
Abstract
Molecular imaging technologies, such as PET imaging, have evolved into powerful tools for tumor diagnosis. Radiolabeled dimeric molecular probes have emerged as a promising strategy in PET imaging due to their outstanding characteristics. IF7, a peptide targeting Anxa1 overexpressed in endothelial cells of various tumor blood vessels, serves as an excellent tumor-targeting agent. This research aims to prepare IF7 dimer and evaluate its imaging performance and biological characteristics. The biological properties of the tracer were evaluated through in vitro experiments using U87 cells. MicroPET imaging and biodistribution were also studied in tumor-bearing mice. MicroPET studies showed a high tumor uptake at 30 and 60 min post-injection with 4.09 ± 0.83%ID/g and 2.66 ± 0.55%ID/g, respectively. Co-injection of excess nonradiolabelled peptide significantly reduced tumor uptake at 30 min (0.33 ± 0.15%ID/g), confirming the tumor-targeting specificity of IF7 dimer. The IF7 dimer tracer demonstrated excellent imaging performance as a tumor-targeting molecular probe, indicating potential clinical applicability and offering new possibilities for future early tumor diagnosis.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Molecular nuclear medicine employs radioactive isotopes as tracers to investigate intricate molecular-level biological processes within the human body. Utilizing nuclear medicine imaging technology, it offers real-time insights into cellular activity, metabolism, and functionality. This capability facilitates accurate disease diagnosis, ongoing treatment monitoring, and the delivery of personalized medical care [1, 2]. Positron emission tomography (PET) stands as an advanced nuclear medical imaging technology, distinguished by its significant advantages. This method furnishes molecular-level insights into biological organisms, offering valuable data on cellular activities and metabolic processes. Such information is pivotal for early disease diagnosis and treatment monitoring. Additionally, PET plays a substantial role in various fields, including oncology, neuroscience, and cardiology, thereby enhancing medical diagnostics' accuracy and personalized treatment strategies [3]. Fluorine-18 (18F) stands out as the optimal positron-emitting radionuclide for positron emission tomography (PET) probes. With a well-suited half-life of 109.8 min that aligns with various in vivo biological ligands, low positron energy (Emean = 0.25 MeV), and convenient accessibility via cyclotrons, 18F enhances spatial resolution in PET/CT imaging. Its widespread use as a PET imaging tracer is attributed to these advantageous characteristics [4, 5].
Annexins constitute a widely distributed class of calcium (Ca2+)-dependent phospholipid-binding proteins. They have specific affinities for phosphatidylserine (PS) and play vital roles in diverse Ca2+-dependent membrane-related processes, such as cell exocytosis, endocytosis, vesicle transport, blood clot regulation, and inflammatory responses. These activities significantly contribute to the pathogenesis and progression of many human diseases [6, 7]. Annexin-A1 (Anxa1), a member of the Annexin family, is detected on the endothelial cell surface of tumor vasculature in various mouse and human tumor types, making it a crucial tumor vascular marker [8]. Anxa1 is a multifunctional protein involved in various biological processes, including tumor angiogenesis, inflammation, neuroendocrine signaling, apoptosis, and membrane transport [9]. These attributes render Anxa1 a suitable target for tumor imaging and therapy.
Research conducted by Shingo Hatakeyama and colleagues identified a unique set of peptides capable of mimicking the binding of heparin to Anxa1 [10]. Among these peptides, IF7 (IFLLWQR) exhibits a specific affinity for the tumor vascular receptor Anxa1. IF7 is a concise 7-amino acid peptide proven valuable for imaging mouse tumors[11]. Moreover, it can bind to anticancer drugs, inhibiting the growth of various tumors in mouse models [10, 12]. In our previous research, we developed 68Ga-NOTA-IF7 and 18F-AlF-NOTA-IF7 molecular probes for imaging mouse tumors [13, 14]. The findings indicated that while these imaging agents did accumulate at the tumor site, they also displayed elevated background signals and lacked precise targeting in the tumor-bearing mice. Additionally, while maintaining the biological activity of IF7, we implemented targeted modifications to the peptide molecule. Employing Gly-Gly-Gly-Arg-Asp-Asn (GGGRDN) as a novel hydrophilic peptide linker, we subsequently synthesized 99mTc-p-SCN-Bn-DTPA-GGGRDN-IF7 and 18F-Al-NODA-Bn-p-SCN-GGGRDN-IF7. Imaging results showed a substantial accumulation of the molecular probes at the tumor site, indicating a favorable target-to-background ratio [15, 16].
Recent studies indicate that dimeric tracers demonstrate superior tumor uptake and prolonged retention when compared to their monomeric counterparts [17,18,19,20,21]. Guided by the concept of dimeric PET probes our objective is to create a dimer by linking two molecules, GGGRDN and IF7, using lysine as a connector. This dimer will maintain the peptide's biological activity, improve in vivo stability, and enable radioisotope labeling.
In this study, we synthesized 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys and employed it as a novel radiotracer for imaging in tumor-bearing mice. We conducted in vitro cell binding studies with U87 cells and performed in vivo evaluations using xenograft U87 tumor-bearing mice.
Materials and methods
General
The custom-synthesized GGGRDN-IF7 dimer (GGGRDN-IF7)2-Lys, obtained from Apeptide Co. Ltd, (Shanghai, China), exhibited a chemical purity exceeding 95%. p-SCN-Bn-NOTA was purchased from Macrocy-clics Co. Ltd, (USA). All commercially available chemicals were of analytical grade and required no further purification. The U87 tumor cell line was sourced from the Cell Bank of the Shanghai Institute for Biological Sciences. Nude mice were sourced from Shanghai SLAC Laboratory Animal Co. Ltd. Radiolabeled compounds were analyzed using RP-HPLC on a Waters Breeze system, equipped with a Radiomatic 610TR flow scintillation analyzer (Perkin-Elmer) and a Waters 2487 dual λ absorbance detector. Analytical HPLC employed a C18 column (Luna 5 μm, 250 × 4.6 mm, Phenomenex) with a flow rate of 1 mL/min. The buffer system consisted of buffer A (0.1% v/v trifluoroacetic acid in H2O) and buffer B (0.1% v/v trifluoroacetic acid in acetonitrile). A gradient from 95% buffer A at 0–2 min to 35% buffer A at 35 min was applied.
Statistical analysis was conducted using SPSS 17.0 software, and the results are presented as mean ± standard deviation (SD). The two-sample t-test was employed for statistical analysis, with p-values less than 0.05 considered statistically significant.
Tumor model establishment
Subcutaneously inoculated 1 × 10^7 cells into the right anterior flank of female Balb/c nude mice (SLAC Experimental Animal Co. Ltd) to establish the U87 tumor model. Utilized U87 Balb/c nude mice (6–8 weeks old, female, 16–20 g), housed in specific pathogen-free (SPF) individually ventilated cages (IVCs), with ad libitum access to food and water. Allowed the tumor on the right side of the mouse to reach a diameter of approximately 0.5–1 cm (approximately 2–3 weeks) before conducting imaging and in vivo biodistribution experiments.
Synthesis and radiolabeling
Radiolabeling of the compound was conducted using p-SCN-NOTA-(GGGRDN-IF7)2-Lys. Specifically, 5.57 mg of (GGGRDN-IF7)2-Lys and 1.00 mg of p-SCN-NOTA (in excess) in a molar ratio of 1:1.3 were dissolved in 1 mL of Dimethylsulfoxide (DMSO) and 30 μL N, N-diisopropylethylamine (DIPEA), sealed under nitrogen and stirred at 37 °C overnight. The reaction mixture underwent purification using preparative HPLC (column: C18, 10 × 250 mm; mobile phase: A, water + 0.1% TFA; B, acetonitrile + 0.1% TFA; gradient: 95% A from 0–2 min to 35% A at 35 min). The resulting product, p-SCN-NOTA-(GGGRDN-IF7)2-Lys, was measured and analyzed by mass spectrometry (MS), [M + 4H]4+ = 929, calc: 3712.17 (C166H248N50O46S).
For radiolabeling, 500 μg of p-SCN-NOTA-(GGGRDN-IF7)2-Lys were dissolved in 20 μL of anhydrous DMSO. Then, 5 μL of 2 mmol/L AlCl3, 10 μL of 0.2 M sodium acetate buffer (pH 4), and 1110 MBq (30 mCi, 30 μL) of 18F or a higher amount, dissolved in four times the total volume of acetonitrile (pH = 4.5), were added. The reaction mixture was heated at 100 °C in an oil bath for 10 min. After cooling, the solution was diluted on a Varian Bond Elut C18 column (using 10 mL of anhydrous ethanol and 10 mL of deionized water). The radioactive activity on the column was measured. The C18 column was washed with 10 mL of PBS followed by 5 mL of distilled water to eliminate impurities. This washing process was repeated until the radioactive activity of the C18 column remained stable after two consecutive washes, with the change being less than 3.7 MBq (100 μCi). Finally, the C18 column was eluted with 0.3 mL of hydrochloric acid ethanol to obtain the product. The radiochemical purity of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys was determined via radio-HPLC (C18 column, 4.6 × 250 mm; mobile phase: A, water + 0.1% TFA; B, acetonitrile + 0.1% TFA; gradient: 95% A from 2 min to 35% A at 35 min).
Partition coefficient and in vitro stability
Diluted 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys in PBS to a concentration of 37 kBq (1 μCi)/mL. Took 0.5 mL of the diluted solution and placed it in each of the three centrifuge tubes. Added 0.5 mL of n-octanol to each tube, mixed thoroughly, and centrifuged at 3000 × rpm for 5 min at room temperature. Took 100 μL from the upper layer (organic phase) and 100 μL from the lower layer (aqueous phase). Measured the radioactivity of each tube using a γ counter (CPM). Finally, the partition coefficient (log P) was calculated as the ratio of the average CPM values for the organic and aqueous phases. In vitro stability testing of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys was conducted in healthy human plasma at 10, 30, 60, and 120 min using radio-HPLC. The stability of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys in PBS and human plasma was assessed by measuring radiochemical purity using an HPLC method at various time intervals. The radiolabeled compound (1 mCi/ml) underwent incubation in 2 ml of the corresponding solution/medium at 37 °C for 120 min. At predefined time points, an aliquot of the PBS solution was directly injected into HPLC, while the serum was passed through a Sep-Pak C18 cartridge, washed with 500 μl PBS buffer, and eluted with 500 μl acetonitrile (ACN) containing 0.1% TFA.
Cell uptake inhibition and competitive binding assays
Cell uptake studies involved U87 cells cultured in 24-well plates (4 × 105 cells per well). U87 cells were incubated with 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys at 370 kBq (10 μCi) per milliliter for 10, 30, 60, and 120 min at 37 °C in a 5% CO2 incubator. In the inhibition group, nonradiolabelled (GGGRDN-IF7)2-Lys (1 μg/well) was added and incubated for 30 min before repeating the previous steps. After incubation, the cells were lysed with 200 μL of 0.1 M NaOH, and the radioactivity was measured using a gamma counter. For the competitive binding assay, U87 cells were incubated in 24-well plates with varying concentrations of non-radioactive precursor 250 μL (0.1 nM to 10 μM) and 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys 250 μL (2 μCi) for 2 h at 37 °C. After incubation, the cells were lysed with 200 μL of 0.1 M NaOH, and the radioactivity was measured. IC50 values were calculated using nonlinear regression.
Biodistribution studies
In a biodistribution study, Female Balb/c nude mice bearing U87 tumors (n = 4) received intravenous injections of 200 μL of 740 kBq (20 μCi) of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys. Biodistribution studies were conducted at 30, 60, and 120 min post-injection. In blocking experiment, tumor-bearing mice were coinjected with excess nonradiolabelled (GGGRDN-IF7)2-Lys. Blood, tumor, and major organs were collected and wet-weighed. The radioactivity in these tissues was measured using a gamma counter (1470 Automatic Gamma Counter, PerkinElmer, USA). The results were presented as the percentage injected dose per gram of tissue (%ID/g). For each mouse, the radioactivity of the tissue samples was calibrated against a known aliquot of the injected activity. The mean uptake (%ID/g) for each group of animals was calculated with standard deviations.
MicroPET imaging
Balb/c nude mice bearing tumors were administered 200 μL of 3.7 MBq (100 μCi) 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys. Additionally, three tumor-bearing mice underwent a blocking experiment, receiving a tail vein injection of excess nonradiolabelled (GGGRDN-IF7)2-Lys. 30 min later, they were injected with 3.7 MBq (100 μCi) of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys. The mice were anesthetized and positioned on an imaging bed for microPET imaging using version 1.4 of the Siemens Preclinical Solutions software. Static microPET scans were conducted at 30, 60, 90, and 120 min post-injection, as well as at 30 min post-blocking (n = 3). Analysis was performed using the Inveon Acquisition Workplace software (version 1.4; Siemens Preclinical Solutions). Regions of interest (ROI) were delineated around the tumors and major organs on the microPET images. The SUVmax for both tumor and muscle was calculated, and the tumor-to-muscle ratio was determined.
Results
Synthesis and radiolabeling
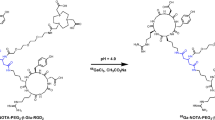
In the presence of DIPEA, p-SCN-NOTA-(GGGRDN-IF7)2-Lys was synthesized by direct coupling (GGGRDN-IF7)2-Lys with activated NOTA, yielding 38%. The p-SCN-NOTA-(GGGRDN-IF7)2-Lys is synthesized at the milligram scale, purified using semi-preparative HPLC to obtain a pure product, and can be further utilized following lyophilization and weighing. The identity of nonradioactive compounds was determined by MALDI-TOF, which observed m/z ions that matched their calculated molecular weights. The synthesis and labeling steps of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys are shown in Fig. 1. The entire labeling process can be accomplished within 20 min, with a decay-corrected yield achieved at 65 ± 3.5% (n = 5). The radiochemical purity of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys was more than 97%. In our preparation of this tracer, 10 nmol of p-SCN-NOTA-(GGGRDN-IF7)2-Lys was used to make 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys. The HPLC chromatogram of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys was presented in Fig. 2. The retention time of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys was 18.5 min.

Synthesis of 18F-Al-p-SCN-NOTA-(GGGRDN-IF7)2-Lys

HPLC radiochromatogram of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys
Partition coefficient and in vitro stability
The oil–water partition coefficient (LogP) for 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys is − 1.86 ± 0.07, indicating a predominant concentration in the aqueous phase and confirming the probe's notable hydrophilicity. Radio-high-performance liquid chromatography (HPLC) results affirm the exceptional stability of the synthesized 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys in vitro. The retention time in HPLC remains consistently unchanged for at least 120 min in both physiological saline and plasma, maintaining a radiochemical purity exceeding 95% (Fig. 3 and Fig. S2).

The HPLC radio chromatogram of radiolabeled 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys compounds incubated in human plasma for 10, 30, 60, and 120 min
Cell uptake inhibition and competitive binding
As illustrated in Fig. 4, the time-dependent increase in cellular uptake values of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys in U87 cells, escalating from 2.19 ± 0.15%AD at 10 min to 3.69 ± 0.12%AD at 120 min (n = 5), which could be blocked by the addition of excess nonradiolabelled (GGGRDN-IF7)2-Lys at 120 min (0.78 ± 0.05 AD%), indicating the specific binding between 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys and U87 cells. The IC50 values of displacement the 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys with (GGGRDN-IF7)2-Lys was 75.1 nM (Fig. 5). This value aligns with other monomeric IF7 molecular probes, indicating that the dimerization of the molecular probe has minimal impact on its affinity.

Tracer uptake in U87 cells at different time points after the addition of.18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys (10 μCi)

Competitive binding of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys in U87 cells. The IC50 of (GGGRDN-IF7)2-Lys was calculated to be 75.1 nM
Biodistribution studies
To assess the in vivo pharmacokinetic characteristics of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys, we conducted a biodistribution study using U87 tumor-bearing mice. As depicted in Fig. 6 and Table 1, the tumor uptake of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys at 30, 60, and 120 min post-injection were 4.97 ± 0.80%ID/g, 2.74 ± 0.11%ID/g, and 1.26 ± 0.12%ID/g, respectively. These values were significantly higher than those observed in normal organs, such as the heart, intestines, muscles, and bones, where uptake remained below 1%ID/g within 120 min post-injection. Hepatic and renal metabolism primarily governed the fate of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys, with the kidney exhibiting the highest uptake at 16.01 ± 0.85%ID/g 30 min post-injection, followed by the liver at 9.78 ± 0.30%ID/g. Co-injection of (GGGRDN-IF7)2-Lys almost entirely blocked tumor uptake at 30 min (0.49 ± 0.18%ID/g).

Biodistribution of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys in U87 tumor bearing mice (n = 4). The uptake of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys in tumors was blocked by co-injection of (GGGRDN-IF7)2-Lys
MicroPET imaging
The representative decay-corrected images at different time points post-injection of the radiotracer are shown in Fig. 7. The U87 tumor is clearly observable, and 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys demonstrates a favorable tumor-to-background ratio. Quantitative analysis of regions of interest (ROI) indicates the following tumor uptake values at different time intervals post-injection: 4.09 ± 0.83%ID/g, 2.66 ± 0.55%ID/g 1.32 ± 0.58%ID/g and 0.55 ± 0.29%ID/g at 30, 60, 90 and 120 min, respectively. The imaging illustrates the primary accumulation of the radiolabeled probe in the liver, kidneys, and the tumor site.

MicroPET imaging of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys in U87 and blocked U87 tumor-bearing mice. Arrows indicate tumors
To evaluate targeting specificity, nonradiolabelled (GGGRDN-IF7)2-Lys was co-injection with 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys. In U87 tumor-bearing mice, there was a significant inhibition of tumor uptake at 30 min, reducing the tumor uptake value to 0.33 ± 0.15% ID/g (Fig. 7).
Discussion
The labeling process for most peptide-based radiotracers is often time-consuming, involves complex multi-step radioactive synthesis, and typically yields low radiochemical yields [22]. Therefore, we selected the Al18F one-step labeling method, offering a straightforward process, reduced processing time, high efficiency, and ease of clinical translation [23, 24]. A study by Carine San et al. in 2023 employed a design of experiments (DoE) approach to determine various relevant chemical reaction conditions for Al18F labeling [25]. These conditions were validated experimentally, including maintaining a pH value of 4.5 and setting the concentration of p-SCN-NOTA-(GGGRDN-IF7)2-Lys at 450 μM with a 1:1 ratio of p-SCN-NOTA-(GGGRDN-IF7)2-Lys to AlCl3. Under these optimized conditions, the radiochemical labeling rate was achieved at 65 ± 3.5% (n = 5).
In microPET imaging, IF7 demonstrates substantial uptake at the tumor site but suffers from poor hydrophilicity, resulting in elevated uptake and slow clearance in organs like the liver, stomach, and intestines [14]. These issues impede imaging effectiveness and diagnostic capabilities. Therefore, we undertook the modification of IF7 to alter its pharmacokinetic properties. The modified peptide, Gly-Arg-Asp-Asn (GGGRDN), serves as a new hydrophilic peptide linker. Compared to IF7, the synthesis of GGGRDN is more straightforward, more accessible, and reduces steric hindrance while enhancing hydrophilicity. This modification significantly improves the in vivo pharmacokinetics of peptides [26].
Previously, we developed 18F-Al-NODA-Bn-p-SCN-GGGRDN-IF7, a PET tracer that demonstrated excellent pharmacokinetic effects and PET imaging results [16]. To further enhance the probe's targeting ability with Anax1 based on multivalency, we connected two GGGRDN-IF7 molecules using lysine (lys). Lysine is a natural amino acid found in living organisms, making it biocompatible in biological systems. This reduces the risk of immune or toxic reactions when lysine is used to link biomolecules. The amino group in the lysine's side chain was used to react with the bifunctional chelator NOTA for labeling. This approach avoids connecting NOTA to the peptide chain of GGGRDN-IF7, preserving its performance.
In vitro, investigations revealed that the compound, when incubated individually in physiological saline and fresh human serum at 37 °C, maintained consistent peak retention time and peak shape on HPLC for up to 120 min. It exhibited remarkable stability, boasting a radiochemical purity (RCP > 97%). The IC50 values for the dimeric molecular probe and the monomer were 75.1 nM and 129.5 nM, respectively [16]. These findings suggest that the dimerized GGGRDN-IF7 molecular probe possesses enhanced receptor binding affinity compared to the monomer.
In Li's research [27], it was observed that the tetrameric RGD peptide labeled with 68Ga exhibited the highest tumor uptake. However, its relatively poor tumor-to-kidney ratio rendered this compound less practical when compared to its dimeric and monomeric counterparts. Moreover, both the dimeric and monomeric peptides displayed similar tumor-to-kidney ratios, but the dimeric peptide demonstrated higher tumor uptake and prolonged tumor retention time.
The 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys was evaluated with microPET imaging and biodistribution studies in the U87 tumor-bearing mice. Results indicated substantial tumor uptake, predominantly cleared through the liver and kidneys. In contrast to the earlier synthesized 18F-AlF-NOTA-IF7, which exhibited notable tumor uptake but elevated radioactive levels in the gastrointestinal tract [14], the novel dimeric molecular probe significantly reduced gastrointestinal radioactive uptake while enhancing tumor uptake. Remarkably, 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys displayed no uptake in the bones, maintaining stable values within 120 min, suggesting excellent in vivo stability with minimal 18F detachment. In contrast to the monomeric GGGRDN-IF7 molecular probe, the dimeric (GGGRDN-IF7)2-Lys showed no significant difference in tumor uptake. However, (GGGRDN-IF7)2-Lys exhibited a slight reduction in uptake in major organs such as the liver and kidneys at 30 min compared to the monomer (10.32 ± 2.72%ID/g vs. 9.78 ± 0.30%ID/g and 17.24 ± 4.56%ID/g vs. 16.01 ± 0.85%ID/g, respectively) [16]. The absolute uptake in tumors and the tumor/non-tumor ratio are vital benchmarks for assessing PET imaging tracers. Typically, the tumor/non-tumor ratio holds greater significance since the primary objective of imaging is lesion visualization. Elevated tumor/non-tumor ratios assist in precisely outlining the position and boundaries of malignant lesions in relation to surrounding tissues. Furthermore, based on imaging and biodistribution data in tumor-bearing mice, the dimeric molecular probe, 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys, appeared to be a superior tracer compared to 18F-AlF-NOTA-IF7 and 18F-Al-NODA-Bn-p-SCN-GGGRDN-IF7 to some extent [14, 16]. Consistent with PET imaging, biodistribution studies showed significant radioactive accumulation in the tumor. At 30, 60, and 120 min post-injection, the tumor uptake rates for 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys in U87 tumor-bearing mice were 4.97 ± 0.80%ID/g, 2.74 ± 0.11%ID/g, and 1.26 ± 0.12%ID/g, respectively. In vivo blocking experiments also demonstrated that co-injection with (GGGRDN-IF7)2-Lys could block over 90% of radioactive uptake in the tumor at 30 min post-injection (4.97 ± 0.80%ID/g vs. 0.49 ± 0.18%ID/g), surpassing the 75% observed in the monomeric 18F-Al-NODA-Bn-p-SCN-GGGRDN-IF7 blocking experiment [16], confirming the specific uptake mediated by Anxa1 of 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys.
Conclusion
In this study, we successfully developed 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys, a radiopharmaceutical probe designed to target Anxa1. This development holds great promise for advancing early cancer diagnostics. The uncomplicated manual radiolabeling process, combined with its high yield and specific activity, underscores the practicality of this probe for clinical applications. In preclinical studies involving U87 tumor-bearing mice, 18F-AlF-p-SCN-NOTA-(GGGRDN-IF7)2-Lys exhibited remarkable imaging efficacy, offering clear tumor visualization. This achievement marks a significant stride towards enhancing cancer diagnosis and management.
References
de Vries EGE, Kist de Ruijter L, Lub-de Hooge MN, Dierckx RA, Elias SG, Oosting SF (2018) Integrating molecular nuclear imaging in clinical research to improve anticancer therapy. Nat Rev Clin Oncol 16:241–255. https://doi.org/10.1038/s41571-018-0123-y
Mankoff DA, Farwell MD, Clark AS, Pryma DA (2017) Making molecular imaging a clinical tool for precision oncology. JAMA Oncol 3:695–701. https://doi.org/10.1001/jamaoncol.2016.5084
Tan H, Gu Y, Yu H, Hu P, Zhang Y, Mao W et al (2020) Total-body PET/CT: Current applications and future perspectives. AJR Am J Roentgenol 215:325–337. https://doi.org/10.2214/ajr.19.22705
Wängler C, Kostikov A, Zhu J, Chin J, Wängler B, Schirrmacher R (2012) Silicon-[18F]fluorine radiochemistry: basics, applications and challenges. Appl Sci 2:277–302. https://doi.org/10.3390/app2020277
Sahnoun S, Conen P, Mottaghy FM (2020) The battle on time, money and precision: Da[18F] id vs. [68Ga]liath. Eur J Nucl Med Mol Imaging 47:2944–2946. https://doi.org/10.1007/s00259-020-04961-1
Parente L, Solito E (2004) Annexin 1: more than an anti-phospholipase protein. Inflamm Res 53:125–132. https://doi.org/10.1007/s00011-003-1235-z
Li L, Xu L, Chen W, Li X, Xia Q, Zheng L et al (2018) Reduced annexin A1 secretion by ABCA1 causes retinal inflammation and ganglion cell apoptosis in a murine glaucoma model. Front Cell Neurosci 12:347. https://doi.org/10.3389/fncel.2018.00347
Oh P, Li Y, Yu J, Durr E, Krasinska KM, Carver LA et al (2004) Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature 6992:629–635. https://doi.org/10.1038/nature02580
Yi M, Schnitzer JE (2009) Impaired tumor growth, metastasis, angiogenesis and wound healing in annexin A1-null mice. Proc Natl Acad Sci U S A 42:17886–17891. https://doi.org/10.1073/pnas.0901324106
Hatakeyama S, Shibata TK, Tobisawa Y, Ohyama C, Sugihara K, Fukuda MN (2013) Tumor targeting by a carbohydrate ligand-mimicking peptide. Methods Mol Biol 1022:369–386. https://doi.org/10.1007/978-1-62703-465-4_28
Nonaka M, Suzuki-Anekoji M, Nakayama J, Mabashi-Asazuma H, Jarvis DL, Yeh J-C et al (2020) Overcoming the blood–brain barrier by Annexin A1-binding peptide to target brain tumours. Br J Cancer 123:1633–1643. https://doi.org/10.1038/s41416-020-01066-2
Chen Q, Liang H, Sun Y, Chen Y, He W, Fang X et al (2019) A carbohydrate mimetic peptide modified size-shrinkable micelle nanocluster for anti-tumor targeting and penetrating drug delivery. Int J Nanomed 14:7339–7352. https://doi.org/10.2147/ijn.S213455
Gu X, Cai G, Zhang R, Jiang M, Zhou Y, Pan D et al (2014) Synthesis and preliminary evaluation of 68Ga-NOTA-IF7 as a tumor imaging agent. J Radioanal Nucl Chem 303:777–782. https://doi.org/10.1007/s10967-014-3459-5
Gu X, Jiang M, Pan D, Cai G, Zhang R, Zhou Y et al (2020) Preliminary evaluation of novel 18F-AlF-NOTA-IF7 as a tumor imaging agent. J Radioanal Nucl Chem 308:851–856. https://doi.org/10.1007/s10967-015-4533-3
Chen F, Pu X, Xiao Y, Shao K, Wang J, Hu W et al (2019) Preparation and SPECT imaging of the novel Anxa 1-targeted probe 99mTc-p-SCN-Bn-DTPA-GGGRDN-IF7. J Radioanal Nucl Chem 320:525–530. https://doi.org/10.1007/s10967-019-06500-1
Chen F, Xiao Y, Shao K, Zhu B, Jiang M (2020) Positron emission tomography imaging of a novel Anxa1-targeted peptide 18F-Al-NODA-Bn-p-SCN-GGGRDN-IF7 in A431 cancer mouse models. J Labelled Comp Radiopharm 63:494–501. https://doi.org/10.1002/jlcr.3865
Li Y, Liu Z, Lozada J, Wong MQ, Lin KS, Yapp D et al (2013) Single step 18F-labeling of dimeric cycloRGD for functional PET imaging of tumors in mice. Nucl Med Biol 40:959–966. https://doi.org/10.1016/j.nucmedbio.2013.08.001
Kaeopookum P, Petrik M, Summer D, Klinger M, Zhai C, Rangger C et al (2019) Comparison of 68Ga-labeled RGD mono- and multimers based on a clickable siderophore-based scaffold. Nucl Med Biol 78–79:1–10. https://doi.org/10.1016/j.nucmedbio.2019.09.002
Li D, Zhao X, Zhang L, Li F, Ji N, Gao Z et al (2014) 68Ga-PRGD2 PET/CT in the evaluation of glioma: a prospective study. Mol Pharm 11:3923–3929. https://doi.org/10.1021/mp5003224
Shao T, Chen Z, Belov V, Wang X, Rwema SH, Kumar V et al (2020) [18F]-Alfatide PET imaging of integrin αvβ3 for the non-invasive quantification of liver fibrosis. J Hepatol 73:161–169. https://doi.org/10.1016/j.jhep.2020.02.018
Zheng K, Liang N, Zhang J, Lang L, Zhang W, Li S et al (2015) 68Ga-NOTA-PRGD2 PET/CT for integrin imaging in patients with lung cancer. J Nucl Med 56:1823–1827. https://doi.org/10.2967/jnumed.115.160648
Liu S, Liu Z, Chen K, Yan Y, Watzlowik P, Wester HJ et al (2010) 18F-labeled galacto and PEGylated RGD dimers for PET imaging of αvβ3 integrin expression. Mol Imaging Biol 12:530–538. https://doi.org/10.1007/s11307-009-0284-2
Laverman P, McBride WJ, Sharkey RM, Eek A, Joosten L, Oyen WJG et al (2010) A novel facile method of labeling octreotide with 18F-fluorine. J Nucl Med 51:454–461. https://doi.org/10.2967/jnumed.109.066902
McBride WJ, Sharkey RM, Karacay H, D’Souza CA, Rossi EA, Laverman P et al (2020) A novel method of 18F radiolabeling for PET. J Nucl Med 50:991–998. https://doi.org/10.2967/jnumed.108.060418
San C, Hosten B, Vignal N, Beddek M, Pillet M, Sarda-Mantel L et al (2023) Optimization study of a NODA derivative radiofluorination by Al18F complexation using a design of experiments approach. Chem-Eur J. https://doi.org/10.1002/chem.202302745
Wan W, Guo N, Pan D, Yu C, Weng Y, Luo S et al (2013) First experience of 18F-alfatide in lung cancer patients using a new lyophilized kit for rapid radiofluorination. J Nucl Med 54:691–698. https://doi.org/10.2967/jnumed.112.113563
Li ZB, Chen K, Chen X (2008) 68Ga-labeled multimeric RGD peptides for microPET imaging of integrin αvβ3 expression. Eur J Nucl Med Mol Imaging 35:1100–1108. https://doi.org/10.1007/s00259-007-0692-y
Acknowledgements
The authors are grateful to the Jiangsu Institute of Nuclear Medicine (NHC Key Laboratory of Nuclear Medicine).
Funding
We are grateful that this work is supported by the Wuxi Municipal Health Commission General Project (M202230) and the Wuxi Medical Centre Project (WMCG202333).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Du, Z., Xue, X., Liao, W. et al. Evaluation of 18F-AlF-labeled IF7 dimer as a promising molecular probe for tumor targeting PET imaging in mice. J Radioanal Nucl Chem 333, 2059–2068 (2024). https://doi.org/10.1007/s10967-024-09391-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-024-09391-z