
Abstract
N,N′-bis(2,4-di(acryloyloxyethyl)-[1,3,5]-triazin-2-yl)-hexane-1,6-diamine(BDAETH) and 2,2-dimethyl-1,3-propanediol glycerol-methacrylate phosphate (PGMH) were synthesized from cyanuric chloride and phosphorus oxychloride. BDAETH and PGMH were further blended in different ratios to prepare UV-curable intumescent flame-retardant coatings. The thermal behaviors of their cured films were studied by thermal gravimetric analysis, and the results showed that degradation process of cured blend films could be divided into three degradation regions. The degradation mechanism was further investigated by in situ Fourier transform infrared spectra. The flame-retardant properties and the expansion process of the intumescent flame-retardant coatings were further monitored by limiting oxygen index and a self-made equipment, which indicated the synthetic effect between BDAETH and PGMH.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
UV-curable coatings developed rapidly in recent years and have been widely used in many fields [1–3]; however, monomers and oligomers, commonly used in UV curing coatings such as polyester acrylate, epoxy acrylate, polyurethane and unsaturated polyester, have the same drawback of flammable, which limits their application to some extent [4, 5]. Therefore, how to improve the flame-retardant properties of UV-curable resin becomes more and more important [6, 7].
As thin films, the most effective method is intumescent flame-retardant coating [8–10], which can form expansion char inhibition heat and oxygen during combustion. Thus, intumescent flame-retardant additives are becoming more and more popular in flame-retardant coatings industry [11–13]. Professor Shi prepared an UV-curable intumescent flame-retardant coating based on tri(acryloyloxyethyl) phosphate (TAEP) and methacrylated phenolic melamine (MAPM), in which degradation mechanism was studied by DP-MS and scanning electron microscopy [12, 14, 15]. Professor Hu and coworkers prepared UV-curable intumescent flame-retardant coatings by blending TAEP in certain ratios with star poly(urethane acrylate; SPUA), in which combustion properties were studied by microscale combustion calorimetry (MCC) experiments [16]. Moreover, TAEP was blended with triglycidyl isocyanurate acrylate (TGICA) in different ratios to obtain a series of UV-curable intumescent flame-retardant resins. The flame-retardant properties of the cured films were characterized by limiting oxygen index (LOI), UL 94 and cone calorimeter [17]. Little work has been performed to monitor the expansion process with increasing temperature and further study the expansion mechanism, which are very important for UV-curable intumescent flame-retardant coatings.
In this work, two novel flame-retardant monomers used for UV curing systems N,N′-bis(2,4-di(acryloyloxyethyl)-[1,3,5]-triazin-2-yl)-hexane-1,6-diamine(BDAETH) and 2,2-dimethyl-1,3-propanediol glycerol-methacrylate phosphate (PGMH) were synthesized, which were blended in different ratios to prepare a series of UV-curable intumescent flame-retardant coatings. The thermal degradation mechanisms were studied by thermal gravimetric analysis (TG) and in situ FTIR. A self-made equipment containing heating furnace, temperature controller and a camera, which was connected with computer, was set up and was used to monitor the expansion process of UV-cured intumescent flame-retardant coating.
Experimental
Materials
Phosphorus oxychloride and triethylamine were distilled before use. Cyanuric chloride, 2,2-dimethyl-1,3-propanediol and 1,6-diamino-hexane were used as received. Hydroxypropyl acrylate and glycidyl methacrylate were dried with 4 Å molecular sieve before use. All the chemicals mentioned above were purchased from the Aladdin Co. of Shanghai, China. Toluene, sodium hydroxide and anhydrous sodium sulfate, purchased from the Xi-long chemical Co. of Guangdong, China, were used as received. 2-Hydroxy-2-methyl-1-phenyl-1-propanone (Darocur 1173) as a photoinitiator was purchased from the Ying-li chemical Co. of Tianjin, China.
Synthesis
Synthesis of N,N′-bis(2,4-dichloro-[1,3,5]-triazin-2-yl)-hexane-1,6-diamine (BDCTH)
In a 100-mL three-necked flask equipped with an addition funnel and a mechanical stirrer, cyanuric chloride (36.8 g) and dichloromethane (80 mL) were introduced and stirred at 0 °C for 1 h. 1,6-Diamino-hexane (11.6 g) and sodium hydroxide (8 g) were then added dropwise in 2 h. The reaction mixture was stirred vigorously and cooled to 0–5 °C for 4–5 h. The product was filtered and washed with ice water for 3–4 times. The obtained solid was then dried in desiccator under 20 °C for 48 h to give a white product, named BDCTH, with a 93 % yield.
Synthesis of N,N′-bis(2,4-di(acryloyloxyethyl)-[1,3,5]-triazin-2-yl)-hexane-1,6-diamine(BDAETH)
In a 100-mL three-necked flask equipped with an addition funnel, BDCTH (41.2 g), triethylamine (80 mL), toluene (60 mL) and butylene oxide (15 mL) were introduced and stirred at room temperature for 1 h. Hydroxypropyl acrylate (57.2 g) was then added dropwise in 2 h. The reaction mixture was stirred vigorously at room temperature for 3–4 h and then warmed to 80 °C for 16 h in oxygen atmosphere. The solvent was removed in vacuum, and then the filtrate was dissolved in 100 mL of toluene. Using a 500 mL liquid–liquid extractor, this solution was extracted with brine (3*500 mL) until it became clear. The organic phase was dried over Na2SO4 for 24 h to give a light yellow product, named BDAETH, with a 74.8 % yield.
Synthesis of 2,2-dimethyl-1,3-propanediol phosphoryl chloride (DPPC)
In a 100-mL three-necked flask equipped with an addition funnel, CHCl3 (40 mL) and phosphorus oxychloride (30.6 g) were introduced and 2, 2-dimethyl-1,3-propanediol (20.8 g) was then added dropwisely in 2.5 h. The reaction mixture was stirred vigorously at room temperature for 3–4 h and then warmed to 65 °C for 16 h in nitrogen atmosphere. The solvent was removed in vacuum, to give a white product, named DPPC, with 83.0 % yield.
Synthesis of 2,2-dimethyl-1,3-propanediol phosphoryl alcohol (DPPA)
In a 100-mL three-necked flask equipped with an addition funnel, DPPC (36.8 g) 1,4-dioxane (80 mL), triethylamine (30 mL) and water (3.6 mL) were introduced. The reaction mixture was stirred vigorously at 50 °C for 4 h. The volatile components were removed by evaporation. This solution was washed with brine for three times, the organic phase was dried over Na2SO4, and the solvent was evaporated to dryness to give the desired product DPPA with 78.0 % yield.
Synthesis of 2,2-dimethyl-1,3-propanediol glycerol-methacrylate phosphate(PGMH)
In a 100-mL three-necked flask equipped with an addition funnel, DPPA (33.2 g), 1,4-dioxane (80 mL) and glycidyl methacrylate (26.5 mL) were introduced. The reaction mixture was stirred vigorously at 80 °C for 8 h (until the acid value was reduced to the lowest). The volatile components were removed by evaporation. This solution was washed with water for three times, the organic phase was dried over Na2SO4, and the solvent was evaporated to dryness to give the desired product PGMH as a white solid.
Sample preparation
The mixtures of BDAETH with PGMH in different ratios were stirred at 60 °C for 20 min to get various homogenous blends. TAEP, MAPM and their blends were UV-cured with a UV irradiation equipment (80 W cm−1; Run-wo Co., Shenzhen) in the presence of 2 mass% Darocur 1173. The formulations of blends are listed in Table 1.
Measurements
The FTIR and in situ FTIR spectra were recorded with a Bruker VERTEX70 spectrometer. The in situ FTIR data were measured using the KBr disk method with a heating rate of 10 °C min−1 in the range of RT-550 °C. The 1H-NMR spectra were recorded with a Bruker AVANCE III 400 MHz spectrometer using DMSO as an internal reference and CDCl3 as a solvent. The TG was carried out on a Perkin-Elmer Diamond TG/DTA apparatus using a heating rate of 10 °C min−1 in air. The limiting oxygen index (LOI) values of the cured films were measured using a HC-2 LOI instrument (made in China) on the sheets of 120 × 60 × 3 mm3 according to ASTM D2863-77.
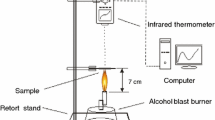
The expansion process of the cured sample was measured by a self-made equipment as shown in Fig. 1, which included heating furnace, temperature controller and a camera, which was connected with computer. The data of expansion degree was figured out by MATLAB, which include four main steps: first to read the videos, then handled it to gray mode, the third step was the most important step that calculation of the area (threshold > 200) and the last step was output data to figure.

Schematic of the self-made equipment for monitoring the expansion process
In the paper, the expansion degree that we define is the ratio between the expansion char area at different temperature (A T) and the original area (A 0) with the increasing temperature of coating. Following the expansion degree, the calculation method is as follows:
Results and discussion
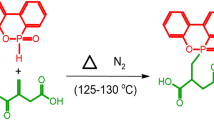
Synthesis of BDAETH and PGMH
Characterization of BDAETH
BDAETH was synthesized via two-step reaction, which was illustrated in Scheme 1. The FTIR spectra of BDCTH and BDAETH are shown in Fig. 2, and the spectra are consistent with the expected structures, through the presence of the relevant peaks, the existence of -NH amine bands at 3267 cm−1, the triazine bands (1406, 1441, 1550, 1616 cm−1) and the C–Cl band at 844 cm−1, which disappeared for BDAETH accompanied the appearance of C=O ester band at 1729 cm−1.

Schematic outline of the synthesis of BDAETH and PGMH

FTIR spectra of the BDCTH and BDAETH
The 1H-NMR spectrum of BDAETH is shown in Fig. 3. The chemical shifts of H with different chemical environment have been marked in the picture, which indicates that the expected structure is successfully synthesized.

1H-NMR spectrum of BDAETH
Characterization of PGMH
PGMH was synthesized via three-step reactions, which was illustrated in Scheme 1. The FTIR spectra of DPPC, DPPA and BDAETH are shown in Fig. 4, and the spectra are consistent with the expected structures, through the presence of the relevant peaks, the existence of P–O–C bands (1053, 1004 cm−1), P–Cl (547 cm−1), P=O (1305 cm−1), –OH (3315, 2230, 1714 cm−1), C=O (1720 cm−1), C=C (1637 cm−1), C–O–C (1177 cm−1). The P–Cl bands at 547 cm−1 and the P–O–H (2230, 1717 cm−1) disappeared in DPPC and DPPH, respectively.

FTIR spectra of the DPPC, DPPH and PGMH
The 1H-NMR spectrum of PGMH is shown in Fig. 5. The chemical shifts of H with different chemical environment have been marked in the picture, which indicates that the expected structure is successfully synthesized.

1H-NMR spectrum of PGMH
Thermal degradation behavior
Figure 6 shows the TG and the mass loss (DTG) curves of the UV-cured samples in air. The TG and DTG data of the films are listed in Table 2.

TG curves of the UV-cured samples
As we can see, cured PGMH and BDAETH films show two characteristic degradation regions, and cured P1N5, P1N2, P1N1, P2N1 and P5N1 films show three characteristic degradation regions. The 5.0 % mass loss temperatures of cured BDAETH, P1N5, P1N2, P1N1, P2N1, P5N1 and PGMH films were 244, 241, 227, 221, 195, 196 and 217 °C, respectively. The worsening of thermal stability with PGMH addition is mainly attributed to the unstable phosphate structure. Obviously, the first degradation temperature regions shift to lower temperature with increasing PGMH content. Meanwhile, the cured PGMH and blends with BDAETH films show more mass loss than cured BDAETH film below 350 °C. This is mainly due to the catalyzation of the degradation products of phosphate structures, in which mechanism will be further studied by in situ FTIR.
From the TG curves of Fig. 6, the cured P1N5, P1N2, P1N1, P2N1 and P5N1 show another degradation stage in the range of 350–450 °C, while degradation temperature range for cured films is shifted to lower temperature with PGMH addition, such as 376–448 °C (P1N5), 362–442 °C (P1N2), 359–438 °C (P1N1), 353–432 °C (P2N1) and 348–412 °C (P5N1). The degradation of PGMH/BDAETH blends would be caused by the degradation of triazine structure and thus the formation of nitrogen volatiles. This will be further studied by the following in situ FTIR measurements.
As we can see, the cured films with PGMH addition are more stable over 400 °C. The second degradation temperature range for cured BDAETH film is 378–401 °C, which is shifted to higher temperature with PGMH addition. The 80.0 % mass loss temperatures show the same trend. Moreover, the char residue over 600 °C increases from 2.5 % (BDAETH) to 19.5 % (P2N1). All these results indicate that the cured films can form stable char with PGMH addition at high temperature, in which mechanism will be further studied by in situ FTIR.
Thermal degradation mechanism
Figure 7 shows the FTIR spectra of residual products after degradation of cured BDAETH film at different temperatures. The peaks around 1500 cm−1 of triazine bands decrease over 400 °C and almost disappear at 500 °C, which indicate that the triazine bands in cured BDAETH film mainly degrade between 400 and 500 °C. The peak at 1713 of O=C–O–C bands decreases over 250 °C and disappears at 300 °C, which indicates that the O=C–O–C bands mainly degrade between 250 and 300 °C.

In situ FTIR spectra for the degradation of UV-cured BDAETH film
Figure 8 shows the FTIR spectra of residual products after degradation of cured PGMH film at different temperatures. The peaks at 1266, 1035 and 780 cm−1 of P–O–C bands decrease over 250 °C and almost disappear at 350 °C with the new peaks at 1155, 1007 and 885 cm−1 attributed to P–O–P functionality. These indicate that phosphate group degrades and forms poly(phosphoric acid)s over 250 °C, which can further catalyze the degradation of ester to form char. As a result, the peak at 1730 cm−1 of ester group disappears completely over 350 °C.

In situ FTIR spectra for the degradation of UV-cured PGMH film
Figure 9 shows the FTIR spectra of residual products after degradation of cured P2N1 films at different temperatures. The peaks at 1093 and 1008 cm−1 of the P–O–C group decrease over 200 °C and disappear at 350 °C with the new peaks at 1085 and 978 cm−1 assigned to the stretching vibration of P–O–C and PO2/PO3 in phosphate–carbon complexes, respectively, which indicated the degradation of P–O–C to form poly(phosphoric acid)s [18]. However, the peaks at 1549 and 1659 cm−1 were attributed to triazine degrade over 300 °C and almost disappear at 400 °C, which were greatly lower than that pure BDAETH. This can be attributed to the catalyzation of poly(phosphoric acid)s. Moreover, new peaks at 910, 1260 and 1400 cm−1 appear at 400 °C were attributed to P–N, P=O and P–O–N, respectively [19]. These three peaks further disappear at 500 °C. These results indicate that the poly(phosphoric acid)s catalyze the triazine structure to form phosphorus–nitrogen–carbon complexes, which further degrades to form char and nonflammable gas. Meanwhile, the nonflammable gas is wrapped by the char and causes the char to expand.

In situ FTIR spectra for the degradation of UV-cured P2N1 film
Flame-retardant properties
LOI was used to investigate the flame-retardant properties of the intumescent systems, and the results are listed in Table 2. The LOI value increases from 27.0 % (PGMH) to 29.0 % (P2N1) and then decreases to 24.5 % (P1N5). More importantly, the formation of expanding during combustion is shown in Fig. 10 and it can be seen that the degree of expansion during combustion is greatly changed by the ratio of PGMH to BDAETH. As mentioned in TG and in situ FTIR results, the obvious synthetic effect between PGMH and BDAETH causes the cured film to form expanding char during combustion and thus improves the flame retardant.

Photographs of the UV-cured samples after and before combustion
Expansion process in situ monitoring
The expanding process with the increasing temperature of the UV-cured instrument flame-retardant coatings was in situ monitored by video camera using a self-made equipment, and the results are shown in Fig. 11. The maximum unidirectional expansion degree, expansion staring temperature and the char residue are listed in Table 3. As we can see, the expansion staring temperature decreases with increasing PGMH addition, which was mainly attributed to the catalyzation of poly(phosphoric acid)s to form stable char as being indicated by TG and in situ FTIR results.

Curves for the expansion process of UV-cured samples with increasing temperature
Moreover, the expansion behaviors are greatly changed from one stage for P1N5 and P1N2 to two stages for P1N1, P2N1 and P5N1. The first stage is the softening process caused by poly(phosphoric acid)s to catalyze the degradation of coating to form phosphorus–nitrogen–carbon complexes. And the second stage is degradation of the formed complexes to form nonflammable gas, which could lead the char to further expand. For P1N5 and P1N2, the PGMH content is too low to form enough poly(phosphoric acid)s to form phosphorus–nitrogen–carbon complexes. Thus, the cured sample P1N5 and P1N1 just had one expanding stage. The maximum unidirectional degree of expansion first increases from 15.0 % (P1N5) to 21.5 % (P1N1) and then decreases to 9.8 % (P5N1). For cured sample P1N2 and P1N5, the PGMH content is too low to catalyze the coating to form enough char for wrapping the nonflammable gas. As listed in Table 3, the char residues for P1N2 and P1N5 are 14.6 and 13.2 %, respectively, which are greatly lower than other samples. On the contrary, cured sample P5N1 has the highest char residue 35.3 %, while the low content of BDAETH cannot form enough nonflammable gas to enforce the char effectively expanding. For cured sample P2N1 and P1N1, the suitable ratio of PGMH to BDAETH balances the char residue and the formation of nonflammable gas. As a result, cured P2N1 and P1N1 have higher expansion degree, 20.3 and 21.5 %, respectively. Moreover, P2N1 has a higher char residue 35.0 %, which indicates lower flammable gas formation during degradation. Thus, P2N1 has the highest LOI value among all samples.
Conclusions
The results indicate that phosphate group in PGMH first degrades to form poly(phosphoric acid)s, which then catalyze the degradation of triazine to form phosphorus–nitrogen–carbon complexes. The complexes further degrade over 400 °C to form nonflammable gas and lead the char to expand. The ratio of BDAETH to PGMH greatly changes the expansion process, and there was a distinct synergistic effect between them. The P2N1 blend shows the best flame-retardant properties with maximum unidirectional degree of expansion of 21.5 %, LOI value of 29.0 % and char residue of 35.0 % among all samples.
References
Jiao CM, Zhang CJ, Dong J, et al. Combustion behavior and thermal pyrolysis kinetics of flame-retardant epoxy composites based on organic–inorganic intumescent flame retardant. J Therm Anal Calorim. 2015;119(3):1759–67.
Wang XF, Wang BB, Hu Y, et al. Flame retardancy and thermal property of novel UV-curable epoxy acrylate coatings modified by melamine-based hyperbranched polyphosphonate acrylate. Prog Org Coat. 2013;77(1):94–100.
Xing WY, Song L, Lv P, et al. Preparation, flame retardancy and thermal behavior of a novel UV-curable coating containing phosphorus and nitrogen. Mater Chem Phys. 2010;123(481):486.
Wang F, Hu JQ, Tu WP. Research progress of UV-curable oligomer acrylate and its coatings. Thermosetting Resin. 2007;22(3):41–6.
Chen XL, Liu L, Zhuo JL, et al. Influence of iron oxide green on smoke suppression properties and combustion behavior of intumescent flame retardant epoxy composites. J Therm Anal Calorim. 2015;119(1):625–33.
Ding J, Shi WF. Thermal degradation and flame retardancy of hexaacrylated/hexaethoxyl cyclophosphazene and their blends with epoxy aerylate. Polym Degrad Stab. 2004;84(1):159–65.
Wang XF, Wang BB, Xing WY, et al. Flame retardancy and thermal property of novel UV-curable epoxy acrylate coatings modified by melamine-based hyperbranched polyphosphonate acrylate. Prog Org Coat. 2014;77(1):94–100.
Wang HL, Xu SP, Shi WF. Photopolymerization behaviors of hyperbranched polyphosphonate acrylate and properties of the UV cured film. Prog Org Coat. 2009;65(4):417–24.
Chen YZ, Peng HQ, Li JH, et al. A novel flame retardant containing phosphorus, nitrogen, and sulfur. J Therm Anal Calorim. 2014;115(2):1639–49.
Agrawal S, Narula AK. Curing and thermal behaviour of a flame retardant cycloaliphatic epoxy resin based on phosphorus containing poly (amide–imide) s. J Therm Anal Calorim. 2014;115(2):1693–703.
Beheshti A, Heris SZ. Experimental investigation and characterization of an efficient nanopowder-based flame retardant coating for atmospheric-metallic substrates. Powder Technol. 2015;269:22–9.
Liang HB, Shi WF, Gong M. Expansion behaviour and thermal degradation of tri(acryloyloxyethyl) phosphate/methacrylated phenolic melamine intumescent flame retardant system. Polym Degrad Stab. 2005;90(1):1–8.
Gao M, Wu WH, Liu S, et al. Thermal degradation and flame retardancy of rigid polyurethane foams containing a novel intumescent flame retardant. J Therm Anal Calorim. 2014;117(3):1419–25.
Huang ZG, Shi WF. Synthesis and properties of a novel hyperbranched polyphosphate acrylate applied to UV curable flame retardant coatings. Eur Polymer J. 2007;43(4):1302–12.
Wang HL, Xu SP, Shi WF, et al. Photopolymerization behaviors of hyperbranched polyphosphonate acrylate and properties of the UV cured film. Prog Org Coat. 2009;65(4):417–24.
Chen XL, Hu Y, Song L, et al. Thermal properties and combustion behaviors of UV-cured phosphate triacrylate/star poly(urethane acrylate) oligomer blends. Polym Adv Technol. 2008;19(5):393–8.
Xing WY, Song L, Hu Y, et al. Combustion and thermal behaviors of the novel UV-cured intumescent flame retardant coatings containing phosphorus and nitrogen. e-Polymers. 2010;10(1):684–94.
Liang HB, Shi WF. Thermal behaviour and degradation mechanism of phosphate di/triacrylate used for UV curable flame-retardant coatings. Polym Degrad Stab. 2004;84(3):525–32.
Zhou S. Preparation and characterization of intumescent flame retardant and silane-crosslinked PP and its EPDM composites. Dissertation for doctor’s degree. Hefei: University of Science and Technology of China; 2009.
Acknowledgements
The authors gratefully acknowledge the financial support of the National Natural Science Foundation of China (51103069 and 51463017), Jiang’xi Educational Committee (GJJ12423) and Foundation of Aeronautics of China (2014ZF56018).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bu, J., Liang, H., Zhang, D. et al. Preparation and expansion properties of a novel UV-curable intumescent flame-retardant coating. J Therm Anal Calorim 122, 329–338 (2015). https://doi.org/10.1007/s10973-015-4733-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-4733-7