
Abstract
Main conclusion
Chinese medicinal herbs have a similar appearance and are easily confused, complicating identification via traditional methods. This study provided a scientific approach, based on DNA barcoding, to accurately and rapidly identify Anoectochilus roxburghii and its adulterants. This technology complements traditional methods of identification of medicinal herbs.
A comparison of the DNA barcodes matK, psbA-trnH and ITS2 was performed to verify that the ITS2 sequence is an effective marker for rapidly and accurately identifying A. roxburghii and its closely related species. Genomic DNA extracted from A. roxburghii and its adulterants were used as templates and the ITS2 sequence was amplified using PCR amplification and sequencing. Species identification was conducted using BLAST1 and neighbor-joining trees. The 12 samples were successfully classified into four species based on the ITS2 sequence. The ITS2 sequence length of A. roxburghii was 253 bp. The average intra-specific genetic distance of A. roxburghii was 0.0021, markedly lower than the inter-specific genetic distance between A. roxburghii and its adulterants (0.0380). Our findings illustrate that ITS2 sequence can accurately and efficiently distinguish A. roxburghii and its adulterants. In addition, the results provided reference for molecular identification of other Chinese herbal medicine.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Anoectochilus roxburghii, a member of the Orchidaceae, is a valued plant species in many Asian countries where it is used for ornamental and medicinal purposes (Tseng et al. 2006; Shao et al. 2014a). Because of its unique medicinal properties, such as its notable curative effects of breaking fevers and cooling blood, as well as eliminating dampness and detoxifying, A. roxburghii has been called “the king of medicine” (Zhang et al. 2007; Du et al. 2008). The main chemical composition of A. roxburghii includes polysaccharides, amino acids, alkaloids, flavonoids and organic acids (He et al. 2004; Zheng et al. 2013; Shao et al. 2014b). It has been used to treat diabetes, tumors, hyperliposis and hepatitis (Chang et al. 2000; Du et al. 2008; Cui et al. 2013). However, because of the plant’s demanding growth conditions, slow rate of growth, low seed germination rate and long-term excavation, A. roxburghii resources have become limited (Luo et al. 2012). Adulteration has been observed because of the supply shortage and high price. Consequently, the rapid and accurate authentication of A. roxburghii is required to ensure safety in the medicinal herb trade. A. roxburghii and its adulterants vary greatly in pharmacological activities but are similar in profile. Therefore, it is difficult to distinguish A. roxburghii based solely on morphological characteristics (Liu et al. 2012). Microscopy and TLC have been used to distinguish A. roxburghii from its adulterants to ensure quality and therapeutic effects (Zheng et al. 1997). However, both of the ways are time-consuming and inaccurate because A. roxburghii has varied shapes and a complex chemical composition. Therefore, discovering an easy and accurate method for the identification of A. roxburghii has attracted attention.
DNA barcoding uses a short DNA sequence from a standard locus as a species identification tool (Hebert and Gregory 2005; Kress et al. 2005; Miller 2007). This method is not affected by species development stage (leaves, seeds, flowers, etc.) and medicine materials (raw herbs or powder) and it has been widely used in identification of medicinal plants and herbs (Han et al. 2008). The third International Barcode of Life Conference (CBOL) presented that matK and rbcL sequences as the international common bar code sequence, ITS/ITS2 sequence and psbA-trnH as complementary sequence. However, DNA barcodes have not been used to identify the medicinal plant A. roxburghii or its adulterants. In this study, we chose psbA-trnH, matK and ITS2 as DNA barcodes and discovered that the ITS2 sequence was effective in identifying A. roxburghii and its adulterants.
Materials and methods
Plant materials
In this study, all 12 samples were collected from Fujian, Zhejiang, Jiangxi, Guizhou, Guangxi and Taiwan (Table 1), of which eight samples were A. roxburghii, two were Anoectochilus formosanus, one was Ludisia discolor and one was Goodyera schlechtendaliana. All plant species were identified by Professor Runhuai Hu (The Nurturing Station for the State Key Laboratory of Subtropical Silviculture, Zhejiang A & F University). A total of 15 individuals were selected randomly from each of the 12 populations, and about 0.5 g of fresh leaves per plant was collected and immediately dried in silica gel. All samples were stored at −20 °C until being processed. In addition, Anoectochilus geniculatus, Anoectochilus albolineatus and Anoectochilus lylei ITS2 sequences were downloaded from GenBank.
DNA extraction, amplification and sequencing
Genomic DNA was extracted from silica gel-dried leaves according to the protocol associated with the Plant Genomic DNA Kit (Tiangen Biotech Co, China). For the candidate barcodes, ITS2, psbA-trnH and matK, PCR reaction conditions and universal DNA barcode primers were used (Table 2). PCR amplification of the ITS2 region was carried out in a Peltier Thermal Cycler PTC0200 (BioRad Lab Inc, USA) using approximately 30 ng of genomic DNA as the template in a 25-μl reaction mixture (1× PCR buffer without MgCl2, 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.1 μM of each primer (synthesized by Sangon Co, China), and 1.0 U of Taq DNA Polymerase (TaKaRa). The PCR products were run on a 1.0 % agarose gel in 0.5× TBE buffer and purified with the TIANGel Midi Purification Kit (Tiangen Biotech Co, China). The PCR products were purified using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). The purified PCR products were sequenced on an ABI 3730XL sequencer (Applied Biosystems Inc.) using the amplification primers.
Sequence alignment and analysis
Consensus sequences and contig generation were performed by CodonCode Aligner V 2.06 (CodonCode Co, USA). The ITS2 sequences were subjected to HMM model analysis to remove the conserved 5.8S and 26S (or equivalent) rRNA sequences (Keller et al. 2009). The sequences were then aligned using Clustal W (Thompson et al. 1994) and bootstrap tests were conducted using 1000 resampling to assess the confidence of the phylogenetic relationships using MEGA 4.0 (Tamura et al. 2011). Moreover, the bootstrap values were displayed on the branches of the NJ tree. The inter/intra-specific variation of the samples was calculated according to Kress et al. (2005) and Song et al. (2009). Two methods of species identification, including BLAST1 and nearest distance method, were performed as described previously (Ross et al. 2008; Chen et al. 2010).
Results
Amplification and sequencing success
Total genomic DNA was isolated from the samples and the PCR amplification rate of the ITS2 sequences from plants of A. formosanus was 100 %. High-quality bidirectional sequences were obtained using the PCR of the ITS2 and psbA-trnH barcodes. The sequencing quality of matK was inferior to that of the other two loci, but a complete sequence was still produced by the assembly. The alignment results indicated that the eight populations of A. roxburghii and two of A. formosanus psbA-trnH sequences did not harbor any variation sites. For these two species, the psbA-trnH sequence was less divergent and thus not suitable for species identification. The matK sequences also lacked a variation site, which indicated that this locus was not suitable to differentiate among the two species. However, all 12 samples were successfully classified into four species based on the ITS2 sequence. For this reason, only the ITS2 barcode was used in further analyses.
Sequence analysis and inter/intra-specific variations
In this study, all 12 ITS2 sequences were obtained from the A. roxburghii, A. formosanus, L. discolor and G. schlechtendaliana species. The A. geniculatus A. albolineatus and A. lylei ITS2 sequence was downloaded from GenBank (JN166059, JN166058, JN166060). All 15 ITS2 sequences were included in the final analysis. The characteristics of these sequences are summarized in Table 3. The length of the ITS2 sequences of A. roxburghii, A. formosanus, A. geniculatus, A. albolineatus and A. lylei were 253 bp and the average GC content were 48.37, 48.62, 47.83, 47.83 and 48.62 %, respectively. The length of the ITS2 sequences of L. discolor and G. schlechtendaliana were 250 and 252 bp; their average GC contents were 50.00 and 49.60 %, respectively. 43 nucleotide variation sites were found in the ITS2 sequence, contained 21 single base-point mutations. The point mutations (48.84 %) were either purine substitutions (A by G, and G by A; n = 9), or pyrimidine substitutions (C by T, and T by C; n = 8), or purine/pyrimidine substitutions (G by T, T by G, T by A and A by T; n = 4) (Table 4). When calculated according to the K2P model, the average intra-specific genetic distance (0.0021) is far less than the average inter-specific genetic distance (0.0380). The results demonstrated that the differences between the inter-specific and intra-specific divergences were significant.
Species identification capability of the ITS2 barcode and NJ tree analysis
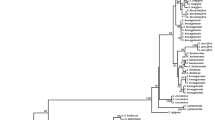
BLAST1 and the nearest distance as two discrimination methods were used to evaluate the ability of the barcoding sequences to distinguish among species in the given samples. The results showed that ITS2 performed well when using either BLAST1 or distance discrimination method at the species level. The NJ tree is an ideal analytical method that can generate a graphical representation of the ITS2 sequence results, especially when they are closely related. It can effectively determine the power of a given locus combination to discriminate among species (Zhang et al. 2012). In this study, the NJ tree demonstrated that the A. roxburghii samples clustered into one clade, whereas A. formosanus, A. geniculatus, A. albolineatus, A. lylei, L. discolor and G. schlechtendaliana clustered into their own clades (Fig. 1). Thus, the NJ tree clearly distinguished between A. roxburghii and closely related species. Overall, this study demonstrated that ITS2 was efficient and effective.

The NJ tree of Anoectochilus roxburghii and its closely related species with the ITS2 sequences. The bootstrap scores (1000 replicates) are shown (≥50 %) for each branch
Secondary structure of the ITS2 sequence
To identify the species, we focused not only on the divergence of primary sequences of ITS2, but also on the use of variations in the secondary structures of ITS2. According to the ITS2 database built by Koetschan et al. (2010), the ITS2 secondary structure of A. roxburghii and its adulterants contain a central ring and four similar helices: Helix I, II, III, and IV. Moreover, Helix III was relatively longer than the others which were shown in Fig. 2. Secondary structures of different species formed the diversified helices in number, size and loci of loops, degree of angles from the center of the spiral arm. It revealed that their differences were mainly present in Helix I and III after analyzing the ITS2 secondary structure. The secondary structure of A. formosanus shows that Helix I was different from the others and the secondary structure of L. Discolor is close to A. roxburghii, but it can still distinguish them by Helix IV. The secondary structure of G. schlechtendaliana contains a small center ring but a big ring in Helix I. Therefore, we could distinguish A. roxburghii and its adulterants at the molecular level by comparing ITS2 secondary structures.

The secondary structure of ITS2 of Anoectochilus roxburghii and its adulterants. Their differences were mainly present in Helix I and III, so we could distinguish them at the molecular level
Discussion
DNA barcoding using short DNA region to identify A. roxburghii and its close species was quick and accurate, which was a more precise identification method at the molecular level. In this study, three DNA regions, ITS2, psbA-trnH and matK, had been recommended as primary DNA barcodes for plants according to the rules of CBOL. Then, we tested the ability of these three DNA regions through PCR amplification to discriminate between A. roxburghii and its adulterants, but only ITS2 sequence was capable of being used to distinguish at the inter-specific level to a greater extent than psbA-trnH or matK. DNA barcodes must simultaneously contain enough variability to be used for species identification and adequate conservative regions for the design of universal primers (Stoeckle 2003). The matK sequence belongs to the chloroplast genome sequences, via maternal uniparental inheritance; thus, constructed branch relations often cannot truly reflect the evolutionary direction within the species (Han et al. 2011). Meanwhile, the matK sequences amplified success rate is very low and the psbA-trnH sequences lack a variation site so we have not chosen them for further experiments. However, in the identification of other species, it should utilize three different DNA barcodes. The results indicated that ITS2 sequences proved to fully comply with these requirements as a new technical means and basis.
New methods for identifying traditional Chinese medicines should focus on accuracy, digitization, repeatability, simplicity and practicality (Zhao et al. 2013). The DNA sequence identification technique has important significance comparing with the traditional methods. Our results highlighted the advantages of using the ITS2 sequence as a DNA barcode from three aspects: first, the ITS2 sequence provides a precise and simple method for the identification of medicinal herbs as a complement to traditional identification methods. Second, it provides an important reference value in the exploration of the relationships between different species of rare herbs. Third, DNA sequencing technology can be applied to control the quality of herbal sources. Several studies used DNA barcoding methods to identify Chinese herbal medicine, such as Dendrobium officinale Kimura et Migo (Zhang et al. 2013), Ajuga ciliata Bunge (Han et al. 2008) and Paeoniae Radix Rubra (Sun et al. 2011), have verified the stability and conservation of the ITS2 sequence. The current study was the first to use a DNA barcode molecular identification technology to distinguish A. roxburghii and its adulterants and this study expanded the application of the ITS2 sequence to the medicinal plant field and assured drug safety on clinical use. In conclusion, ITS2 as a DNA barcode would broaden our understanding of plant resource classification and phylogenetics. We could collect and sort out the related data about ITS2 for further experiments and build a DNA library of species classification and evolutionary information. Therefore, this technology still has much room for development and prospects.
Conclusion
In summary, we confirmed that the ITS2 sequence can be used as a standard DNA barcode to distinguish A. roxburghii and its adulterants at the molecular level. Cluster analysis using the ITS2 barcode in A. roxburghii was basically consistent with traditional plant morphology. Also, this study not only broadened the application of the ITS2 sequence in the medicinal field, but also provided an efficient method to solve classification problems in a wide range of plant at the species levels.
Author contribution statement
TW Lv, QS Shao designed research and wrote the paper, HZ Wang performed research, RD Teng and MY Li helped in collection of plant materials and WS Zhang, LL Zhang helped in data analysis. All authors read, reviewed and approved the manuscript.
Abbreviations
- ITS2:
-
Internal transcribed spacer 2
- PCR:
-
Polymerase chain reaction
- TBE:
-
Tris-borate-EDTA
- NJ:
-
Tree neighbor-joining tree
- TLC:
-
Thin layer chromatography
- HMM:
-
Hidden Markov model
References
Chang CJ, Wu SM, Yang PW (2000) High-pressure carbon dioxide and co-solvent extractions of crude oils from plant materials. Innov Food Sci Emerg 1(3):187–191
Chen SL, Yao H, Han JP, Liu C, Song JY, Shi LC, Zhu YJ, Ma XY, Gao T, Pang XH, Luo K, Li Y, Li XW, Jia XC, Lin YL, Leon C (2010) Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS One 5(1):e8613
Cui SC, Yu J, Zhang XH, Cheng MZ, Yang LW, Xu JY (2013) Antihyperglycemic and antioxidant activity of water extract from Anoectochilus roxburghii in experimental diabetes. Exp Toxicol Pathol 65(5):485–488
Du XM, Irino N, Furusho N, Jun HS, Shoyama YH (2008) Pharmacologically active compounds in the Anoectochilus and Goodyera species. J Nat Med-Tokyo 62(2):132–148
Han JP, Shi LC, Chen SL, Qian J, Luo K, Chen KL (2008) Authentication of Ajuga ciliate and its close species using ITS2 sequence. World Sci Technol Mod Tradit Chin Med Mater Med 10(6):86–89
Han JP, Li MN, Luo K, Liu MZ, Chen XC (2011) Chen SL (2011) Identification of Daturae Flos and its adulterants based on DNA barcoding technique. Acta Pharm Sin 46(11):1408–1412
He CN, Wang CL, Guo SX, Xiao PG (2004) Advances on chemical compositions and pharmacological studies of Anoectochilus Blume (Orchidaceae). Chin Pharm J 39(2):81–84
Hebert PDN, Gregory TR (2005) The promise of DNA barcoding for taxonomy. Syst Biol 54(5):852–859
Keller A, Schleicher T, Schultz J, Müller T, Dandekar T, Wolf M (2009) 5.8S-28S rRNA interaction and HMM-based ITS2 annotation. Gene 430(1–2):50–57
Koetschan C, Förster F, Keller A, Schleicher T, Ruderisch B, Schwarz R, Müller T, Wolf M, Schultz J (2010) The ITS2 Database III-sequences and structures for phylogeny. Nucl Acids Res 38(1):275–279
Kress WJ, Wurdack KJ, Zimmer EA, Weigt LA, Janzen DH (2005) Use of DNA barcodes to identify flowering plants. Proc Natl Acad Sci USA 102(23):8369–8374
Liu ZH, Zeng X, Yang D, Ren GM, Chu GY, Yuan ZR, Chen S (2012) Identification of medicinal vines by ITS2 using complementary discrimination methods. J Ethnopharmacol 141(1):242–249
Luo AX, Meng ZX, Chen XM, Guo SX (2012) Seed germination and young seedling propagation of Anoectochilus roxburghii, China. Pharm J 47(15):1199–1203
Miller SE(2007)DNA barcoding and the renaissance of taxonomy. Proc Natl Acad Sci USA 104(12):4775–4776
Ross HA, Murugan S, Li WLS (2008) Testing the reliability of genetic methods of species identification via simulation. Syst Biol 57(2):216–230
Shao QS, Deng YM, Liu HB, Zhang AL, Huang YQ, Xu GZ, Li MY (2014a) Essential oils extraction from Anoectochilus roxburghii using supercritical carbon dioxide and their antioxidant activity. Ind Crop Prod 104:104–112
Shao QS, Wang HZ, Guo HP, Zhou AC, Huang YQ, Sun YL, Li MY (2014b) Effects of shade treatments on photosynthetic characteristics, chloroplast ultrastructure, and physiology of Anoectochilus roxburghii. PLoS One 9(2):e85996
Song JY, Yao H, Li Y, Li XW, Lin YL, Liu C, Han JP, Xie CX, Chen SL (2009) Authentication of the family Polygonaceae in Chinese pharmacopoeia by DNA barcoding technique. J Ethnopharmacol 124(3):434–439
Stoeckle M (2003) Taxonomy, DNA, and the barcode of life. Bioscience 53:796–797
Sun ZY, Song JY, Yao H, Sun C, Chen SL (2011) Molecular identification of Paeoniae Radix Rubra and its adulterants based on ITS2 DNA barcode. World Sci Technol Mod Tradit Chin Med Mater Med 13(2):407–411
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res 22:4673–4680
Tseng CC, Shang HF, Wang LF, Su B, Hsu CC, Kao HY, Chen KT (2006) Antitumor and immunostimulating effects of Anoectochilus formosanus Hayata. Phytomedicine 13(5):366–370
Zhang YH, Cai JY, Ruan HL, Pi HF, Wu JZ (2007) Antihyperglycemic activity of kinsenoside, a high yielding constituent from Anoectochilus roxburghii in streptozotocin diabetic rats. J Ethnopharm 114(2):141–145
Zhang CY, Wang FY, Yan HF, Hao G, Hu CM, Ge XJ (2012) Testing DNA barcoding in closely related groups of Lysimachia L. (Myrsinaceae). Mol Ecol Resour 12(1):98–108
Zhang L, Qiu DS, Cai SK, Deng RY, Luo HM, Liu XJ (2013) The genuine and adulterants identification of Dendrobium officinale by rDNA ITS sequences analysis. J Anhui Agric Sci 41(7):2872–2874
Zhao S, Xin TY, Hou DY, Pang XH, Chen RY, Gao JP (2013) Identification of Codonopsis Radix and its adulterants using the ITS/ITS2 barcodes. World. Sci Technol.-Mod. Tradit Chin Med. Mater Med 15(3):421–428
Zheng C, Huang YZ, Pan X, Ji LF (1997) Pharmacognostical identification of Anoectochilus roxburghii. Chin Herb Med 20(11):552–554
Zheng CF, Pan YT, Cai WY, Yuan XN, Gao F (2013) Identification and determination of flavonoids in Anoectochilus roxburghii (Wall.) Lindl by HPLC-US-MS/MS. Nat Prod Res Dev 25(10):1381–1386
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 81303167), the Zhejiang Provincial Natural Science Foundation (Grant No. LY13H280010, LY14H280008), Zhejiang Province Public Technology Applied Research Projects (Grant No. 2014C32017), Ningbo Agricultural Science and Technology Research Projects (Grant No. 2014C10013) and Science and Technology Innovation Program for Students of Zhejiang (Xinmiao Talent Scheme) (2015R412003). We thank the anonymous reviewers for carefully reviewing our manuscript and making many valuable suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lv, T., Teng, R., Shao, Q. et al. DNA barcodes for the identification of Anoectochilus roxburghii and its adulterants. Planta 242, 1167–1174 (2015). https://doi.org/10.1007/s00425-015-2353-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-015-2353-x