
Abstract
Background
Aconitum species, belonging to Ranunculaceae, have high medicinal importance but due to their overexploitation come under IUCN (International Union for Conservation of Nature) red list. The precise identification of the Aconitum species is equally important because they are used in herbal formulations. The present study aimed to develop an efficient DNA barcode system for the authentic identification of Aconitum species.
Methods and results
A set of 92 barcode gene sequences (including 12 developed during the present study and 80 retrieved from NCBI) of 5 Aconitum species (A. heterophyllum, A. vialoceum, A. japonicum, A. napellus, and A. stapfianum) were analyzed using three methods (tree-based, distance-based, and similarity-based) for species discrimination. The PWG-distance method was found most effective for species discrimination. The discrimination rate of PWG- distance ranged from 33.3% (rbcL + trnH-psbA) to 100% (ITS, rbcL + ITS, ITS + trnH-psbA and rbcL + ITS + trnH-psbA). Among DNA barcodes and their combinations, the ITS marker had the highest degree of species discrimination (NJ-40%, PWG-100% and BLAST-40%), followed by trnH-psbA (NJ-20%, PWG-60% and BLAST-20%). ITS also had higher barcoding gap as compared to other individual barcodes and their combinations. Further, we also analyzed six Aconitum species (A. balfourii, A. ferox, A. heterophyllum, A. rotundifolium, A. soongaricum and A. violaceum) existing in Western Himalaya. These species were distinguished clearly through tree-based method using the ITS barcode gene with 100% species resolution.
Conclusion
ITS showed the best species discrimination power and was used to develop species-specific barcodes for Aconitum species. DNA barcodes developed during the present study can be used to identify Aconitum species.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The correct identification and distinction of different plant species are challenged by the vegetative characteristics when they are quite similar to one another and reproductive parts are not available. This is especially required in the case of medicinal plants; whose authenticity needs to be determined before these are utilized for important formulations. Additionally, other challenges that put off the safety and herbal efficacy of medicinal plants are the inability to accurately separate them from their similar relatives, adulterants, and counterfeits [1]. Further, medicinal plants have an ever-growing market for herbal-based drugs, and uncontrolled harvesting has resulted in many of them becoming rare or threatened. The Himalayan range is a storehouse of plant biodiversity with more than 8,000 species reported in the eco-region, of which about 1748 species have been said to possess medicinal properties [2]. In this eco-region, medicinal plants are continuously exploited due to the demand by pharmaceutical companies, and being harvested from the wild in large quantities. This has resulted in most of these plant species being listed in IUCN (International Union for Conservation of Nature) red list and many of them finding place in different Appendices of CITES (Convention on International Trade in Endangered Species of Wild Fauna and Flora), prohibiting their trade. However, their trade keeps continuing due to demands, although not through legalized channels. Further, there is a probability that the traded material is not of the actual species but adulterated with other species with similar morphological characteristics of traded part(s). The inability to identify them either as a collected specimen or in fragmented form (plant parts as medicines) makes it challenging to enforce the prohibitions placed on their unsustainable harvesting and illegal trafficking. At times the identity is also faked with other plant species which are legalized for trade to avoid detection of prohibited plant species. Thus, there are a large number of plants which are extracted from the Himalaya which have become threatened in recent years due to unsustainable harvesting. Lack of organized cultivation and indiscriminate harvesting has caused the wild germplasm of these species to be depleted. Hence, conservation efforts are also required for the correct identification of medicinal plant species, as a first step. Therefore, there is a significant need for biotechnology-based interventions such as DNA barcoding, in order to ensure true identification of these species and their conservation. Each barcode exhibits a distinct genetic variation which is specific to each species and is able to differentiate between closely related species. Thus, DNA barcoding techniques have proved a promising approach for identifying plant species, and databases of DNA sequences of the nuclear or organelle genome of different plant species are now readily available and continuously being updated [3].
In DNA barcoding, short, varied, and uniform DNA sections are used to identify and distinguish between different species [4,5,6]. It was introduced as a revolutionary method for rapid and extensive species identification employing unique gene sequences as molecular species-specific marks two decades ago [4]. Three fundamental requirements must be fulfilled for a DNA sequence to be useful as a barcode: (i) significant genetic variation at the species level to allow species discrimination, (ii) a short sequence length to support DNA extraction and amplification, and (iii) primer universality across highly divergent taxa [5, 7, 8]. It was initially proposed in animals and extensively used [4]. The cytochrome c oxidase 1 (CO1), which has been proven to be universally relevant in animal barcoding, is not acceptable for most species of plants because of a substantially slower rate of cytochrome c oxidase 1 gene mutation in higher plants than in animals [5, 9]. All of the existing loci do not operate across all species, despite the fact that numerous researchers have looked for a universal plant barcode [10, 11]. The matK and rbcL two-locus combination has been selected as the best plant barcode by the Consortium for the Barcode of Life-Plant Working Group (CBOL) [12, 13] while other researchers proposed that differentiating between plant species may need the use of a multi-locus technique. There are four standard barcodes which are being utilized as the core barcode markers for the molecular identification of plants: the ribulose-bisphosphate/carboxylase Large-subunit gene (rbcL), the maturase-K gene (matK), the trnH-psbA intergenic spacer, and the nuclear ribosomal internal transcribed spacers (nrITS) [14,15,16,17]. Combining plastid (matK, rbcL, and psbA-trnH) and nuclear (ITS) regions has been suggested as a potential way to categorise different plant species.
Aconitum, a member of the Ranunculaceae family, contains about 350 species, are found primarily in Asia, accompanied by Europe and North America. Many species of Aconitum plants were employed in traditional medicine, primarily have the properties analgesic, anti-inflammatory, antirheumatic, and cardiotonic while some are poisonous [18]. Due to their exploitation for high medicinal uses, the Aconitum species, under the present study, are included in the IUCN Red List [19,20,21], with A. heterophyllum and A. violaceum categorized as ‘Endangered’ and ‘Vulnerable’ respectively. Microsatellite markers were used to study genetic diversity and population structure in order to gain access to its conservation and population status [22]. Further, the precise identification of these Aconitum species is equally important particularly if they are used in herbal formulations. Some efforts have been made in the development of DNA barcode of Aconitum for species identification, but with a limited number of species, and only single barcode gene was explored (psbA-trnH [23], ITS2 [24], ITS and matK [25], and rbcL [26]).

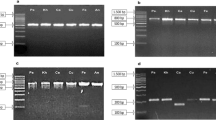
Representative images showing (A) flowering stage of Aconitum heterophyllum (a, b) and A. violaceum (c,d) in their natural habitat of Western Himalaya (B) intact genomic DNA and successful amplification of four DNA barcodes in Aconitum samples collected during the present study. The expected product size of rbcL = 554 bp, ITS = 707 bp, trnH-psbA = 450 bp, and matK = 900 bp. In case of rbcL, different sizes of bands are amplified in different samples suggested that the sequence variations among the samples, but the variation is not species-specific. In other barcode genes like ITS, trnH-psbA and matK, some non-specific bands also amplified, but sequencing was done for specific bands after purification. The sequencing was successfully done in the case on rbcL, ITS and trnH-psbA, however, in case of matK, sequencing failed
The present study focused to develop efficient DNA barcodes for species identification of Aconitum, with particular emphasis on the species of Western Himalaya. Fresh samples of Aconitum species were collected from Western Himalaya. Four DNA barcodes were explored in all the Aconitum species (including those collected during the present study, and all available sequences on databases). The present study investigated the effectiveness of individual barcodes and their combination for species differentiation; and develop an efficient barcode system to identify Aconitum species.
Materials and methods
Sampling site and sample collection of Aconitum spp
Multiple accessions were collected from the wild for A. heterophyllum (Trade name: Patish; IUCN Red List: Endangered (EN)) and A. violaceum (trade name: Mitha Patish; IUCN Red List: Vulnerable (VU)) from Himachal Pradesh State of India. This area forms a part of the Western Himalayan region, which is the native distributional range of these species. Both the species have patchy distribution in alpine areas of this eco-region and are included in various IUCN red list threat categories, as per ‘global assessment’. Although there are other species such as A. balfourii, A. ferox, A. rotundifolium, A. soongaricum, A. deinorhizum, A. chasmanthum etc.., but as these are much rare to find in Himachal Pradesh, and thus, were not included in the present study. Further, the 02 species under consideration have been reported to be under severe threat [27] due to rampant extractions in the region and are reported to be highly traded in recent years [28, 29]. Voucher specimens of these species were collected and deposited in CSIR-IHBT’s herbarium, and identified to the species level using local floras and existing specimens.
DNA extraction, PCR amplification and sequencing of collected samples
For DNA extraction, fresh leaves of above-mentioned plant samples were collected in silica gel. High quality genomic DNA were extracted using cetyltrimethylammonium bromide (CTAB) method [30]. Quality and quantity of extracted DNA were checked using 1.0% agarose gel and nanodrop (Thermo Fisher, NanoDrop™ One/Onec microvolume), respectively. The extracted DNA was diluted with nucleus free water to a final concentration of 50 ng/µl for PCR amplification with four most widely used plant DNA barcodes. These barcodes included rbcL, ITS, trnH-psbA and matK; details are provided in Table S1. For each DNA barcode, PCR was performed in final volume 20 µl with following reaction composition- 2 µl of template DNA, 4 µl of 5x Colourless Go Taq® Flexi Buffer, 2 µl of 25mM MgCl2, 0.4 µl dNTPs, 0.1 µl Taq DNA polymerase, 1 µl of each primer (forward and reverse). The amplification was done with following thermal profile- 94℃ for 4 min, 36 cycles of 94℃ for 30 s, 48–55℃ for 30 s, and 72℃ for 1 min followed by a final extension at 72℃ for 5 min using C1000 Touch™ Thermal Cycler (Bio-Rad). The amplified products were resolved on 1.5% agarose gel to examine the single amplicon.
In cases, where multiple bands were amplified, the desired product was eluted from the gel using MinElute® Gel Extraction Kit (Qiagen) following manufacturer’s protocol. The eluted product was re-amplified with corresponding primers to get the single amplicon. The PCR products were cleaned up with ExoSAP-IT™ (Applied Biosystems) following the manufacturer’s protocol and sequenced using ABI 3130xl DNA Analyzer (Applied Biosystems). Sequencing was done in both directions (forward and reverse primers) using BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems).
Sequence retrieval of DNA barcode of Aconitum Sp from NCBI
In order to develop the efficient DNA barcode, all the DNA barcode sequence of Aconitum sp available on National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/) was also searched and retrieved. (Table S2). We excluded the species, where only single sequence of DNA barcode was available due to statistical reasons.
Sequence alignment and nucleotide diversity
The raw sequences were checked for their quality using software FinchTV version 1.4.0 and only high-quality sequences were utilized for downstream analysis. All the sequences (including both, retrieved from NCBI and generated during the present study) of individual barcodes were aligned using MUSCLE in MEGA X software [31] and unaligned nucleotides from both the ends were trimmed manually. Nucleotide base frequency in protein-coding region, candidate nucleotide at particular codon position and nucleotide pair frequency were also calculated for individual barcode and their combinations using MEGA X software. The aligned sequences were analyzed for different parameters of nucleotide variation like number of variable sites, number of informative sites, and number of segregating sites using DnaSP v6.12.03 software [32]. Nucleotide diversity and neutrality tests were performed to assess the genetic diversity. The possibility of recent population growth was examined through two statistical tests- Tajima’s D [33] and Fu’s Fs [34] using DnaSP v6.12.03 software. The populations’ mismatch frequency graphs were produced under a constant population size model to see if they showed signs of a stable population history [33].
Species discrimination
In the present study, three commonly used methods; tree-based, distance-based, and similarity-based (BLAST-Basic Local Alignment Search Tool) were applied to the individual DNA barcode and their potential combinations to assess the effectiveness of species discrimination.
Tree-based approach
In this approach, discrimination effectiveness of individual barcodes or their combinations is determined based on the proportion of monophyletic species in dendrograms. Unrooted neighbour-joining (NJ) trees were built using p-distance and pairwise deletion model for the tree-based technique in MEGA X [35,36,37]. Node support was determined using 1000 bootstrap replicates. Species discrimination was considered successful when every individual of the same species belonged to a single clade [7]. The species discrimination rate for a DNA barcode region was calculated as the number of species which did not cluster with individuals of any other species divided by the total number of species used.
Distance-based approach
In this approach, pairwise-genetic distance (PWG-distance) was calculated using Kimura-2-parameter (K2P) model using MEGA X software. Inter- and intra-specific genetic distances were used to calculate barcoding gaps [7, 38]. No species discrimination was considered when the interspecific genetic distance between the corresponding species was estimated as zero (0.0) [39]. Species discrimination rate was calculated as the number of species having K2P distance zero with any other species divided by the total number of species that have been analyzed for a DNA barcode region.
Similarity-based approach
In this approach, NCBI BLAST program was used and all sequences were queried using the blastn command. Discrimination of a species was considered satisfactory if all the members of a species appeared in top matching hit. If all the sequences of a species showed 100% match (query cover and percent identity) or maximum with only same species, not with others, then species identification was considered successful. The species discrimination rate was calculated as the number of species which matched with same species divided by the total number of species used.
Results
Morphological features of Aconitum spp
The plants of A. heterophyllum are erect, herbaceous perennials, and their height varies from 30 to 100 cm with ovoid or fusiform roots. Stem is simple or sometimes branches arose from the lower part, glabrous with a few very short crispate hairs above. Lower leaves are long petiolate, usually 5 lobed and orbicular-cordate to ovate-cordate in shape. Upper leaves are short petiolate or sessile, lanceolate and irregularly crenate-dentate. Inflorescence is loose raceme, greenish blue in colour with dark purple veins (Fig. 1A). Sepals are petaloid, blue to violet, glabrous and erect; the upper sepals are helmet shape and the lateral sepals are narrow.
A. violaceum is perennial, erect or ascending herbaceous plant, height upto 30 cm. Stem is usually simple, erect in the lower part and rarely branched, glabrous or hairy. Lower leaves are long-petioled, forming cluster near the base, deeply penta- partite towards the base; lobes are further divided into linear segments. Upper leaves are small in size, shortly petioled or sessile. Flowers are large in size, violet to bluish-purple in colour and arranged in corymbs or short racemes (Fig. 1A).
PCR amplification and sequencing success rate
High-quality genomic DNA was extracted from all the collected samples and successfully amplified with all four DNA barcodes including rbcL, ITS, trnH-psbA and matK. Sequencing was successful with three DNA barcodes i.e. rbcL, ITS, trnH-psbA (Supplementary dataset S1). However, in case of matK, sequencing failed repeatedly (Fig. 1B), and thus matK barcode was excluded from downstream analysis.
Sequence retrieval from NCBI
On NCBI database, DNA barcodes were available for total 193 Aconitum species (Table S3). However, out of 193 species, DNA sequences of all the three barcodes (i.e. rbcL, ITS and trnH-psbA) were available for only three species (A. japonicum, A. napellus, and A. stapfianum); and thus included in downstream analysis during the present study. For remaining (190) species, sequences were available for only one or two barcodes.
A total 80 barcode sequences were retrieved from NCBI representing five Aconitum species (Table S2). These species included two species (A. heterophyllum and A. vialoceum) for which sampling has been done from Western Himalaya during the present study; and three species (A. japonicum, A. napellus, A. stapfianum) for which all the DNA barcodes were available on the NCBI. For individual barcode, maximum 41 sequences were retrieved for rbcL, followed by 35 sequences for ITS, and minimum of four (4) sequences for trnH-psbA.
Sequence characteristics
A total 92 sequences (including 12 developed during the present study and 80 retrieved from NCBI) of 5 Aconitum species were used for the development of efficient DNA barcodes for species discrimination during present study. For individual barcode and their combinations, length of aligned sequences varied from 124 bases (rbcL) to 706 bases (rbcL + ITS + trnH-psbA), Table 1. For rbcL, total 45 sequences (length 307–1480 bases) were used for alignment, and 124 bases long aligned sequences were obtained. Similarly, for ITS and trnH-psbA, total 39 (length 361–749 bases) and eight (8) (length 222–411 bases) sequences were aligned, respectively. In case of ITS, final aligned sequence was 450 bases and for trnH-psbA, the final aligned sequence was 157 bases long.
Nucleotide frequency and average AT and GC content at various codon coding sites are given in Table S4. Frequencies of individual nucleotides were found variable for different barcodes and their combinations. For instance, in case of rbcL, the maximum frequency was observed for thymine (T, 36.7%) followed by adenine (A, 28.0%); similar was the case for trnH-psbA where T and A represented 40.5% and 26.7%, respectively. However, in case of ITS, the maximum proportion was covered by cytosine (C) and guanine (G).
As per the individual position of codon (1st, 2nd, and 3rd) is concerned, maximum difference in the proportion of AT and CG contents was observed at 3rd position of the codon in majority of barcodes as well as their combinations; although, the differences in the proportion found variable in different barcodes and their combinations (Table S4). For instance, in case of rbcL, AT content was found higher (78.1%) at third position of codon as compared to the GC content (21.9%); however, opposite was observed for ITS where GC content was higher (61.9%) at third position of codon as compared to the AT content (38.2%).
Nucleotide pair frequency suggested that most of the nucleotide pairs were identical as expected; and few mutation events were also observed in each barcode (Table 2). Maximum 22 mutations (11 each transition and transversion) were observed in trnH-psbA, followed by seven (7) mutations in ITS and four (4) mutations in rbcL. Transition/transversion ratio slightly varied across the individual barcode and ranged 0.9 (ITS) to 1.1 (rbcL). In case of barcode combinations, nucleotide pair frequencies including identical pairs, transition and transversion events varied proportionally (Table 2).
Genetic diversity
Aligned sequences of individual barcodes and their combinations were analysed for different variability parameters (Table 3). As expected, maximum variable sites (102) and maximum informative sites (25), were observed in case of barcode combination rbcL + ITS + trnH-psbA. However, maximum proportion of variable sites (39.49%) and informative sites (5.73%) were observed in trnH-psbA. ITS showed minimum proportion (less than 10%) of indel events; however, other two DNA barcodes showed that all the variability arose due to indels, except one variable event in case of trnH-psbA.
Different parameters of nucleotide diversity and neutrality tests for each DNA barcodes and their combinations are shown in Table 4. The maximum average number of nucleotide difference between pairs of sequences were observed in barcode combination rbcL + ITS + trnH-psbA (39.533) as expected. Among individual barcodes, maximum average number of nucleotide differences between pairs of sequences were observed in trnH-psbA (17.75) and lowest in rbcL (3.002). On the basis of Eta value, rbcL + ITS + trnH-psbA showed highest nucleotide diversity (102) and rbcL showed the lowest (47) among all DNA barcodes and combinations. Total number of mutations (Eta) varied 29 (rbcL + ITS) to 102 (rbcL + ITS + trnH-psbA). Haplotype diversity (Hd) ranged from 0.245 (rbcL) to 1.0 (ITS + trnH-psbA and rbcL + ITS + trnH-psbA). Further, rbcL + ITS had minimum (0.0209) nucleotide substitution rate and ITS showed minimum nucleotide diversity (0.01596); however, maximum nucleotide substitution rate (0.26113) and nucleotide diversity (0.18490) was found in case of trnH-psbA.
Fu’s Fs neutrality test revealed that only ITS marker had a negative value (-0.494). Tajima’s D values for all markers were negative except rbcL + ITS (0.17466) but statistically non-significant (Table 4), indicating a greater number of unusual nucleotide site variations that would be expected by a neutral model of evolution. The nucleotide mismatch distribution analysis was done among various sequences (Fig. S1). DNA sequences were evaluated for population size changes, which expanded the findings of genetic diversity among species. All the sequences showed genetic variations.
Species discrimination using different methods
Three techniques (NJ, PWG-Distance, and Blast) were used to assess the effectiveness of species discrimination for DNA barcodes and combinations.
Tree based
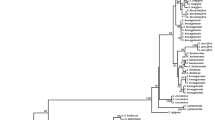
The effectiveness of identification at the species levels was evaluated using the NJ approach. Successful species identification was considered to have occurred when members of a species formed a monophyletic clade or were close to each other (Fig. 2). The marker combinations rbcL + ITS, ITS + trnH-psbA, and rbcL + ITS + trnH-psbA all demonstrated species discrimination, with the exception of rbcL + trnH-psbA. All individuals in the NJ tree of ITS, cluster together across all areas, demonstrating the best species discrimination (Fig. 2).

The neighbor-joining (NJ) tree constructed for Aconitum species under p-distance model using DNA barcode markers using different DNA barcode genes and their available combinations. ITS showed the best discrimination between the Aconitum species among all the barcode genes/combinations. rbcL and trnH-sbA do not discriminate the Aconitum species individually, but species discrimination is enhanced for these barcodes while used in combination with ITS.
Distance-based
The highest pairwise genetic distance (overall mean) observed 28.97% (trnH-psbA) followed by 13.73% (rbcL + trnH-psbA), and 6.35% (ITS + trnH-psbA) whereas lowest 1.59% (ITS) accompanied by 2.21% (rbcL + ITS), 5.62% (rbcL), and 6.09% (rbcL + ITS + trnH-psbA). The intra- and inter-specific genetic distance with each barcode and their combinations for each species are depicted in Table 5. Among all the barcodes and their combination, ITS performed better in order to discriminate the species. In ITS, intraspecific genetic distance was found lower than the interspecific genetic distance of corresponding species in most cases. For instance, intraspecific genetic distance with ITS in A. heterophyllum was observed 0.0140, however, interspecific distances of A. heterophyllum were higher with other species like A. stapfianum (0.0417), A. japonicum (0.0370), A. violaceum (0.0455) and A. napellus (0.0496). Similarly, the intraspecific genetic distance in A. japonicum was found 0.0021; and interspecific genetic distances were higher with other species- A. heterophyllum (0.0370), A. violaceum (0.0291), A. napellus (0.0198) and A. stapfianum (0.0089).
Similarity-based
The discrimination rate of individual barcode and their combinations was highly variable (Table S5). Among individual barcodes, rbcL showed the lowest resolution for species discrimination while ITS showed the highest resolution (Table S5). ITS alone and combined with other barcodes (ITS + rbcL, ITS + trnH-psbA and rbcL + ITS + trnH-psbA), successfully discriminate the A. heterophyllum from other species. However, for other four species (A. violaceum, A napellus, A. stapfianum and A. japonicum) ITS showed low resolution and discriminated the sequences at the genus level only. Other barcodes like rbcL and trnH-psbA and their combination did not discriminate the sequences even at the genus level.
Comparison of species discriminations rate of three methods used
Among the three methods (NJ, PWG-Distance, and Blast) used to assess the effectiveness of species discrimination for DNA barcodes and combinations, the PWG-distance method was found the best (Fig. 3A). The rbcL showed zero discrimination rate in all the three methods used, thus excluded from further comparisons. The discrimination rate of PWG- distance ranged from 33.3% (rbcL + trnH-psbA) to 100% (ITS, rbcL + ITS, ITS + trnH-psbA and rbcL + ITS + trnH-psbA). The NJ method was found second best to discriminate the Aconitum species with a discrimination rate of 20% (trnH-psbA) to 66.6% (rbcL + trnH-psbA), and BLAST was found least effective with a discrimination rate of 20% (trnH-psbA) to 40% (ITS).

DNA barcode analysis in Aconitum species. (A) Comparison of species discrimination success of single barcode region and their combinations based on NJ-tree, PWG-distance and BLAST method. I = ITS, R = rbcL, and T = trnH-psbA. Bar diagram suggested that PWG-distance approach is the best among three approaches used. However, among barcode genes, ITS found best for species discrimination. (B) Barcoding gap assessment for markers rbcL, ITS, trnH-psbA and their possible combinations in Aconitum. x-axes represent K2P distance intervals and y-axes represent the percentage of occurrences. With the increase in K2P distance intervals. ITS performed best with higher barcode gap with interspecies distance as compared to the intraspecies distance (C) Linear DNA barcode of Aconitum spp. based on the ITS gene. The sequence length of the available ITS genes varies and this might be the possible reason for the length variation of developed barcodes
Discrimination efficiency of individual barcode and their combination
With respect to recognition rates for single barcodes, the ITS marker had the highest degree of species discrimination (NJ-40%, PWG-100% and BLAST-40%), followed by trnH-psbA (NJ-20%, PWG-60% and BLAST-20%), but rbcL had the lowest level (0%) (Fig. 3A). The species resolution of the rbcL DNA region is less effective. Among combinations, both ITS + trnH-psbA and rbcL + ITS + trnH-psbA had the same level (NJ-33.3%, PWG-100% and BLAST-33.3%) of species discrimination.
Assessment of DNA barcoding gap
For the assessment of barcoding gap, we calculated mean genetic distance within and between species under Kimura-2-parameter distance (Table 5) and plotted K2P distance against percentage occurrence of single marker and, their combination (Fig. 3B). The range of average genetic distance within/between various species of Aconitum with K2P were 0.0–9.31% / 0.0–7.44% for rbcL, 0.21–1.40% / 0.89–4.96% for ITS, 0.0–54.82% / 0.71–58.20% for trnH-psbA, 0.22–0.54% / 0.8–4.10% for rbcL + ITS, 0.0–21.89% / 4.49–16.42% for rbcL + trnH-psbA, 0.34–8.63% / 2.76–7.26% for ITS + trnH-psbA and 0.29–7.14% / 3.81–7.33% for rbcL + ITS + trnH-psbA (Table 5). The genetic distance method based on histograms exhibited overlap between intra- and interspecific distance. ITS had higher barcoding gap as compared to other individual barcodes and their combinations (Fig. 3B).
Discrimination of Aconitum species present in Western Himalaya
We also analyzed six Aconitum species (A. balfourii, A. ferox, A. heterophyllum, A. rotundifolium, A. soongaricum and A. violaceum) existing in Western Himalaya. For these species total 54 sequences for rbcL and ITS barcode regions retrieved from NCBI database is shown in Table S6. For the trnH-psbA, no sequences were available on NCBI database, thus did not include in the analysis. These species were distinguished clearly through tree-based method using the ITS barcode gene with 100% species resolution, while rbcL showed very poor species resolution (Fig. S2); and suggested the efficient performance of ITS over the rbcL for discrimination of Aconitum species of Western Himalaya.
Moreover, in case of ITS, intraspecific genetic distance was found lower than the interspecific genetic distance of corresponding species in most cases. For instance, intraspecific genetic distance in A. ferox was observed 0.0222, however, interspecific genetic distances of A. ferox with other species ranged 0.0379–0.0686. Likewise, intraspecific genetic distance in A. heterophyllum and A. rotundifolium were 0.0298 and 0 (zero) whereas interspecific genetic distances with other species ranged 0.0399–0.0686 and 0.0265–0.0399 respectively (Table S7). The minimum interspecific mean distance was observed in A. rotundifolium and A. soongaricum (0.0265) and the maximum in A. ferox and A. heterophyllum (0.0686).
Validation of species discrimination of ITS with larger dataset
To validate the species discrimination power of ITS, we used 643 sequences (including ITS sequences available on NCBI database and sequences generated during the present study) representing 64 Aconitum spp (Fig. S3). The Aconitum spp. with ≤ 3 ITS sequences were excluded from the analysis to reduce the confounding. Out of 64 Aconitum spp., 36 spp. were grouped into particular clades, including 13 Aconitum spp. where all the individuals come together. However, remaining 23 spp. were clubbed with other Aconitum spp. but in a single clade. For some Aconitum spp, the majority of sequences grouped together, but few sequences were grouped in different clades. For instance, all sequences the six Aconitum spp. like A. anthoroideum, A. baicalense, A. contortum, A. ciliare, A. liangshanicum, and A. nemorum were grouped together except for one sequence. Similarly, in the case of A. lycoctonum, out of 15 sequences, 12 were grouped together, however, the remaining 3 sequences were little dispersed and intermingled with other spp. but at a short distance.
Further, we also identified some species-specific sequence variation in ITS gene. For instance, in case of A. coreanum, three variations including deletion of 2 nucleotides, insertion of 2 nucleotides and one SNP (C/G) were identified at different positions. Similarly, in case of A. bicalense (one nucleotide deletion) and A. gymnandrum (three nucleotides deletion) species-specific mutations were observed. For A. heterophyllum, one SNP was observed where allele “A” is specifically present in A. heterophyllum while other Aconitum spp. contained allele “C or T”.
Barcode generation
Among all the DNA barcodes and their combinations, ITS showed the best species discrimination power and was used to develop species-specific barcodes for Aconitum species (Fig. 3C). Electronic devices can read information from DNA fragments and can be used to identify species.
Discussion
The Aconitum species, under the present study, are included in the IUCN Red List due to their exploitation for medicinal uses [19,20,21], with A. heterophyllum and A. violaceum categorized as ‘Endangered’ and ‘Vulnerable’ respectively [40]. The development of DNA barcodes is important in the case of Aconitum species because conventional methods mainly focus on floral characteristics for identifying plants. For instance, in Aconitum genus tuberous roots have medicinal values, in that case, it is difficult to identify vegetative or fragmented plant samples [41]. Further, it is difficult to accurately identify the species growing in their native habitats in their vegetative state. Thus, DNA barcodes play a significant role in the identification of species. However, only limited efforts have been done for species discrimination in Aconitum and needs to be further explored. For instance, DNA barcode region psbA-trnH intergeneric spacer region found useful marker for the authentication of 19 taxa of Aconitum which are occurring in China [23]. However, none of them has any occurrence in the Himalayan region. It was suggested that psbA-trnH intergenic spacer analysis can distinguish most of the Aconitum medicinal species, but with limited differentiation power for the variants. In another study, rbcL locus showed the successful discrimination between A. heterophyllum and A. balfourii and thus suggested for the authentication of their traded parts like dried rhizomes obtained from market [26, 42] designed nrDNA ITS sequence-based SCAR (Sequence Characterized Amplified Region) markers to authenticate Aconitum heterophyllum (Ativisha) and Cyperus rotundus (Musta) at the raw drug source and in prepared herbal products. Further [43], developed a 23 bp genus-specific nucleotide signature which they found was unique to Aconitum species and is conserved within the genus and utilized this to differentiate 79 species which are occurring in China [44] undertook complete chloroplast (CP) genome sequencing of 10 species of Aconitum which was found to be beneficial in determining the complex phylogenetic relationships among these species. They also carried out data comparison with other sequenced species in GenBank database. For phylogenetic interference of Aconitum spp., two phylogenetic techniques, namely ITS (632 sequences from 164 spp.) and linked housekeeping proteins present in the chloroplast (rbcL and matK from 29 spp.), have recently been studied [45].
The goal of this work was to create an effective DNA marker that will enable individual DNA barcodes and their combinations to identify the two different species of Aconitum species from Western Himalaya, which are threatened in their native habitats. Different analytical techniques have been used to evaluate the ability to distinguish between species, including tree-based (NJ), distance-based (PWG-distance), and sequence similarity-based (BLAST) techniques on the same data set, each of these techniques had a different level of discrimination power. The present study demonstrated that sequence-based phylogenetic analysis and barcoding gap of ITS showed the best result to identify the Aconitum species. Amplification of matK showed a lower rate of sequence recovery [46].
A total of 45 sequences of rbcL, 39 sequences of ITS, and 8 sequences of trnH-psbA were utilized to construct phylogenetic trees (NJ). Because there were fewer trnH-psbA sequences in the NCBI GenBank database, the number of sequences decreased in two and three gene combinations. A. napellus and A. japonicum were eliminated from further analysis because all the three region’s sequences were not available for the same individual. As a result, the rbcL + ITS phylogenetic tree analysis includes four species, while the other combinations three species. In terms of species discrimination, ITS followed by rbcL + ITS provided a higher resolution and was more effective at identifying medicinal Aconitum species. ITS sequences of A. heterophyllum showed successful results at species level whereas A. violaceum at genus level because of the limited sequences of the A. violaceum ITS region in the NCBI GenBank database. The non-availability of sequences for A. violaceum and A. stapfianum in the NCBI GenBank database may be one of the explanations of genus level discrimination. The rbcL had 124 bases long sequences because of the trimming after alignment at both the ends (5’ and 3’) to get the best alignment.
ITS had 51 variable sites and only five (5) indel events which indicates the highest number of SNPs (Single Nucleotide Polymorphisms) among individual DNA barcodes. SNPs can cause variations in phenotypic characteristics among individuals of a species [47]. Thus, these SNPs might be a possible explanation in support of higher species discrimination through the ITS DNA barcode. trnH-psbA is highly variable DNA barcode of the plastid region [5, 48]. In present study, ITS had 450 bases long aligned sequences and 51 total mutations (Eta) whereas trnH-psbA 157 bases and 65 total mutations. These observations suggested that the number of mutations does not depend on sequence length.
A negative Tajima’s D for all, markers except rbcL + ITS denotes an excess of low frequency polymorphisms in comparison to expectations (Table 4), which indicates population growth. A favourable low amount of both low and high frequencies of polymorphisms are indicated by positive Tajima’s D value, which denotes a shrinking population. The negative Fu’s Fs test statistic in ITS showed recent population growth. In Figure S1 expected values (green line) represent constant population size and observed values (red line) represent the deviation from it.
An optimal DNA barcoding sequence for species identification should demonstrate that genetic variation across species is significantly greater than the genetic variance within species. When performing a within-species mean distance analysis and a between-species mean distance analysis, we excluded species that had a single sequence and those that lacked distinct DNA barcode for the same species. All pairwise genetic distances were estimated for each individual species using MEGA X’s application of the Kimura 2-parameter model (K2P%) [49]. Since DNA barcoding studies typically employ this technique, we employed K2P%. The highest barcoding gap was found in ITS.
In the present study, PWG and NJ trees had greater identification rates compared to BLAST. This is also supported by other studies [50, 51]. The influence of redundant genes may have decreased BLAST’s resolution. The best species resolution among all single candidate DNA barcodes in our research was found in ITS markers. The same outcome was shown in prior studies [46, 52, 53]. In the present study, rbcL + ITS, ITS + trnH-psbA, and rbcL + ITS + trnH-psbA showed increased discrimination power at species level as compared to single rbcL and trnH-psbA. This is the combined effect of ITS with another barcode region. High resolving ability of ITS, either alone in Ficus and Gossypium species [54], Palm [55] and some timber species (Meliaceae) [56] or in combination with rbcL and/or matK in Lysimachia [15] and Palm [55] was also reported. In our study rbcL was not able to show species discrimination using any of the methods used like NJ, PWG-distance and BLAST. The DNA barcode rbcL is frequently used in phylogenetic research and is very helpful at the family and genus levels with good universality across various taxa is one of the explanations [53]. The second explanation is the short length (124 bases) of aligned sequences. In the present study, Aconitum species are more closely related to each other within single genera. It is evident that rbcL is effective at genus-level discrimination, below the genus level their discrimination power decreases [53]. At the species level, the rbcL markers showed only 10% discriminating ability in Alnus [52] and 26% in Arecaceae [55]. In another study, rbcL showed lower (24.7–28.5%) species discrimination power among tropical trees from Xishuangbanna Nature Reserve in China [57]. In contrast, ITS DNA barcode which had maximum aligned sequence length (450 bases) among individual DNA barcodes showed the best level of species discrimination. Based on rbcL and ITS sequence length and discrimination rate we interpreted that it is best to use long aligned sequences for higher species discrimination rate.
After alignment of rbcL + ITS sequence, it showed 100% base composition similarity with ITS, however, there was variation in length. The only difference was rbcL + ITS had more gaps or missing sites as compared to ITS. In Fig. 3B, DNA barcode only contains nucleotide bases not gaps/missing sites, this is one of the explanations that both sequences are of the same length.
For the construction of phylogenetic tree of Aconitum species of Western Himalaya, total 45 sequences of rbcL and 9 sequences of ITS were used. The rbcL and ITS barcode sequences for A. rotundifolium and A. balfourii, respectively, were not available in the NCBI database. Similar to other Aconitum species (A. heterophyllum, A. japonicum, A. napellus, A. stapfianum, and A. violaceum), the ITS barcode region was successful in differentiating between Aconitum species found in the Western Himalaya. In previous studies, the same result was demonstrated where ITS found the most efficient DNA barcode to discriminate plant species like Alnus, Crawfurdia, Hippaphae etc. [46, 52, 53, 58]. The NJ tree and interspecific mean distance analysis suggested that A. rotundifolium and A. soongaricum are closer, while A. ferox and A. heterophyllum are less close as compared to other species of Western Himalaya.
Altogether, ITS found the best DNA barcode to discriminate the Aconitum spp., as it discriminates the majority of the Aconitum spp. (Fig S3). NJ tree of 643 sequences revealed the potential of ITS barcode, where most of the sequences were grouped in a species-specific manner. Although some sequences were clustered with non-specific clades. There might be two possible reasons for this discrepancy- (i) some technical error during sequencing etc. or (ii) wrong nomenclature of the spp. Similar observations were also highlighted and discussed in recent review [45]. Further, species-specific sequence variation is found to be very important in order to develop markers to distinguish a particular species. It becomes even more important if some species are commercially potential and others are toxic in nature. We also observed species-specific sequence variation in ITS gene. Maximum species-specific sequence variation was observed in A. coreanum with one insertion, one deletion and one SNP; suggested that A. coreanum. is most evolved. Similar observation for A. coreanum has also been made while analysing the sequence variation ndhC-trnV region in earlier study [59].
In conclusion, The PWG-distance method was found most effective for species discrimination and ITS showed the best species discrimination power and was used to develop species-specific barcodes for Aconitum species. The DNA barcodes developed during the present study would be a potential tool for the identification of Aconitum species with particular emphasis on species of Western Himalaya.
Data Availability
All data needed to support the conclusions are included in this article. Additional data related to this paper can be requested from the corresponding author.
Abbreviations
- BLAST:
-
Basic Local Alignment Search Tool
- CBOL:
-
Consortium for the Barcode of Life
- CITES:
-
Convention on International Trade in Endangered Species of Wild Fauna and Flora
- CO1:
-
Cytochrome c oxidase 1
- CP:
-
Chloroplast
- CTAB:
-
Cetyl Trimethyl Ammonium Bromide
- EN:
-
Endangered
- IUCN:
-
International Union for Conservation of Nature
- K2P:
-
Kimura-2-parameter
- matK:
-
maturase-K
- NCBI:
-
National Center for Biotechnology Information
- nrITS:
-
nuclear ribosomal Internal Transcribed Spacer
- PWG-distance:
-
Pairwise-Genetic Distance
- rbcL:
-
ribulose-bisphosphate/carboxylase Large-subunit gene
- SCAR:
-
Sequence Characterized Amplified Region
- SNPs:
-
Single Nucleotide Polymorphisms
- VU:
-
Vulnerable
References
Rajphriyadharshini R, Weerasena OV (2020) DNA barcoding of medicinal plant: a systemic review. Int J Pharm Sci Invent 9:06–16. https://doi.org/10.35629/6718-09060616
Badola HK, Aitken S (2003) The himalayas of India: a treasury of medicinal plants under siege. Biodiversity 4:3–13. https://doi.org/10.1080/14888386.2003.9712694
Safhi FA, ALshamrani SM, Jalal AS, El-Moneim DA, Alyamani AA, Ibrahim AA (2022) Genetic characterization of some Saudi Arabia’s accessions from Commiphora gileadensis using physio-biochemical parameters, molecular markers, DNA barcoding analysis and relative gene expression. Genes 13:2099. https://doi.org/10.3390/genes13112099
Hebert PD, Cywinska A, Ball SL, DeWaard JR (2003) Biological identifications through DNA barcodes. Proc R Soc B: Biol Sci 270:313–321. https://doi.org/10.1098/rspb.2002.2218
Kress WJ, Wurdack KJ, Zimmer EA, Weigt LA, Janzen DH (2005) Use of DNA barcodes to identify flowering plants. Proc Natl Acad Sci 102:8369–8374. https://doi.org/10.1073/pnas.0503123102
Kress WJ (2017) Plant DNA barcodes: applications today and in the future. J Syst Evol 55:291–307. https://doi.org/10.1111/jse.12254
Hollingsworth ML, Andra Clark AL, Forrest LL, Richardson J, Pennington RT, Long DG, Cowan R, Chase MW, Gaudeul M, Hollingsworth PM (2009) Selecting barcoding loci for plants: evaluation of seven candidate loci with species-level sampling in three divergent groups of land plants. Mol Ecol Resour 9:439–457. https://doi.org/10.1111/j.1755-0998.2008.02439.x
Hollingsworth PM, Graham SW, Little DP (2011) Choosing and using a plant DNA barcode. PLoS ONE 6:e19254. https://doi.org/10.1371/journal.pone.0019254
Fazekas AJ, Kesanakurti PR, Burgess KS, Percy DM, Graham SW, Barrett SC, Newmaster SG, Hajibabaei M, Husband BC (2009) Are plant species inherently harder to discriminate than animal species using DNA barcoding markers? Mol Ecol Resour 9:130–139. https://doi.org/10.1111/j.1755-0998.2009.02652.x
Chase MW, Fay MF (2009) Barcoding of plants and fungi. Science 325:682–683. https://doi.org/10.1126/science.1176906
Chen S, Yao H, Han J, Liu C, Song J, Shi L, Zhu Y, Ma X, Gao T, Pang X, Luo K, Li Y, Li X, Jia X, Lin Y, Leon C (2010) Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS ONE 5:e8613. https://doi.org/10.1371/journal.pone.0008613
CBOL Plant Working Group 1, Hollingsworth PM, Forrest LL, Spouge JL, Hajibabaei M, Ratnasingham S, Bank MVD, Chase MW, Cowan RS, Erickson DL, Fazekas AJ, Graham SW, James KE, Kim KJ, Kress WJ, Schneider H, AlphenStahl JV, Barrett SCH, Berg CVD et al (2009) A DNA barcode for land plants. Proc Natl Acad Sci 106:12794–12797. https://doi.org/10.1073/pnas.0905845106
Li FW, Kuo LY, Rothfels CJ, Ebihara A, Chiou WL, Windham MD, Pryer KM (2011) rbcL and matK earn two thumbs up as the core DNA barcode for ferns. PLoS ONE 6:e26597. https://doi.org/10.1371/journal.pone.0026597
Xiang XG, Hu HA, Wang WE, Jin XH (2011) DNA barcoding of the recently evolved Genus Holcoglossum (Orchidaceae: Aeridinae): a test of DNA barcode candidates. Mol Ecol Resour 1:1012–1021. https://doi.org/10.1111/j.1755-0998.2011.03044.x
Zhang CY, Wang FY, Yan HF, Hao G, Hu CM, Ge XJ (2012) Testing DNA barcoding in closely related groups of Lysimachia L. (Myrsinaceae). Mol Ecol Resour 12:98–108. https://doi.org/10.1111/j.1755-0998.2011.03076.x
Li X, Yang Y, Henry RJ, Rossetto M, Wang Y, Chen S (2015) Plant DNA barcoding: from gene to genome. Biol Rev 90:157–166. https://doi.org/10.1111/brv.12104
Safhi FA, Alshamrani SM, Bogmaza AF, El-Moneim DA (2023) DNA barcoding of wild plants with potential Medicinal properties from Faifa Mountains in Saudi Arabia. Genes 14:469. https://doi.org/10.3390/genes14020469
Wood C, Coulson J, Thompson J, Bonner S (2020) An intentional aconite Overdose: a case report. J Crit Care Med 6:124–129. https://doi.org/10.2478/jccm-2020-0016
Yun YE, Yu JN, Nam GH, Ryu SA, Kim S, Oh K, Lim CE (2015) Next-generation sequencing identification and characterization of microsatellite markers in Aconitum austrokoreense Koidz., an endemic and endangered medicinal plant of Korea. Genet Mol Res 14:4812–4817. https://doi.org/10.4238/2015.May.11.13
Kong H, Liu W, Yao G, Gong W (2018) Characterization of the whole chloroplast genome of a rare and endangered species Aconitum reclinatum (Ranunculaceae) in the United States. Conserv Genet Resour 10:165–168. https://doi.org/10.1007/s12686-017-0789-y
Wani TA, Kaloo ZA, Dangroo NA (2022) Aconitum heterophyllum Wall. Ex Royle: a critically endangered medicinal herb with rich potential for use in medicine. J Integr Med 20:104–113. https://doi.org/10.1016/j.joim.2021.12.004
Kumar A, Tiwari A, Narendran P, Raturi PP (2023) Genetic diversity and population structure of Aconitum heterophyllum-an endangered plant from North-Western Himalayas using microsatellite markers. Med Plants Int J 15:390–401. https://doi.org/10.5958/0975-6892.2023.00039.4
He J, Wong KL, Shaw PC, Wang H, Li DZ (2010) Identification of the medicinal plants in Aconitum L. by DNA barcoding technique. Planta Med 76:1622–1628. https://doi.org/10.1055/s-0029-1240967
Ren YY (2018) Identification of tibetan medicinal plants of Aconitum Genus by ITS2 sequences. Chin Tradit Herb Drugs 24:4614–4620
Almerekova S, Ivaschenko A, Kaparbay R, Myrzagalieva A, Turuspekov Y (2020) Phylogenetic assessment of tree species of Aconitum L. from Kazakhstan by using ITS and matK markers. Eurasian Union of Scientists 2:4–9. https://doi.org/10.31618/ESU.2413-9335.2020.2.79.1036
Negi RK, Nautiyal P, Bhatia R, Verma R (2021) rbcL, a potential candidate DNA barcode loci for aconites: conservation of himalayan aconites. Mol Biol Rep 48:6769–6777. https://doi.org/10.1007/s11033-021-06675-5
Sharma P, Samant SS, Lal M, Sharma A (2014) Diversity, indigenous uses, threat categorization and conservation prioritization of medicinal plants: a case study from Himachal Pradesh, India. J Biodivers Endanger Species 2:2. https://doi.org/10.4172/2332-2543.1000134
Thakur KS, Kumar M, Bawa R, Bussmann RW (2014) Ethnobotanical study of herbaceous flora along an altitudinal gradient in Bharmour Forest Division, District Chamba of Himachal Pradesh, India. https://doi.org/10.1155/2014/946870. J Evid Based Complementary Altern Med
Jishtu V, Bhondge SW, Bhushan B, Chauhan M, Chauhan A (2021) Threatened ethnomedicinal plants of Dodra-Kwar region of Himachal Pradesh, NW Himalaya. J Med Plants Stud 9:151–159
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Tajima F (1989) Statistical method for testing the Neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595. https://doi.org/10.1093/genetics/123.3.585
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925. https://doi.org/10.1093/genetics/147.2.915
Srivathsan A, Meier R (2012) On the inappropriate use of Kimura-2‐parameter (K2P) divergences in the DNA‐barcoding literature. Cladistics 28:190–194. https://doi.org/10.1111/j.1096-0031.2011.00370.x
Collins RA, Cruickshank RH (2013) The seven deadly sins of DNA barcoding. Mol Ecol Resour 13:969–975. https://doi.org/10.1111/1755-0998.12046
Yan LJ, Liu J, Möller M, Zhang L, Zhang XM, Li DZ, Gao LM (2015) DNA barcoding of Rhododendron (Ericaceae), the largest Chinese plant genus in biodiversity hotspots of the Himalaya–Hengduan Mountains. Mol Ecol Resour 15:932–944. https://doi.org/10.1111/1755-0998.12353
Kress WJ, Erickson DL (2007) A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnh-psba spacer region. PLoS ONE 2:e508. https://doi.org/10.1371/journal.pone.0000508
Krawczyk K, Szczecińska M, Sawicki J (2014) Evaluation of 11 single-locus and seven multilocus DNA barcodes in Lamium L. (Lamiaceae). Mol Ecol Resour 14:272–285. https://doi.org/10.1111/1755-0998.12175
Jabeen N, Kozgar M, Dar GH, Shawl AS, Khan S (2013) Distribution and taxonomy of genus Aconitum in Kashmir: potent medicinal resource of Himalayan Valley. Chiang Mai J Sci 40:173–186
Shyaula SL (2011) Phytochemicals, traditional uses and processing of Aconitum species in Nepal. Nepal J Sci Technol 12:171–178
Seethapathy GS, Balasubramani SP, Venkatasubramanian P (2014) nrDNA ITS sequence-based SCAR marker to authenticate Aconitum heterophyllum and Cyperus rotundus in Ayurvedic raw drug source and prepared herbal products. Food Chem 145:1015–1020. https://doi.org/10.1016/j.foodchem.2013.09.027
Wang G, Liu Y, Bai X, Cao P, Pang X, Han J (2022) Identification and Poisoning diagnosis of Aconitum materials using a genus-specific nucleotide signature. Ecotoxicol Environ Saf 237:113539. https://doi.org/10.1016/j.ecoenv.2022.113539
Xia C, Wang M, Guan Y, Li J (2022) Comparative analysis of the chloroplast genome for Aconitum species: genome structure and phylogenetic relationships. Front Genet 13:878182. https://doi.org/10.3389/fgene.2022.878182
Kakkar RA, Haneen MA, Parida AC, Sharma G (2023) The known, unknown, and the intriguing about members of a critically endangered traditional medicinal plant genus Aconitum. Front Plant Sci 14:1139215. https://doi.org/10.3389/fpls.2023.1139215
Zhang D, Jiang B, Duan L, Zhou N (2016) Internal transcribed spacer (ITS), an ideal DNA barcode for species discrimination in Crawfurdia Wall. (Gentianaceae). Afr J Tradit Complement Altern Med 13:101–106. https://doi.org/10.21010/ajtcam.v13i6.15
Huq MA, Akter S, Nou IS, Kim HT, Jung YJ, Kang KK (2016) Identification of functional SNPs in genes and their effects on plant phenotypes. J Plant Biotechnol 43:1–11. https://doi.org/10.5010/JPB.2016.43.1.1
Shaw J, Lickey EB, Schilling EE, Small RL (2007) Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot 94:275–288. https://doi.org/10.3732/ajb.94.3.275
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. https://doi.org/10.1007/BF01731581
Xie L, Wang YW, Guan SY, Xie LJ, Long X, Sun CY (2014) Prospects and problems for identification of poisonous plants in China using DNA barcodes. Biomed Environ Sci 27:794–806. https://doi.org/10.3967/bes2014.115
Wang J, Zhao J, Yu W, Wang S, Bu S, Shi X, Zhang X (2021) Rapid identification of common poisonous plants in China using DNA barcodes. Front Ecol Evol 9:698418. https://doi.org/10.3389/fevo.2021.698418
Ren BQ, Xiang XG, Chen ZD (2010) Species identification of Alnus (Betulaceae) using nrDNA and cpDNA genetic markers. Mol Ecol Resour 10:594–605. https://doi.org/10.1111/j.1755-0998.2009.02815.x
China Plant BOL Group 1, Li DZ, Gao LM, Li HT, Wang H, Ge XJ, Liu JQ, Chen ZD, Zhou SL, Chen SL, Yang JB, Fu CX, Zeng CX, Yan HF, Zhu YJ, Sun YS, Chen SY, Zhao L, Wang K et al (2011) Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc Natl Acad Sci 108:19641–19646. https://doi.org/10.1073/pnas.1104551108
Roy S, Tyagi A, Shukla V, Kumar A, Singh UM, Chaudhary LB, Datt B, Bag Sk, Singh PK, Nair NK, Husain T, Tuli R (2010) Universal plant DNA barcode loci may not work in complex groups: a case study with Indian Berberis species. PLoS ONE 5:e13674. https://doi.org/10.1371/journal.pone.0013674
Jeanson ML, Labat JN, Little DP (2011) DNA barcoding: a new tool for palm taxonomists? Ann Bot 108:1445–1451. https://doi.org/10.1093/aob/mcr158
Muellner AN, Schaefer H, Lahaye R (2011) Evaluation of candidate DNA barcoding loci for economically important timber species of the mahogany family (Meliaceae). Mol Ecol Resour 11:450–460. https://doi.org/10.1111/j.1755-0998.2011.02984.x
Huang XC, Ci XQ, Conran JG, Li J (2015) Application of DNA barcodes in Asian tropical trees–a case study from Xishuangbanna Nature Reserve, Southwest China. PLoS ONE 10:e0129295. https://doi.org/10.1371/journal.pone.0129295
Liu ZF, Ci XQ, Li L, Li HW, Conran JG, Li J (2017) DNA barcoding evaluation and implications for phylogenetic relationships in Lauraceae from China. PLoS ONE 12:e0175788. https://doi.org/10.1371/journal.pone.0175788
Park I, Kim WJ, Yang S, Yeo SM, Li H, Moon BC (2017) The complete chloroplast genome sequence of Aconitum coreanum and Aconitum carmichaelii and comparative analysis with other Aconitum species. PLoS One 12:e0184257. https://doi.org/10.1371/journal.pone.0184257
Acknowledgements
This study was supported by the Council of Scientific and Industrial Research (CSIR) (MLP-172). AC thanks CSIR for the Junior Research Fellowship. This manuscript represents CSIR-IHBT communication number 5430.
Funding
This study was funded by the Council of Scientific and Industrial Research (CSIR) (MLP-172).
Author information
Authors and Affiliations
Contributions
AC- data acquisition, wet-lab experiments, analysis, writing; DS- data acquisition; JP- data acquisition; KS and SS- plant sample collection; AC- conceptualization, plant material and sampling, editing; VJ-conceptualization, supervision, result interpretation, writing and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11033_2023_8927_MOESM3_ESM.pdf
Supplementary Material 3: Fig. S1. Pairwise mismatch distribution based on rbcL, ITS, trnH-psbA and the combinations. Pairwise differences are displayed on the x-axis, while frequency is displayed on the y-axis. R2= Ramos-Onsins and Rozas statistics; r= Raggedness statistic; Tau= Tau Date of the Growth or Decline measured of mutational time; C.V.= Coefficient of variation
11033_2023_8927_MOESM5_ESM.pptx
Supplementary Material 5: Fig. S2 The neighbor-joining (NJ) tree constructed under the p-distance model for Aconitum species of Western Himalaya using ITS and rbcL barcodes.
11033_2023_8927_MOESM6_ESM.pptx
Supplementary Material 6: Fig. S3 The neighbor-joining (NJ) tree constructed under the p-distance model for Aconitum species using ITS.
11033_2023_8927_MOESM7_ESM.docx
Supplementary Material 7: Table S1. Primer details including sequences, annealing temperature (Tm) and product size used to amplify the four DNA barcodes in Aconitum species. Table S2. List of sequences (accession number) of rbcL, ITS, and trnH-psbA barcode in five Aconitum species retrieved from NCBI database. Table S4. Nucleotide distribution of individual barcodes and their combinations in protein-coding regions and three positions of the codon. Table S6. List of sequences (accession number) of rbcL and ITS barcode of six Aconitum species present in Western Himalaya, retrieved from NCBI database. Table S7. Comparison of the genetic distances at three different loci under K2P distance between and within six different species of Aconitum present in Western Himalayan region.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Choudhary, A., Shekhawat, D., Pathania, J. et al. Exploring DNA barcode for accurate identification of threatened Aconitum L. species from Western Himalaya. Mol Biol Rep 51, 75 (2024). https://doi.org/10.1007/s11033-023-08927-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-023-08927-y