
Abstract
Purpose
Our goal was to demonstrate that suitably derivatized monomeric RGD peptide-based PET tracers, targeting integrin αvβ3, may offer advantages in image contrast, time for imaging, and low uptake in nontarget tissues.
Methods
Two cyclic RGDfK derivatives, (PEG)2-c(RGDfK) and PEG4-SAA4-c(RGDfK), were constructed and conjugated to NOTA for 64Cu labeling. Their integrin αvβ3-binding properties were determined via a competitive cell binding assay. Mice bearing U87MG tumors were intravenously injected with each of the 64Cu-labeled peptides, and PET scans were acquired during the first 30 min, and 2 and 4 h after injection. Blocking and ex vivo biodistribution studies were carried out to validate the PET data and confirm the specificity of the tracers.
Results
The IC50 values of NOTA-(PEG)2-c(RGDfK) and NOTA-PEG4-SAA4-c(RGDfK) were 444 ± 41 nM and 288 ± 66 nM, respectively. Dynamic PET data of 64Cu-NOTA-(PEG)2-c(RGDfK) and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) showed similar circulation t 1/2 and peak tumor uptake of about 4 %ID/g for both tracers. Due to its marked hydrophilicity, 64Cu-NOTA-PEG4-SAA4-c(RGDfK) provided faster clearance from tumor and normal tissues yet maintained excellent tumor-to-background ratios. Static PET scans at later time-points corroborated the enhanced excretion of the tracer, especially from abdominal organs. Ex vivo biodistribution and receptor blocking studies confirmed the accuracy of the PET data and the integrin αvβ3-specificity of the peptides.
Conclusion
Our two novel RGD-based radiotracers with optimized pharmacokinetic properties allowed fast, high-contrast PET imaging of tumor-associated integrin αvβ3. These tracers may facilitate the imaging of abdominal malignancies, normally precluded by high background uptake.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The expression of several integrins is essential for the induction and sustainment of tumor angiogenesis [1, 2]. In particular, integrin αvβ3 is closely associated with aggressiveness and a poor prognosis in several malignancies including breast cancer, ovarian cancer, melanoma, and glioblastoma [2–5]. Integrin αvβ3 is a membrane protein that strongly binds to extracellular matrix proteins (e.g. fibronectin and vimentin) through its interaction with arginine-glycine-aspartic acid (RGD) tripeptides. Over the last two decades, peptides containing the RGD motif have been widely employed for the imaging and targeted therapy of tumors overexpressing integrin αvβ3 [6–11]. The body of work on tumor targeting using RGD peptides is extensive, the radiolabeling of such vectors for noninvasive PET and SPECT imaging of in vivo integrin αvβ3 expression being predominant.
The success attained using these radiotracers has led to several human trials employing 18F-radiolabeled RGD peptides for PET imaging [12, 13]. However, the clinical implementation of these agents has been halted by several difficulties including their synthesis and unfavorable pharmacokinetic (PK) profiles. In an effort to improve the overall tumor uptake of these tracers, several strategies have been adopted including the cyclization of the peptides – to enhance enzymatic instability – and/or the synthesis of multimeric RGD analogs that show improved tumor accumulation/retention. The latter enhancement of tumor accumulation has been credited to the binding of the multimeric peptide to more than one target, or more plausibly, to a statistical effect given the increased local concentration of RGD moieties. However, such improvement comes at the expense of increasing nontarget uptake of the tracer in significant organs such as the liver, spleen, intestines, and muscle, which negatively impacts image contrast and result in unnecessary radiation exposure.
In this work, we synthesized and evaluated two structurally modified RGD peptides with enhanced PK properties that allow excellent tumor targeting, while keeping off-target uptake at negligible levels. The peptides featured a monomeric cyclic RGDfK motif with a combination of 8-amino-3,6-dioxaoctanoic acid (PEG), 15-amino-4,7,10,13-tetraoxapentadecanoic acid (PEG4), and/or 7-amino-l-glycero-l-galacto-2,6-anhydro-7-deoxyheptanamide (SAA) linkers. Subsequently, the modified peptides (PEG)2-c(RGDfK) and PEG4-SAA4-c(RGDfK) were conjugated to the chelator 1,4,7-triazacyclononane-triacetic acid (NOTA) for radiolabeling with 64Cu. The acquisition of dynamic PET scans allowed us to evaluate and compare the in vivo PK and tumor targeting properties of NOTA-c(RGDfK), NOTA-(PEG)2-c(RGDfK), and NOTA-PEG4-SAA4-c(RGDfK), in athymic nude mice bearing integrin αvβ3-positive human glioblastoma (U87MG) tumors. Finally, competitive cell binding, receptor blocking, and biodistribution studies were also performed to confirm that the integrin αvβ3 binding affinity and specificity of the modified peptides was conserved.
Materials and methods
Reagents
All chemicals employed were of the highest purity available and used without further purification. The catalog peptides c(RGDfK) and (PEG)2-c(RGDfK), along with the custom synthesis product PEG4-SAA4-c(RGDfK), were all supplied by Peptides International, Inc. (Louisville, KY). Chelex 100 resin (50 – 100 mesh) was obtained from Sigma-Aldrich (St. Louis, MO) and 2-(p-isothiocyanatobenzyl)-NOTA (p-SCN-Bn-NOTA) was purchased from Macrocyclics (Dallas, TX). When not indicated otherwise, materials and reagents were obtained from Thermo Fisher Scientific (Fair Lawn, NJ). Water and all buffers were of Milli-Q grade (resistivity >18.2 MΩ · cm) and were treated with Chelex 100 resin to remove heavy metal contaminants.
NOTA conjugation and 64Cu radiolabeling
The conjugation of NOTA was performed using a previously described method with slight modifications [14]. Briefly, in a 1.5-mL Eppendorf vial, 2 mg of each peptide (about 3.3 nmol, 2.2 nmol, and 1.25 nmol of c(RGDfK), (PEG)2-c(RGDfK) and PEG4-SAA4-c(RGDfK), respectively) were dissolved in phosphate-buffered saline (PBS) and the pH adjusted to 9.0 with 0.1 M Na2CO3. Subsequently, a freshly prepared solution of p-SCN-Bn-NOTA in DMSO (about 20 mg/mL) was added to the peptide solution for a peptide/p-SCN-Bn-NOTA ratio of 1:2, and the reaction was carried out for 2 h at room temperature; the concentration of DMSO was kept below 5 % v/v. The conjugated peptides were separated using semipreparative HPLC (Phenomenex Luna C18 column, 5 μm, 10 × 250 mm; flow 5 mL/min; mobile phase 5 − 65 % acetonitrile/water linear gradient in 40 min) and the product lyophilized to yield a white powder. MALDI-TOF-MS was performed to confirm the identity of the purified product.
For 64Cu radiolabeling, 10 μL of a peptide stock solution (1 mg/mL) was reacted with 74 MBq (2 mCi) of 64CuCl2 in 300 μL of NaOAc buffer (0.1 M, pH 4.5) at 37 °C for 15 min under constant shaking. The radiolabeled peptides were then separated by analytical HPLC (Acclaim 120 C18 column, 5 μm, 4.6 × 250 mm; flow 1 mL/min; mobile phase 5 − 65 % ethanol/water linear gradient in 40 min), the radioactive fractions collected, diluted in PBS for a final EtOH concentration of <10 %, and filtered through a 20-μm syringe filter. The radiochemical purity and labeling yields were estimated from the radiochromatograms.
Octanol–water partition coefficient
The hydrophilicity of 64Cu-NOTA-c(RGDfK), 64Cu-NOTA-(PEG)2-c(RGDfK), and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) was evaluated through an octanol–water distribution study. The radiolabeled peptide of interest (15 μCi, about 0.6 MBq) was added to 2 mL of 1:1 n-octanol/water mixture. The mixture was vigorously mixed and allowed to reach equilibrium for 1 h. After reaching equilibrium, the mixture was centrifuged to separate the two phases (5 min, 5,000 rpm). Lastly, the radioactivity in each phase was quantified in an automated γ-counter (Perkin Elmer) and logP values for each compound were determined in triplicate.
Cell lines and animal model
Human glioblastoma U87MG cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), fetal bovine serum (10 %; Sigma-Aldrich, St. Louis, MO) and incubated at 37 °C in an atmosphere containing 5 % CO2. Cells were used for in vitro experiments and tumor induction once about 80 % confluence had been reached. All animal studies were conducted under a protocol approved by the University of Wisconsin Institutional Animal Care and Use Committee. U87MG tumor xenografts were induced in 5-week-old female/male athymic nude mice (Harlan, Indianapolis, IN) by subcutaneous injection of 5 × 106 cells suspended in 100 μl of 1:1 mixture of DMEM and Matrigel (BD Biosciences, Franklin Lakes, NJ), into a lower flank. Tumor size was visually assessed every other day and the animals were used for in vivo imaging experiments when tumors reached 5 – 10 mm in diameter about 3 weeks after cell implantation.
Competitive cell binding assay
Following our previously reported method, the integrin αvβ3 specificity and binding affinity of NOTA-c(RGDfK), NOTA-(PEG)2-c(RGDfK), and NOTA-PEG4-SAA4-c(RGDfK) were evaluated via a competitive binding assay using 125I-echistatin (PerkinElmer, Waltham, MA) as the integrin-specific radioligand [15]. Briefly, 1 × 105 U87MG cells where seeded into 96-well filter plates (EMD Millipore Corporation, Billerica, MA) and incubated with 125I-echistatin (about 10,000 cpm) for 2 h at room temperature in the presence of increasing concentrations of the RGD-based peptides. Subsequently, wells were washed with PBS to remove unbound activity, the plates were blow-dried, and the PVDF filters were removed and counted in an automated γ-counter (PerkinElmer, Waltham, MA). Each data point was replicated three times, plotted in GraphPad Prism (GraphPad Software, San Diego, CA) and one-site binding curves were fitted to determine the 50 % inhibition concentrations (IC50).
Small-animal PET imaging
The PET images were acquired in an Inveon microPET/microCT scanner (Siemens Preclinical Solutions, Knoxville, TN). For in vivo dynamic PET studies, two U87MG tumor-bearing mice per group were anesthetized with isoflurane 2 %, and after catheterization of the tail vein placed in the scanner in a prone position. Simultaneously with the injection of 5.5 MBq (150 μCi) of 64Cu-NOTA-c(RGDfK), 64Cu-NOTA-(PEG)2-c(RGDfK), or 64Cu-NOTA-PEG4-SAA4-c(RGDfK), a 30-min dynamic scan was performed and framed into 26 frames: 5 × 6 s, 7 × 30 s, 6 × 60 s, 6 × 120 s, and 2 × 240 s; the last frame was considered equivalent to a static scan 30 min after injection (p.i.). An additional mouse was added to each group to complete (three mice per group), and longitudinal static scans were recorded at 30 min, 2 h, and 4 h p.i. In a fourth group of four U87MG bearing mice corresponding to a receptor blocking experiment, mice received 5.5 MBq of 64Cu-NOTA-PEG4-SAA4-c(RGDfK) and a coadministered blocking dose (10 mg/kg) of c(RGDyK), then sequential scans were performed at 30 min, 2 h, and 4 h after administration. For every static PET emission scan 20 × 106 coincidence events per mouse were acquired. Image reconstructions were performed on an Inveon Acquisition Workplace (Siemens Preclinical Solutions, Knoxville, TN) workstation using an ordered subsets expectation maximization 3D/maximum a posteriori (OSEM3D/MAP) reconstruction algorithm. Tissue uptake was quantified from a region-of-interest (ROI) analysis of the PET images and expressed as percentage of the injected dose per gram (%ID/g).
Biodistribution studies
To confirm the accuracy of the quantitative PET data and obtain a detailed tissue distribution of the tracers, a biodistribution study was performed. Immediately after the last PET scan at 4 h p.i., mice were killed by CO2 asphyxiation and blood, U87MG tumor, and other major organs collected and weighed. The radioactivity contained in each tissue was measured in an automated γ-counter (Perkin Elmer), and the %ID/g calculated and reported as mean ± SD.
Stability
Serum and metabolic stability of peptides was determined using radio-HPLC. For serum stability, radiolabeled peptides were incubated with reconstituted mouse serum at 37 °C for 1 and 4 h. After incubation, an equal amount of acetonitrile was added to the mixture to precipitate serum proteins. Samples were centrifuged at 5,000 rpm for 5 min and the supernatant was collected, filtered through a 0.2 μm filter, and analyzed by HPLC. For metabolic studies, normal ICR mice were injected with about 300 μCi of tracer and placed under shallow anesthesia. After 1 h urine samples were collected, mixed with acetonitrile 1:1, centrifuged, and the supernatant collected, filtered, and analyzed by HPLC.
Statistical analysis
To ensure the statistical power of the studies, all groups had a minimum of three animals. Quantitative data are presented as means ± SD. Means were compared using the two-sample Student’s t test; a P value <0.05 was considered statistically significant.
Results
Synthesis, radiolabeling, and characterization
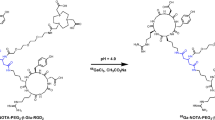
The conjugates NOTA-c(RGDfK), NOTA-(PEG)2-c(RGDfK), and NOTA-PEG4-SAA4-c(RGDfK) were synthesized via standard isothiocyanate chemistry and separated using semipreparative reverse-phase HPLC. Figure 1 shows the structure of the purified peptides whose identity was confirmed by MALDI-TOF mass spectrometry (Online Resource Fig. S1): NOTA-c(RGDfK) ([M+H]+ calc = 1,054.5 vs. [M+H]+ det = 1,054.4), NOTA-(PEG)2-c(RGDfK) ([M+H]+ calc = 1,344.6 vs. [M + H]+ det = 1,344.6), and NOTA-PEG4-SAA4-c(RGDfK) ([M+H]+ calc = 2,057.9 vs. [M+H]+ det = 2,057.8). Radiolabeling of the three peptides with 64Cu was accomplished within 15 min at room temperature, and 64Cu-NOTA-c(RGDfK) (R t = 20.4 min), 64Cu-NOTA-(PEG)2-c(RGDfK) (R t = 20.7 min), and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) (R t = 17.9 min) were purified by radio-HPLC using a biocompatible water/ethanol mobile phase (Online Resource Fig. S1). Excellent yields (>90 %), as determined by radio-HPLC, and specific activities well above 15 MBq/nmol were obtained. For in vivo studies, the purified fractions were diluted with PBS for a final ethanol concentration below 10 %.

Chemical structures of NOTA-c(RGDfK), NOTA-(PEG)2-c(RGDfK), and NOTA-PEG4-SAA4-c(RGDfK)
The hydrophilicity of the three 64Cu-labeled compounds was determined using an octanol–water partition assay. The recorded logP values for 64Cu-NOTA-PEG4-SAA4-c(RGDfK), 64Cu-NOTA-(PEG)2-c(RGDfK), and 64Cu-NOTA-c(RGDfK) were −3.40 ± 0.05, −2.82 ± 0.06, and −2.65 ± 0.01, respectively (n = 3).
Competitive cell binding
We investigated and compared the integrin αvβ3-binding affinities of c(RGDfK), NOTA-c(RGDfK), NOTA-(PEG)2-c(RGDfK), and NOTA-PEG4-SAA4-c(RGDfK) via a competitive binding assay using 125I-echistatin as radioligand in U87MG cells (Fig. 2). Upon incubation with increasing concentrations of the peptides, the U87MG-bound radioactivity decreased indicating displacement of the radioligand (Fig. 2). Similar IC50 values of 254 ± 48, 507 ± 62, 444 ± 41, and 288 ± 66 nM were recorded for c(RGDfK), NOTA-c(RGDfK), NOTA-(PEG)2-c(RGDfK), and NOTA-PEG4-SAA4-c(RGDfK), respectively. These results indicate that neither the structural modification of the peptide nor the conjugation of NOTA had a significant impact on the binding affinities of the peptides.

Concentration-dependent inhibition of 125I-echistatin binding to integrin αvβ3 in U87MG cells by c(RGDfK), NOTA-c(RGDfK), NOTA-(PEG)2-c(RGDfK), and NOTA-PEG4-SAA4-c(RGDfK). Circles c(RGDfK) (IC50 = 254 ± 48 nM); squares NOTA-c(RGDfK) (IC50 = 507 ± 62 nM); triangles NOTA-(PEG)2-c(RGDfK) (IC50 = 444 ± 41 nM); diamonds NOTA-PEG4-SAA4-c(RGDfK) (IC50 = 288 ± 66 nM)
Dynamic PET
Dynamic PET studies were performed to determine and compare the effects of the each structural modification on the early PK properties of the peptides. Mice bearing integrin-positive U87MG tumors were injected with one of the 64Cu-labeled peptides and the temporal in vivo biodistribution of the tracer was recorded and quantified. ROI analysis of the dynamic PET images was performed to determine the time–activity curves of the blood pool, liver, kidneys, muscle, and U87MG tumor (Fig. 3). Given the high correlation between blood tracer concentrations determined by invasive arterial blood sampling and those determined from PET images of the heart left ventricle [16], an image-based approach was used to calculate the circulation PK of the tracers. Analysis of the left ventricular activity curves showed a similar, rapid, clearance of the peptides from the blood circulation. The circulation half-life of each tracer was then calculated by biexponential fitting of the data. The half-lives of 64Cu-NOTA-(PEG)2-c(RGDfK) and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) (4.76 min and 4.07 min, respectively) were longer than the half-life of 64Cu-NOTA-c(RGDfK) (2.56 min).

Dynamic PET-derived time–activity distribution of 64Cu-NOTA-c(RGDfK), 64Cu-NOTA-(PEG)2-c(RGDfK), and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) in the blood pool (a), liver (b), kidneys (c), muscle (d), and tumor (e) in nude mice bearing U87MG xenografts, during the first 30 min after injection of 5.5 MBq of the tracers. f Time progression of the early tumor-to-muscle ratios for each of the radiolabeled peptides
The liver activity curves showed comparable trends for all three peptides, but 64Cu-NOTA-c(RGDfK) showed the highest absolute %ID/g values in this organ. The kidney time–activity curves confirmed renal clearance as the main excretory pathway of the peptides; however, the kidney uptake of 64Cu-NOTA-(PEG)2-c(RGDfK) was markedly different from that of the other peptides. Consistent with its longer circulation half-life, 64Cu-NOTA-(PEG)2-c(RGDfK) showed a slower kidney clearance which resulted in a prolonged higher accumulation of the tracer in this organ.
The U87MG tumor accumulation of the three peptides peaked at comparable values (about 4 %ID/g) approximately 10 min after injection; however, 64Cu-NOTA-PEG4-SAA4-c(RGDfK) exhibited a faster reduction in the magnitude of uptake reflecting its faster clearance/degradation. Similarly, the radioactivity in muscle showed rapid decline, especially for 64Cu-NOTA-PEG4-SAA4-c(RGDfK) which showed the lowest uptake values. Nonetheless, 64Cu-NOTA-PEG4-SAA4-c(RGDfK) showed the highest tumor/muscle ratios (Fig. 3f), reaching values of more than 16 30 min after administration.
Static PET
Longitudinal static PET scans (n = 3), acquired at 0.5 h, 2 h, and 4 h after injection of 64Cu-NOTA-c(RGDfK), 64Cu-NOTA-(PEG)2-c(RGDfK) and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) were performed to evaluate and compare the “long-term” tumor homing and imaging properties of each peptide. PET images in coronal planes intersecting the tumor (Fig. 4a) showed sharp delineation of the tumor contours in all groups as a result of the elevated tumor-to-background ratios attained (Fig. 4b). The results of the ROI quantitative analysis of tracer uptake in the blood pool, liver, kidneys, muscle, and U87MG tumors are summarized in Online Resource Table S1. Compared to the other two peptides, a significantly lower (P < 0.05) tumor accumulation of 64Cu-NOTA-PEG4-SAA4-c(RGDfK) was observed at all time-points (2.50 ± 0.18, 1.77 ± 0.38, 1.67 ± 0.38 %ID/g at 0.5, 2, and 4 h p.i., respectively; n = 3). A similar trend was noted in nontarget tissues such as muscle and blood, in which 64Cu-NOTA-PEG4-SAA4-c(RGDfK) also showed significantly lower uptake resulting in comparable (P > 0.05) contrast ratios. Concurrently with its higher hydrophobicity, the nonspecific accumulation of 64Cu-NOTA-c(RGDfK) in the liver, muscle, and blood was considerably higher throughout the study. Overall, 64Cu-NOTA-PEG4-SAA4-c(RGDfK) exhibited excellent properties for imaging integrin αvβ3 that included faster accumulation in tumor tissue, and lower overall uptake in nontarget organs without compromising the tumor-to-background ratios.

Noninvasive microPET images of tumor-associated integrin αvβ3 in athymic nude mice bearing U87MG tumors. a Representative coronal PET images in planes containing U87MG tumors at 0.5, 2, and 4 h after intravenous injection of 5.5 MBq of 64Cu-NOTA-c(RGDfK), 64Cu-NOTA-(PEG)2-c(RGDfK) and 64Cu-NOTA-PEG4-SAA4-c(RGDfK), and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) coinjected with a blocking dose (10 mg/kg) of c(RGDyK) (arrowheads tumor). b Quantitative analysis of the PET images showing the time-course of accumulation of the tracers in U87MG tumor, blood pool, liver, kidneys, and muscle (n = 3). The graph bottom right shows the tumor-to-muscle ratios attained with each of the radiolabeled peptides
We also demonstrated the specific character of the in vivo integrin αvβ3-binding of 64Cu-NOTA-PEG4-SAA4-c(RGDfK) through a receptor blocking experiment. In this study, mice were treated with a large dose (10 mg/kg) of c(RGDyK)) coinjected with the tracer, and sequential PET scans were acquired. As clearly noticeable in the PET images (Fig. 4a bottom right), coinjection of c(RGDyK) led to a dramatic reduction (P < 0.01) in 64Cu-NOTA-PEG4-SAA4-c(RGDfK) tumor uptake values to 0.42 ± 0.14, 0.24 ± 0.06, 0.18 ± 0.03 %ID/g at 0.5, 2, and 4 h p.i., respectively (n = 4; Fig. 4b and Online Resource Table S1). An overall reduction in uptake in some of the nontarget tissues indicated faster clearance of the tracer that is typical of this type of experiment.
Biodistribution
To validate the accuracy of the PET data and to provide a more comprehensive biodistribution profile of each peptide, we carried out ex vivo biodistribution experiments. Immediately after the last PET scan at 4 h p.i., mice were killed and tracer uptake in the blood pool, U87MG tumors, and other major tissues/organs recorded (Fig. 5 and Online Resource Table S2). The U87MG tumor uptake values were 2.98 ± 0.52 %ID/g for 64Cu-NOTA-c(RGDfK), 2.36 ± 0.31 %ID/g for 64Cu-NOTA-(PEG)2-c(RGDfK), and 1.14 ± 0.26 %ID/g for 64Cu-NOTA-PEG4-SAA4-c(RGDfK), which closely resembled the microPET data. Very low blood pool activities were noted for all three peptides, confirming the rapid clearance of these radiolabeled compounds. Accumulation in nontarget tissues was low, typically under 1 %ID/g, except in the organs involved in the systemic clearance of the peptides (kidney, liver, intestine, spleen). Of note was that background/residual uptake of 64Cu-NOTA-c(RGDfK) was much higher than for the other peptides, which can be explained by its higher hydrophobicity. The tumor blocking effect of the injection of a high dose of c(RGDyK) was shown in U87MG tumors by a significant decrease in 64Cu-NOTA-PEG4-SAA4-c(RGDfK) uptake, but not in the kidneys where the uptake was not affected (0.96 ± 0.12 without blocking vs. 1.78 ± 1.04 %ID/g with blocking; Fig. 5b and Online Resource Table S2). Also upon blocking, a clear decrease in uptake in the remaining organs was observed, an effect that can be attributed to the presence of basal levels of integrin αvβ3 expression in these organs. Nonetheless, the in vivo avidity and specificity of the tracers for integrin αvβ3 was confirmed.

a Ex vivo biodistribution of 64Cu-NOTA-c(RGDfK), 64Cu-NOTA-(PEG)2-c(RGDfK), and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) in U87MG-bearing nude mice 4 h after injection. b Comparison of the biodistribution profile at 4 h of 64Cu-NOTA-PEG4-SAA4-c(RGDfK) with and without coinjection of a blocking dose (10 mg/kg) of c(RGDyK). Data are presented as mean ± SD %ID/g (n = 3)
Stability
Serum and urine stability experiments were performed to determine the in vitro/in vivo degradability of 64Cu-NOTA-(PEG)2-c(RGDfK) and 64Cu-NOTA-PEG4-SAA4-c(RGDfK). Online Resource Fig. S2 shows representative radio-HPLC chromatograms of the tracers after incubation in complete mouse serum or after extraction from mouse urine 1 h after administration. Neither peptide showed significant levels of degradation after 1 and 4 h incubation in serum (>95 % remained intact). Radioactive metabolites of 64Cu-NOTA-(PEG)2-c(RGDfK) and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) were not found in the urine samples, confirming in vivo peptide stability for 1 h.
Discussion
A significant volume of work has shown some of the key factors influencing the PK and pharmacodynamics of RGD-based peptides [17]. Several strategies to improve aspects such as circulation half-life, binding affinity, and enzymatic stability of these probes have been implemented. For example, it is now known that increasing the hydrophilic character of RGD peptides improves their circulation half-life, whereas cyclization reduces sensitivity toward enzymatic degradation [18, 19], and that multimerization significantly increases integrin αvβ3 binding affinity of these antagonists due to a polyvalence effect [20, 21]. A clear tendency towards focusing on increasing the absolute tumor uptake of RGD-based tracers through the synthesis of dimeric, trimeric, tetrameric, and even octameric versions of the cyclic RGD can be identified in the recent literature [22–27]. However, these strategies result in an increased nonspecific uptake of the peptides in nontarget tissues. For example, RGD multimers show significant accumulation in the liver and gut [28], which limits the applicability of these tracers to the detection of tumors in the abdominal cavity. Additionally, the claimed polyvalence effect would require elevated levels of integrin αvβ3 expression within the tumor in order to provide a significant targeting advantage over monomers [29].
In this study, we sought to revisit the imaging of integrin αvβ3 using monomeric c(RGDfK) peptides with enhanced imaging properties. Several studies have demonstrated that alteration in RGD peptides using polyethylene glycol and/or galactose linkers positively influences their PK and pharmacodynamic properties [17, 20, 21, 28–31]. Based on this finding, we developed two peptides (PEG)2-c(RGDfK) and PEG4-SAA4-c(RGDfK) by derivatization of a c(RGDfK) monomer with the hydrophilic linkers PEG and PEG4-SAA4. The two derivatives and the unmodified c(RGDfK) were conjugated to NOTA for 64Cu-labeling and PET imaging. The lower logP (−3.40 ± 0.05) value and shorter retention time of 64Cu-NOTA-PEG4-SAA4-c(RGDfK) (R t = 17.9 min in C18 chromatographic columns) indicates higher hydrophilicity than the other tracers. The integrin αvβ3 binding properties of the NOTA-conjugated peptides were compared to unmodified c(RGDfK) through an in vitro cell binding assay, which revealed binding affinities in the nanomolar range for all compounds. The highest receptor affinity, which was close to that of the c(RGDfK) peptide (254 ± 48 nM), was observed for NOTA-PEG4-SAA4-c(RGDfK) (288 ± 66 nM), presumably due to its higher conformational freedom and improved hydrophilicity.
Both 64Cu-NOTA-(PEG)2-c(RGDfK) and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) showed reduced in vivo accumulation in nontarget tissues including the intestines, liver, kidneys, and muscle compared to the native 64Cu-NOTA-c(RGDfK) peptide. The PK advantage of the modified peptides in terms of background distribution was expected given their higher molecular weight and hydrophilic character, which was shown by their clear tendency to remain within the blood pool compartment (both showed slightly longer blood circulation half-lives). For the same reasons, uptake of the derivatized peptides was lower in U87MG tumors: 3.62 ± 0.21 %ID/g, 2.76 ± 0.04 %ID/g and 4.78 ± 0.74 %ID/g at 30 min p.i. for 64Cu-NOTA-(PEG)2-c(RGDfK), 64Cu-NOTA-PEG4-SAA4-c(RGDfK) and 64Cu-NOTA-c(RGDfK), respectively. However, the observed tumor/muscle, tumor/blood, tumor/liver, and tumor/kidney contrast ratios (Online Resource Tables S3 and S4) were either superior or remained comparable to those of 64Cu-NOTA-c(RGDfK) at 30 min, 2 h, and 4 h after injection. Other 64Cu-labeled monomeric RGD peptides have been reported to show similar tumor-to-normal tissue ratios [32], but with these peptides imaging at delayed time-points was required to attain such high contrast ratios. Altogether, these findings demonstrate the benefits of the addition of the PEG and PEG4-SAA4 linkers for improving the imaging properties of c(RGDfK) peptides.
The ability of 64Cu-NOTA-(PEG)2-c(RGDfK) and 64Cu-NOTA-PEG4-SAA4-c(RGDfK) to target integrin αvβ3 in vivo seemed to contradict the results of the in vitro binding affinity studies. In spite of its higher integrin αvβ3-binding affinity, 64Cu-NOTA-PEG4-SAA4-c(RGDfK) displayed lower in vivo accumulation in U87MG tumor xenografts. Again, it is likely that hydrophilicity played a central role in enhancing the overall excretion of tracer from the mouse body, which resulted in reduced bioavailability of the compound. Nonetheless, 64Cu-NOTA-PEG4-SAA4-c(RGDfK) provided a faster, superior tumor/muscle contrast (16.6 ± 5.6; Fig. 3f) within 30 min of administration. Renal excretion was the main excretory pathway for both compounds, but analysis of kidney time–activity curves (Fig. 3c) showed a lower integral 64Cu-NOTA-PEG4-SAA4-c(RGDfK) kidney uptake. This translates into lower procedural radiation doses and a reduction in the likelihood of developing renal radiotoxicity upon repeated imaging (kidneys are commonly the dose-limiting organ for peptide-based radiopharmaceuticals). Furthermore, the specificity of 64Cu-NOTA-PEG4-SAA4-c(RGDfK) tumor uptake was confirmed in a receptor blocking study in which coinjection of an excess of c(RGDyK) (10 mg/kg) decreased tumor uptake to about 10 % of the %ID/g value without blocking. A partial blocking effect was also seen in several nontarget tissues (liver, lung, spleen, kidney, and intestine), consistent with the basal levels of integrin αvβ3 expression reported for these tissues in both rodents and humans [33–35].
Over the last decade, the study of integrin αvβ3 expression, as a sensitive indicator of tumor angiogenesis, using RGD peptides has shown great success [9, 36]. Without doubt, nuclear diagnostic imaging, which has become an indispensable tool for the exploration of tumor biology in the clinical setting, has been one of the areas where the potential of RGD peptides has been widely exploited. A myriad of RGD-based radiotracers for PET imaging have emerged, several of which have been clinically evaluated [12, 13, 37–39]. However, these studies have shown that significant improvements are still required for successful clinical implementation. An ideal molecular imaging radiotracer should be able to accumulate rapidly and specifically within the target tissue to provide a high target-to-background contrast, while being quickly excreted from the rest of the body, minimizing radiation burden and permitting repeated imaging with few adverse effects [40]. Additionally, it should be easy to synthesize and must feature a radionuclide with properties (decay mode, energy, t 1/2, etc.) that allow its rapid and sensitive detection within a short time after administration. Hence, our results strongly indicate that 64Cu-NOTA-PEG4-SAA4-c(RGDfK) possesses the necessary traits to be considered an excellent radiotracer to provide fast, specific, and high-contrast imaging of integrin αvβ3 expression with potentially minimal adverse effects. Moreover, its fast kinetics facilitate the implementation of analogous radiotracers using isotopes with a short half-life such as 18F and 68Ga.
In conclusion, we present the facile radiosynthesis and evaluation of two novel RGD-based radiotracers with enhanced PK properties for the noninvasive imaging of tumor-associated integrin αvβ3 using PET. The synthesized compounds displayed strong and specific integrin αvβ3 binding, and very high metabolic stability both in vitro and in vivo. Both tracers showed an enhanced overall clearance resulting in lower uptake in background tissues including the kidneys, lungs, liver, intestine, and spleen. These results demonstrate the benefits of the PEG and PEG4-SAA4 derivatization of RDG peptides for high-contrast noninvasive PET imaging of integrin αvβ3 expression, especially in abdominally located malignancies in which high nontarget uptake makes detection difficult.
References
Hwang R, Varner J. The role of integrins in tumor angiogenesis. Hematol Oncol Clin North Am. 2004;18:991–1006.
Robinson SD, Hodivala-Dilke KM. The role of beta3-integrins in tumor angiogenesis: context is everything. Curr Opin Cell Biol. 2011;23:630–7.
Puduvalli VK. Inhibition of angiogenesis as a therapeutic strategy against brain tumors. Cancer Treat Res. 2004;117:307–36.
Sengupta S, Chattopadhyay N, Mitra A, Ray S, Dasgupta S, Chatterjee A. Role of alphavbeta3 integrin receptors in breast tumor. J Exp Clin Cancer Res. 2001;20:585–90.
Felding-Habermann B, Fransvea E, O'Toole TE, Manzuk L, Faha B, Hensler M. Involvement of tumor cell integrin alpha v beta 3 in hematogenous metastasis of human melanoma cells. Clin Exp Metastasis. 2002;19:427–36.
Jin H, Varner J. Integrins: roles in cancer development and as treatment targets. Br J Cancer. 2004;90:561–5.
Kumar CC. Integrin alpha v beta 3 as a therapeutic target for blocking tumor-induced angiogenesis. Curr Drug Targets. 2003;4:123–31.
Kumar CC, Armstrong L, Yin Z, Malkowski M, Maxwell E, Ling H, et al. Targeting integrins alpha v beta 3 and alpha v beta 5 for blocking tumor-induced angiogenesis. Adv Exp Med Biol. 2000;476:169–80.
Cai W, Niu G, Chen X. Imaging of integrins as biomarkers for tumor angiogenesis. Curr Pharm Des. 2008;14:2943–73.
Schottelius M, Laufer B, Kessler H, Wester HJ. Ligands for mapping alphavbeta3-integrin expression in vivo. Acc Chem Res. 2009;42:969–80.
Gaertner FC, Kessler H, Wester HJ, Schwaiger M, Beer AJ. Radiolabelled RGD peptides for imaging and therapy. Eur J Nucl Med Mol Imaging. 2012;39 Suppl 1:S126–38.
Kenny LM, Coombes RC, Oulie I, Contractor KB, Miller M, Spinks TJ, et al. Phase I trial of the positron-emitting Arg-Gly-Asp (RGD) peptide radioligand 18F-AH111585 in breast cancer patients. J Nucl Med. 2008;49:879–86.
Beer AJ, Haubner R, Sarbia M, Goebel M, Luderschmidt S, Grosu AL, et al. Positron emission tomography using [18F]Galacto-RGD identifies the level of integrin alpha(v)beta3 expression in man. Clin Cancer Res. 2006;12:3942–9.
Jeong JM, Hong MK, Chang YS, Lee YS, Kim YJ, Cheon GJ, et al. Preparation of a promising angiogenesis PET imaging agent: 68Ga-labeled c(RGDyK)-isothiocyanatobenzyl-1,4,7-triazacyclononane-1,4,7-triacetic acid and feasibility studies in mice. J Nucl Med. 2008;49:830–6.
Hernandez R, Valdovinos HF, Yang Y, Chakravarty R, Hong H, Barnhart TE, et al. (44)Sc: an attractive isotope for peptide-based PET imaging. Mol Pharm. 2014;11:2954–61.
Kim SJ, Lee JS, Im KC, Kim SY, Park SA, Lee SJ, et al. Kinetic modeling of 3'-deoxy-3'-18F-fluorothymidine for quantitative cell proliferation imaging in subcutaneous tumor models in mice. J Nucl Med. 2008;49:2057–66.
Haubner R, Decristoforo C. Radiolabelled RGD peptides and peptidomimetics for tumour targeting. Front Biosci (Landmark Ed). 2009;14:872–86.
Bogdanowich-Knipp SJ, Chakrabarti S, Williams TD, Dillman RK, Siahaan TJ. Solution stability of linear vs. cyclic RGD peptides. J Pept Res. 1999;53:530–41.
Samanen J, Ali F, Romoff T, Calvo R, Sorenson E, Vasko J, et al. Development of a small RGD peptide fibrinogen receptor antagonist with potent antiaggregatory activity in vitro. J Med Chem. 1991;34:3114–25.
Haubner R, Wester HJ, Burkhart F, Senekowitsch-Schmidtke R, Weber W, Goodman SL, et al. Glycosylated RGD-containing peptides: tracer for tumor targeting and angiogenesis imaging with improved biokinetics. J Nucl Med. 2001;42:326–36.
Haubner R, Wester HJ, Weber WA, Mang C, Ziegler SI, Goodman SL, et al. Noninvasive imaging of alpha(v)beta3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001;61:1781–5.
Li ZB, Cai W, Cao Q, Chen K, Wu Z, He L, et al. (64)Cu-labeled tetrameric and octameric RGD peptides for small-animal PET of tumor alpha(v)beta(3) integrin expression. J Nucl Med. 2007;48:1162–71.
Liu S. Radiolabeled multimeric cyclic RGD peptides as integrin alphavbeta3 targeted radiotracers for tumor imaging. Mol Pharm. 2006;3:472–87.
Auzzas L, Zanardi F, Battistini L, Burreddu P, Carta P, Rassu G, et al. Targeting alphavbeta3 integrin: design and applications of mono- and multifunctional RGD-based peptides and semipeptides. Curr Med Chem. 2010;17:1255–99.
Dijkgraaf I, Yim CB, Franssen GM, Schuit RC, Luurtsema G, Liu S, et al. PET imaging of alphavbeta(3) integrin expression in tumours with 68Ga-labelled mono-, di- and tetrameric RGD peptides. Eur J Nucl Med Mol Imaging. 2011;38:128–37.
Notni J, Pohle K, Wester HJ. Be spoilt for choice with radiolabelled RGD peptides: preclinical evaluation of 68Ga-TRAP(RGD)(3). Nucl Med Biol. 2013;40:33–41.
Knetsch PA, Zhai C, Rangger C, Blatzer M, Haas H, Kaeopookum P, et al. [(68)Ga]FSC-(RGD)3 a trimeric RGD peptide for imaging alphavbeta3 integrin expression based on a novel siderophore derived chelating scaffold-synthesis and evaluation. Nucl Med Biol. 2015;42:115–22.
Shi J, Kim YS, Zhai S, Liu Z, Chen X, Liu S. Improving tumor uptake and pharmacokinetics of (64)Cu-labeled cyclic RGD peptide dimers with Gly(3) and PEG(4) linkers. Bioconjug Chem. 2009;20:750–9.
Ji S, Czerwinski A, Zhou Y, Shao G, Valenzuela F, Sowinski P, et al. (99m)Tc-Galacto-RGD2: a novel 99mTc-labeled cyclic RGD peptide dimer useful for tumor imaging. Mol Pharm. 2013;10:3304–14.
Liu S, Liu Z, Chen K, Yan Y, Watzlowik P, Wester HJ, et al. 18F-labeled galacto and PEGylated RGD dimers for PET imaging of alphavbeta3 integrin expression. Mol Imaging Biol. 2010;12:530–8.
Jung KH, Lee KH, Paik JY, Ko BH, Bae JS, Lee BC, et al. Favorable biokinetic and tumor-targeting properties of 99mTc-labeled glucosamino RGD and effect of paclitaxel therapy. J Nucl Med. 2006;47:2000–7.
Dumont RA, Deininger F, Haubner R, Maecke HR, Weber WA, Fani M. Novel (64)Cu- and (68)Ga-labeled RGD conjugates show improved PET imaging of alpha(nu)beta(3) integrin expression and facile radiosynthesis. J Nucl Med. 2011;52:1276–84.
Zhang X, Xiong Z, Wu Y, Cai W, Tseng JR, Gambhir SS, et al. Quantitative PET imaging of tumor integrin alphavbeta3 expression with 18F-FRGD2. J Nucl Med. 2006;47:113–21.
Wu Z, Li ZB, Chen K, Cai W, He L, Chin FT, et al. microPET of tumor integrin alphavbeta3 expression using 18F-labeled PEGylated tetrameric RGD peptide (18F-FPRGD4). J Nucl Med. 2007;48:1536–44.
Behr TM, Goldenberg DM, Becker W. Reducing the renal uptake of radiolabeled antibody fragments and peptides for diagnosis and therapy: present status, future prospects and limitations. Eur J Nucl Med. 1998;25:201–12.
Cai W, Chen X. Multimodality molecular imaging of tumor angiogenesis. J Nucl Med. 2008;49 Suppl 2:113S–28.
Doss M, Kolb HC, Zhang JJ, Belanger MJ, Stubbs JB, Stabin MG, et al. Biodistribution and radiation dosimetry of the integrin marker 18F-RGD-K5 determined from whole-body PET/CT in monkeys and humans. J Nucl Med. 2012;53:787–95.
Wan W, Guo N, Pan D, Yu C, Weng Y, Luo S, et al. First experience of 18F-alfatide in lung cancer patients using a new lyophilized kit for rapid radiofluorination. J Nucl Med. 2013;54:691–8.
Liu Z, Wang F. Development of RGD-based radiotracers for tumor imaging and therapy: translating from bench to bedside. Curr Mol Med. 2013;13:1487–505.
Torchilin VP, editor. Handbook of targeted delivery of imaging agents. Boca Raton: CRC Press; 1995.
Compliance with ethical standards
Funding
This study was funded by the University of Wisconsin–Madison, the Department of Defense (W81XWH-11-1-0644 & W81XWH-11-1-0648), the National Science Foundation (DGE-1256259), the National Institutes of Health (NIBIB/NCI 1R01CA169365, P30CA014520, T32CA009206, and 5T32GM08349), the US Department of States sponsored Fulbright Scholar Program (1831/FNPDR/2013), and the American Cancer Society (125246-RSG-13-099-01-CCE).
Conflicts of interest
Andrzej Czerwinski and Francisco Valenzuela are employees of Peptides International, Inc. The other authors declare that they have no conflicts of interest.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the University of Wisconsin Institutional Animal Care and Use Committee.
This article does not describe any studies with human participants performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 483 kb)
Rights and permissions
About this article

Cite this article
Hernandez, R., Czerwinski, A., Chakravarty, R. et al. Evaluation of two novel 64Cu-labeled RGD peptide radiotracers for enhanced PET imaging of tumor integrin αvβ3 . Eur J Nucl Med Mol Imaging 42, 1859–1868 (2015). https://doi.org/10.1007/s00259-015-3085-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-015-3085-7