal DK, Badoni R, Semwal R, Kothiyal SK, Singh GJP, Rawat U (2010) The genus Stephania (Menispermaceae): chemical and pharmacological perspectives. J Ethnopharmacol 132:369–383
Aota K, Yamanoi T, Kani K, Azuma M (2018) Cepharanthine inhibits IFN-γ-Induced CXCL10 by suppressing the JAK2/STAT1 signal pathway in human salivary gland ductal cells. Inflammation 41(1):50–58
Co-edited by the Ministry of Environment Protection of the People’s Republic of China and the Chinese Academy of Sciences (2019) China Biodiversity Red list. Higher Plant. http://rep.iplant.cn/protlist/4?key=stephania. Accessed 10 Aug 2019
Shangguan Y, He JY, Kang Y, Wang YQ, Yang P, Guo JX, Huang JM (2018) Structural characterisation of alkaloids in leaves and roots of Stephania kwangsiensis by LC-QTOF-MS. Phytochem Anal 29:101–111
Xie DT, He JY, Huang JM, Xie H, Wang YQ, Kang Y. Jabbour F, Guo JX (2015) Molecular phylogeny of Chinese Stephania (Menispermaceae) and reassessment of the subgeneric and sectional classifications. Aust Syst Bot 28(4):246–255
Ma YS, Yu H, Li YY, Yan H, Cheng X (2008) A study of genetic structure of Stephania yunnanensis (Menispermaceae) by DALP. Biochem Genet 46:227–240
Xiao J, Song NN, Lu T, Pan YN, Song JY, Chen G, Sun L, Li N (2018) Rapid characterization of TCM Qianjinteng by UPLC-QTOF-MS and its application in the evaluation of three species of Stephania. J Pharm Biomed 156:284–296
Li JF, Huang J, Cheng FT, Du Y, Liu ZL (2017) Comparison on DNA barcodes of nuclear and chloroplast gene fragments in Rehmannia Libosch. ex Fisch. et Mey. (Rehmanniaceae). Chin Tradit Herbal Drugs 48(1):165–171
Yang J, Vázquez L, Chen XD, Li HM, Zhang H, Liu ZL, Zhao GF (2017) Development of chloroplast and nuclear DNA Markers for ChineseOaks (Quercus Subgenus Quercus) and assessment of their utility as DNA barcode. Front Plant Sci 8:816
Gao T, Yao H, Song JY, Liu C, Zhu YJ, Ma XY, Pang XH, Xu HX, Chen SL (2010) Identification of medicinal plants in the family Fabaceae using a potential DNA barcode ITS2. J Ethnopharmacol 130:116–121
Yu M, Jiao LC, Guo J, Wiedenhoeft AC, He T, Jiang XM, Ying YF (2017) DNA barcoding of vouchered xylarium wood specimens of nine endangered Dalbergia species. Planta 246:1165–1176
Sun ZY, Gao T, Yao H, Shi LC, Zhu YJ, Chen SL (2011) Identification of Lonicera Japonica and its related species using the DNA barcoding method. Planta Med 77:301–306
Clement WL, Donoghue MJ (2012) Barcoding success as a function of phylogenetic relatedness in Viburnum, a clade of woody angiosperms. BMC Evol Biol 12:73
Lee SY, Ng WL, Mahat MN, Nazre M, Mohamed R (2016) DNA Barcoding of the endangered Aquilaria (Thymelaeaceae) and its application in species authentication of agward products traded in the market. PLoS ONE 11(4):e0154631
Mishra P, Kumar A, Nagireddy A, Shukla AK, Sundaresan V (2017) Evaluation of single and multilocus DNA barcodes towards species delineation in complex tree genus Terminalia. PLoS One 12(8):e0182836
Yan HF, Hao G, Hu CM, Ge XJ (2011) DNA barcoding in closely related species: A case study of Primula L. sect. Proliferae Pax (Primulaceae) in China. J Syst Evol 49(3):225–236
Yu N, Gu H, Wei YL, Zhu N, Wang YL, Zhang HP, Zhu Y, Zhang X, Ma C, Sun AD (2016) Suitable DNA barcoding for identification and supervision of Piper kadsura in Chinese medicine markets. Molecules 21:1221
Storchová H, Olson MS (2007) The architecture of the chloroplast psbA-trnH non-coding region in angiosperms. Plant Syst Evol 268:235–256
Pang XH, Liu C, Shi LC, Liu R, Liang D, Li H, Cherny SS, Chen SL (2012) Utility of the trnH-psbA intergenic spacer region and its combinations as plant DNA barcodes: a meta-analysis. PLoS ONE 7:e48833
Lv TW, Teng RD, Shao QS, Wang HZ, Zhang WS, Li MY, Zhang LL (2015) DNA barcodes for the identification of Anoectochilus roxburghii and its adulterants. Planta 242:1167–1174
CBOL Plant Working Group, Li DZ, Gao LM, Li HT, Wang H, Ge XJ, Liu JQ, Chen ZD, Zhou SL, Chen SL, Yang JB, Fu CX, Zeng CX, Yan HF, Zhu YJ, Sun YS, Chen SY, Zhao L, Wang K, Yang T, Duan GW (2011) Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should beincorporated into the core barcode for seed plants. Proc Natl Acad Sci USA 108:19641–19646
Kress WJ, Wurdack KJ, Zimmer EA, Weigt LA, Janzen DH (2005) Use of DNA barcodes to identify flowering plants. Proc Natl Acad Sci USA 102:8369–8374
Liu J, Yan HF, Ge XJ (2016) The use of DNA barcoding on recently diverged species in the genus Gentiana (Gentianaceae) in China. PLoS ONE 11(4):e0153008
Liu JX, Shi LC, Han JP, Li GN, Lu H, Hou JY, Zhou XT, Meng FY, Downie Stephen R (2014) Identification of species in the angiosperm family Apiaceae using DNA barcodes. Mol Ecol Resour 14:1231–1238
Meyer CP, Paulay G (2005) DNA barcoding: error rates based on comprehensive sampling. PLoS Biol 3:2229–2238
CBOL Plant Working Group (2009) A DNA barcode for land plants. Proc Natl Acad Sci USA 106:12794–12797
Chen SL, Pang XH, Song JY, Shi LC, Yao H, Han JP, Christine L (2014) A renaissance in herbal medicine identification: from morphology to DNA. Biotechnol Adv 32:1237–1244
Xiong C, Sun W, Li JJ, Yao H, Shi YH, Wang P, Huang BS, Shi LC, Liu D, Hu ZG, Chen SL (2018) Identifying the species of seeds in traditional Chinese Medicine using DNA Barcoding. Front Pharmacol 9:701
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, Mcwilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Darriba D, Taboada GL, Doallo R, Posada D (2012) JmodelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772
Ronquist F, Huelsenbeck JP (2003) Mrbayes 3: Bayesian phylogenetic inference under mixed models. J Bioinform 19:1572–1574
Meier R, Shiyang K, Vaidya G, Ng PK (2006) DNA barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Syst Biol 55:715–728
Acknowledgements
We thank Daotao Xie, Weiyu Weng, Xuesong Tang, Xin Weng, Xiaotian Dou, Weiyu Yang, Shun Liu, and Long Lin for assistance with field work.
Funding
This work was financially supported by the grant from the National Natural Science Foundation of China (31100238).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, X., Xue, J., Zhang, Y. et al. DNA barcodes for the identification of Stephania (Menispermaceae) species. Mol Biol Rep 47, 2197–2203 (2020). https://doi.org/10.1007/s11033-020-05325-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05325-6