
Abstract
The new category of high-density polyethylene HDPE/WO3 was synthesized via compression molding technique. Various amounts (10, 15, 25, and 35 wt%) of bulk micro-sized WO3 (Sample A) and two selected WO3 nanoparticles 45 nm (Sample B) and 24 nm (Sample C) obtained through the mechanical process were used as filler to HDPE. The prepared composites with filler 15 and 35 wt% were characterized by X-ray diffraction (XRD), Fourier transforms infrared spectroscopy (FTIR), scanning electron microscope, and energy-dispersive X-ray. XRD and FTIR results exhibited that filler does not affect either the crystal or the chemical structure of HDPE, confirming the adsorption of the filler on the surface polymer matrix. The morphology exhibited a more uniform dispersion of WO3 nanoparticles in the HDPE polymer matrix at low filler weight fractions. The mechanical properties of the composites at all filler contents were investigated using Vickers microhardness and tensile stress–strain tests. The variation of Vickers microhardness Hv showed a reverse indentation size effect (RISE) and exhibited a noticeable increase with increasing the WO3 NPs(C) filler loading up to 25 wt%. The tensile test revealed an increase in Young’s modulus, ultimate tensile strength, and yield stress by the addition of either WO3 nanoparticles (B or C) to 25 wt%. Otherwise, any further increase in nano wt% filler induced a decrease in the elongation at break. This improvement, deduced in the mechanical properties of the nanocomposites, suggests promising applications in radiation shielding and industry such as coating, barriers, catalysis, photo corrosion, and photothermal conversion.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The advanced technology in materials science and engineering prompted researchers to generate new composite materials with enhanced functional properties and high-performance structures for various industrial applications [1,2,3]. Polymer nanocomposites are considered the most valuable and interesting materials in terms of their functionality, durability, and easy processing [4]. However, the high performance of the synthesis of the nanocomposites depends on many aspects including the selection of nanofiller and polymer matrix, the filler loading, dispersion, and adhesion to polymer matrix [5, 6]. Furthermore, the manufacturing techniques and conditions are essential in improving the properties of synthesized nanocomposites [7].
High-density polyethylene (HDPE) [8] is a thermoplastic polymer produced in abundance due to its low cost, unique characteristics, durability, sustainability, and easy processing. However, HDPE has restricted mechanical characteristics such as stiffness, yield stress, and hardness. The reinforcement of a specific amount of inorganic filler into the polymer matrix significantly enhances the HDPE mechanical properties and functionality [9]. Among these fillers, tungsten oxide WO3 was chosen due to its proficient characteristics [10, 11]. WO3, which consists of perovskite units, is a unique multi-functional material that attracted considerable attention including many fields of research such as thermo/pyroelectricity, water evaporation, near-infrared rays (NIR) shielding, water splitting [12, 13], and gas sensing [14]. WO3 has many unique characteristics as being a photocatalyst that has an indirect bandgap varying from 2.5 to 3.0 eV, with the functional group (−W6 + =O), large surface area, and active sites [15]. Also, WO3, possessing resistance to photo corrosion and high physicochemical characteristics, can be easily prepared and processed with high stability and electron storage capability [16].
Many studies focused on the impact of adding metal oxides, especially WO3 nanoparticles (NPs) in enhancing the properties of the synthesized composites. Xavier et al. [17] investigated the effect of adding surface-modified WO3 NPs to the epoxy coatings by scanning electrochemical microscopy (SECM) and electrochemical impedance spectroscopy methods. Epoxy coatings allow water molecules to penetrate the coating and cause the degradation of coatings. The incorporation of (3-glycidyloxypropyl) trimethoxysilane (GPTMS)-treated WO3 nanoparticles decreased the corrosion rate nanoparticle in the epoxy-coated sample as the WO3 nanoparticles protected the metal surface. Henaish et al. [18] studied the effect of adding (0, 0.05, 0.1, 0.2, and 0.3 wt%) of WO3 nanoparticles prepared by the sol–gel method on the physical properties of Poly(vinyl chloride) (PVC) polymer via the solution casting method. They found that by increasing the amount of WO3 NPs, both the optical and electrical conductivities increased. Also, as frequency increased, the values of dielectric permittivity (ε′) decreased. However, high porosity in the polymer films appeared with some agglomerations at high concentrations of WO3 content. Toyen et al. [19] studied the mechanical properties of natural rubber (NR) system with various amounts of oxides, including iron (II, III) oxide (Fe3O4), tungsten (III) oxide (W2O3), or bismuth (III) oxide (Bi2O3). The results showed that tensile modulus at 100% elongation, and hardness values were increased by increasing the content of oxides. On the other hand, the tensile strength and the elongation-at-break increased initially and then dropped when the contents of metal oxide increased. The metal oxides act as co-activators and increase the cross-linking density while at very large content of metal oxide could obstruct the action of activators and accelerators leading to weaker particle dispersion and cause agglomeration of the metal oxide particles. Rahmani et al. [20] investigated the effect of double-action compacted at different pressures including (300, 500, 700 MPa) on the hardness of Mg-based nanocomposite blended with various vol.% (0, 1.5, 3, 5%) of WO3 nanoparticles. The results showed that the relative density decreases and the voids and porosities increase in the samples with an increasing percentage of nanoparticles. The sample containing Mg-5 vol.% WO3 NPs showed the highest hardness value. Whereas, the highest compressive strength was attributed to the sample Mg-1.5 vol.% WO3 nanoparticles. Mahdi et al. [21] reinforced Al7075—5 vol % graphite self-lubricating composite which is frequently used in engineering applications by 0, 1.5, 2.5, 3.5, and 4.5 vol % WO3 NPs using powder metallurgy route to study the mechanical characteristics. The obtained results revealed that by increasing the WO3 NPs content until 3.5 vol%, the microhardness and diametral compressive strength were enhanced. The deterioration of the mechanical properties after the 3.5 vol % content of WO3 nanoparticles is due to the agglomeration and clustering of the nanoparticles and to the increase in the porosity formation as increasing the reinforcement content. Besozzi et al. [22] showed that compact a-WO3−x films possessed the best mechanical properties (moderate stiffness and compressive residual stresses) compared to other tungsten oxide coatings. Also, the mechanical properties turn out to be strictly correlated to the morphology and the O/W ratio: as the films become less dense and rich in oxygen, the stiffness and residual stresses linearly decrease.
Few studies were conducted on the synthesis of HDPE composite by adding different filler sizes of WO3 at various weight fractions. HDPE has excellent resistance to chemicals and oxidation and it is insoluble in common solvents which can be easily mixed with different amounts of WO3 filler through molding technique at a specific temperature. A large amount of WO3 filler has been used to study the attenuation ability of the synthesized composites in the X-ray diagnostic energy range to investigate their effectiveness as radiation shields among various filler loadings and sizes [23]. This work aims to investigate the effect of 10, 15, 25, and 35 wt% of different particle sizes of WO3 (samples A, B, and C) as filler on the structural and mechanical properties of HDPE. In this regard, the new category of HDPE/WO3 polymer composite of different filler sizes and weight fractions provides useful information for developing the composite properties to effectively withstand harsh environments for further applications in many fields such as research and industry including barriers, radiation shields, coating, catalysis, and photothermal conversion [17, 18, 24, 25].
2 Experimental Work
2.1 Materials
Commercial HDPE (Egyptene HD5403EA grade) supplied by SIDPEC (Sidi-Kerir Petrochemicals Company, Egypt), with density 0.955 g/cm3 and a melt flow index (MFI) of about 0.35 g/10 min was used as a polymer matrix. Tungsten (VI) oxide (WO3) in powder form with (purity ≥ 99%, particle size ≤ 25 µm, MW = 231.84 g/mol, and density = 7.16 g/cm3) was purchased from Sigma Aldrich (USA) and used without further purification.
2.2 WO3 Nanoparticles Preparation
The mechanical method of a high-energy planetary ball mill (Retsch, PM 100, Germany) in the dry state was used for the preparation of WO3 NPs. The ball milling process was performed at room temperature in a 250-ml zirconium oxide grinding jar under the following conditions: the ratio of ball to powder weight 10:1, rotation speed about 400 rounds per minute (rpm) and break time = 1 min. Four different milling times were studied where a specific amount of the bulk micro-sized WO3 was milled for 15 min and 30 min separately with interval function = 1 min and the same approach was applied for 60 min and 120 min with interval function = 5 min. Based on the resultant size of NPs achieved, the two milling times at 30 min and 120 min were chosen to give remarkable nano sizes for this study and identified as WO3 NPs(B) for the size range 30–65 nm (average size = 45 nm size) and WO3 NPs(C) for the size range 5–35 nm (average size = 24 nm size).
2.3 Composite Synthesis
Composites with a filler weight fraction (10%, 15%, 25%, and 35%) of WO3 Bulk(A), WO3 NPs(B), and WO3 NPs (C) were prepared using the compression molding technique. Pellets of HDPE were accurately weighed and poured in a two-roll mill mixer (XK400, Shandong, China) heated at about 170 °C for 15 min and at a speed of 40 rpm. Specific amounts of either WO3 Bulk(A) or WO3 NPs (B or C) were added to the mixing chamber after a complete melting of the polymer matrix with continued mixing for 10 min to prevent their agglomeration. The prepared samples were shaken for 10 min to promote a complete and homogeneous mixing. The samples were gathered, grinded, and molded in a rectangular stainless steel mold of (25 × 25 × 0.3 cm3), then layered between Teflon to get a smooth surface. After that, the hot press was applied at 10 MPa and 170 °C for 10 min. Progressively, the pressure was increased to 20 MPa for another 10 min. The shaped composite sample was water-cooled gradually to ambient temperature at the rate (20 °C/min). The pure HDPE sample was prepared by a similar method without any addition of WO3.
3 Characterization Techniques
3.1 XRD Analysis
The X-ray diffraction patterns of the synthesized composites were obtained by XRD (Bruker, D8 advance, USA) using Cu-kα radiation source (λ = 0.154 nm) within a range of 10° ≤ 2θ ≤ 70° and energy resolution < 380 eV at 8 keV. The running conditions for the X-Ray tube were 40 kV and 40 mA, recorded with increment step = 0.02° and exposure time = 1 s. X-rays were directed at the sample where a proportion of them was diffracted to produce a pattern. The reference for fitting the peaks was taken from the International Centre for Diffraction Data (ICDD) PDF-2/2013 database.
3.2 FTIR Analysis
FTIR analysis was done using a Bruker, Vertex 70, USA to identify the functional groups of the synthesized composites. The beam from an IR source passes through a monochromatic controller, ensuring that only specified wavelengths are emitted, with a varying range (4000–500 /cm). Specimens disc-shaped of the diameter of 1 cm from the synthesized composites are placed in a holder in the path of the IR source. The data acquisition of the fully digital FT-IR spectrometer is based on parallel running dual-channel delta-sigma ADC´s with a 24-bit dynamic range that is integrated into each detector. This advanced DigiTect™ technology prevents external signals disturbance and guarantees a PEAK signal-to-noise ratio.
3.3 SEM and EDX Analysis
Scanning electron microscope and energy dispersive X-ray analysis of the synthesized composites were observed using (JEOL, JCM-6000PLUS, Japan) with an optimal spatial resolution limited to about 0.5 nm and operated under low vacuum at 15 kV PC-high and magnification order of 1500. The SEM samples were fixed by double-coated carbon tape which was also used in the dissipation of the electron beam charge and heat buildup. All samples were sputtered with a fine layer of Pt/Pd to prevent the charging of the specimen using an ion sputtering coating device (Quorum, Q150T ES).
4 Mechanical Properties
The microhardness measurements were applied on various specimens of the synthesized composites at room temperature using a Vickers hardness tester (SIOMM, MHVD-1000IS, China) characterized by a diamond pyramid indenter. The applied forces ranged from 0.1 N to 3 N at a load time of 30 s. For each load, three trials of indentation were carried out at different locations on the sample surface where the average values were taken. Vickers microhardness was calculated using:
where F is the load (N), and d is the diagonal indentation (mm).
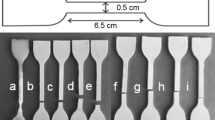
The mechanical tensile tests of the synthesized composite samples were studied using Tension/Compression Force Tester with MESURgaugePlus software for data acquisition (Mark-10, ESM303, USA) based on the international standard ASTM: D638 (Type I) [26] at a crosshead speed of 14 mm/min. The examined samples were carved automatically into the dumbbell shape of the hollow die punch, having dimensions of 13 mm width, 3.5 mm thickness, 160 mm overall length, and 50 mm gauge length as shown in Fig. 1. Using the same conditions and to guarantee the reproducibility of the results, three samples of the same wt% were tested, and average values with deviations were illustrated.

Dumbbell shape of tensile tests specimens with W Width of narrow section, LO Length overall, and G Gauge length
5 Results and Discussion
5.1 Characterization of HDPE/WO3 Composites
5.1.1 XRD
The XRD patterns of HDPE and HDPE incorporated with 15 wt% and 35 wt% WO3 Bulk(A), WO3 NPs(B), and WO3 NPs(C) are illustrated in Fig. 2a, b. HDPE exhibited one strong reflection peak at 21.54˚, followed by a less intensive peak at 23.90˚ related to (110) and (200) lattice planes, respectively. These peaks are compatible with the ICDD (PDF file 00–060-0986) which indicates that HDPE exhibits an orthorhombic structure. The XRD patterns of the synthesized composites in Fig. 2a, b show the blends of the characteristic peaks of HDPE and different particle sizes of WO3. The main diffraction peaks of WO3 at 2θ values of 23.118°, 23.583°, 24.365°, 26.592°, 28.979°, 33.262°, 34.166°, 41.886°, 47.251°, 48.247°, 49.929°, 50.728°, 53.469°, 54.770° and 55.929°, correspond to planes (002), (020), (200), (120), (121), (022), (220), (222), (004), (040), (400), (114), (024), (240), and (420). They displayed nearly the same position with no tangible shift and were found to be highly matched with the ICDD (PDF file 01–083-0951) which indicated the monoclinic structure of WO3.

XRD patterns of a HDPE and 15 wt% and 35wt% of WO3 Bulk(A) composite, and b 15 wt% and 35wt% of WO3 NPs(B) and WO3 NPs(C) composites
The XRD patterns of HDPE/WO3 bulk(A) composites showed no alteration in the position peaks, confirming that there is no effect on either the chemical and crystal structure of HDPE [27, 28]. Whereas, the intensity was changed depending on the weight fraction of WO3 bulk(A) in the HDPE composite. This behavior is illustrated in Fig. 2a, where the corresponding intensity of the HDPE characteristic peak (2θ = 21.542°) was declined when the wt% increases. While the three characteristic peaks of WO3 Bulk (2θ = 23.118°, 23.583°, and 24.365°) appeared more clearly as the wt% increases indicating fine dispersion of the filler inside the polymer [29]. Similar observations to the characteristic peaks of HDPE/WO3 bulk(A) composites are shown in Fig. 2b for both HDPE/WO3 NPs(B) and (C) composites, where the characteristic peak of HDPE (2θ = 21.542°) was declined with increasing the filler wt%, while the three characteristic peaks of WO3 NPs(B) and (C) (2θ = 23.118°, 23.583°, and 24.365°) appeared more clearly as the wt% increases. Moreover, the XRD patterns of HDPE composites exhibited a clear difference which appeared as a broadening and weak intensity for all peaks as decreasing the filler particle size from Bulk(A) to NPs(C) at the same wt%, indicating a diminish in the degree of crystallinity of the composites, which is consistent with the decrease of WO3 particle size [30, 31].
As shown in the results, the X-ray diffraction pattern is distinct for each different phase of the composites. This confirms that the metal oxide was physically adsorbed on the surface polymer matrix without any chemical interaction. In addition, the main characteristic peaks of the composite don’t overlap, leading to a very accurate identification of the crystalline structure of the composites. Similar phenomena was reported by Mahmoud et al. [32] for high-density polyethylene HDPE filled with 10% and 50% weight fraction of lead oxide nanoparticles and bulk lead oxide. They observed that no alternation occurred in the position of the blends of the peaks of HD-PE and PbO at all, while, the width and intensity of the peaks were different depending on the weight fraction of the sample. The relative intensity of the characteristic peaks of HDPE was attenuated with increasing the particle loading and became even weaker at 50 wt% of PbO filler. Also, they found that the incorporation of PbO does not affect either the main chemical structure or the crystal structure of HD-PE.
5.1.2 FTIR
For better perceiving of the functional properties of the synthesized composites, the FTIR spectra of HDPE, 15 and 35 wt% of HDPE/WO3 Bulk(A) composite, HDPE/WO3 NPs (B) composite, and HDPE/WO3 NPs (C) composite were analyzed and illustrated in Fig. 3a, b. In Table 1, all the peaks are presented showing the main characteristic peaks of HDPE 2915 and 2848 cm−1, corresponding to C–H stretching vibrations. The other peaks 1463 and 729–717 /cm were assigned to the methylene CH2 group in-plane rocking band. The slight peak which appeared at 1121 was attributed to skeletal vibration. The spectrum showed a large band at 3367 /cm which was related to the lattice water having H–O-H stretching vibration. Moreover, as a result of its commercial preparation, the chemical structure of polyethylene may contain double bonds, assigned to C = C stretching band at 1650–1583 /cm and C=O stretching band at 1710 /cm [33,34,35]. As illustrated in Fig. 3a, the functional groups corresponding to (O–W–O) stretching vibrations, exhibited at 833–820 and 773–768 /cm [34, 36], appears very weak in the FTIR spectra of HDPE/WO3 Bulk(A) composite and seems undetectable which could be explained by the absence of the Bulk tungsten oxide from the analyzed spots. This is consistent with the SEM results of HDPE/WO3 Bulk(A) composites. Whereas, the same peak near 800 /cm was more evident in the FTIR spectra of HDPE/WO3 NPs(B) and (C) composites, and the intensity increased while increasing the filler weight fraction to 35 wt% as shown in Fig. 3b. However, the characteristic peaks of HDPE in all the samples, bulk and/or NPs, didn’t show any shifting or alterations regardless of the particle size and the filling weight, implying thus, physical adsorption of the metal oxide on the surface of the polymer matrix without any chemical interaction.

FTIR spectra of a HDPE and 15 wt% and 35wt% of WO3 Bulk(A) composite, and b 15 wt% and 35wt% of WO3 NPs(B) and WO3 NPs(C) composites
5.1.3 SEM
The SEM images of pure HDPE, 15 wt%, and 35 wt% HDPE/WO3 Bulk(A), HDPE/WO3 NPs(B), and HDPE/WO3 NPs(C) composites are illustrated in Fig. 4a–d. HDPE shows a compact surface with a continuous phase of the matrix in the presence of lines that may belong to the preparation process [27]. The SEM results were based on atomic number contrast to differentiate between low and high atomic number elements for which the fillers WO3 appear brighter (higher atomic number) than the HDPE polymeric matrix (low atomic number). Based on the back-scattered images of HDPE/WO3 Bulk(A), HDPE/WO3 NPs(B), and HDPE/WO3 NPs (C) composites shown in Fig. 4b–d, respectively, the distribution of WO3 NPs appears clearly on the top surface of the samples as compared to bulk WO3 particles. Also, the obtained results confirm the fact that WO3 NPs are more dispersive within the polymer matrix especially for low filler loading, and show good interfacial adhesion due to their high surface-to-volume ratio compared to bulk WO3 [37]. On the other hand, several aggregates or chunks are arbitrarily dispersed on the surface morphology of the composites with 35 wt% which in turn create voids that affect the properties of the composites [38]. Besides, some cracks appeared in the high wt% composites which may be attributed to the excess heat liberated during the process of water cooling of the samples under the hot press. The insets of Fig. 4b–d display the SEM elemental mapping of W in the composites loaded with 35 wt% WO3. The agglomerated W particles are revealed in contrast colors according to their tendency to aggregate so that the white color indicates the bigger size of agglomerates. This elemental mapping demonstrated the growth of the agglomerated W NPs by increasing its wt% in the polymer matrix.

SEM images of a HDPE, and 15 wt% and 35 wt% of b WO3 Bulk(A) c WO3 NPs(B) and d WO3 NPs(C) composites with the insets of the elemental mapping of W in the composites filled with 35 wt% of WO3 Bulk(A), WO3 NPs(B) and WO3 NPs(C)
5.1.4 EDX
EDX analysis revealed by Fig. 5a–g was performed for HDPE, and its composites filled with 15 wt%, and 35 wt% WO3 bulk(A), WO3 NPs (B), and NPs(C) to detect the chemical elements and homogeneity of the surface polymer composites. Figure 5a shows the EDX spectra of HDPE without surface coating and reveals that it is mainly composed of C elements. Furthermore, the EDX spectra of the HDPE composites show the existence of W elements from the filler together with peaks confirming the presence of carbon from the HDPE matrix in addition to Pt and Pd which were used as sputter coating for the specimen during the process of spectroscopy, as illustrated by Fig. 5b–g. The presence of oxygen peak could mainly be attributed to the ionization caused by the electron beam path generated to interact with the specimen since a low vacuum is used as a condition for the EDX spectroscopy. The very weak peak of Al presented in the spectrum could be attributed to the coating process and the specimen pedestal used during the process of spectroscopy. The intensity of the W peak at 1.774 keV is appeared to be increased to the lowest particle size indicating that WO3 NPs(C) occupy the largest surface of the quantitative analysis due to the large surface-to-volume ratio of the nanoparticles. Table 2 presents the EDX results of the Mass% distribution of tungsten and carbon in all investigated composite samples.

EDX spectra of a HDPE, and HDPE incorporated with b 15 wt% WO3 Bulk(A) c 35 wt% WO3 Bulk(A) d 15 wt% WO3 NPs(B) e 35 wt% WO3 NPs(B) f 15 wt% WO3 NPs(C) g 35 wt% WO3 NPs(C)
5.2 Mechanical Properties
5.2.1 Vickers Microhardness
The values of Vickers microhardness Hv for HDPE, HDPE/WO3 NPs(B), and HDPE/WO3 NPs(C) composites are determined by changing the applied load varying between 0.10 and 3 N as illustrated in Fig. 6a, b. For both composites, Hv showed a non-linear increase for F ≤ 1 N followed by nearly a constant plateau saturation region F > 1 N, indicating the best fitting to reverse indentation size effect (RISE) [39] which attributes the indentation-induced specimen cracking to the applied load. To better understand the effect of wt% on the variation of Hv, a plot of Hv values versus wt% for HDPE and nanocomposites WO3 NPs(B) and WO3 NPs(C) is prevailed by Fig. 7.

Hv versus F(N) for a HDPE and HDPE/WO3 NPs (B) composites, and b HDPE and HDPE/WO3 NPs(C) composites

Hv values as function of wt% for HDPE, WO3 NPs(B) and NPs(C) composites
The results indicate that an increase in the WO3 NPs wt% increased the Hv values. However, a reduction in Hv values is apparent starting from 25 wt% filler loading. On the other hand, it is observed that there is an enhancement in Hv values for HDPE/WO3 NPs(C) with filler up to 25 wt% compared to HDPE/WO3 NPs(B) and HDPE. The initial increase in Hv values is attributed due to the uniform dispersion of nanofillers within the polymer matrix, besides to the fact that HDPE/WO3 NPs(C) composites consist of more number of WO3 per mass compared to HDPE/WO3 NPs(B) due to the smaller size of NPs(C). Whereas, the drop in Hv values can be explained as a result of the undesirable agglomeration of the fillers which act as voids trapped in the polymer matrix in addition to the existence of cracks at high filler loading as confirmed also by SEM. RISE behavior is described through the application of Meyer’s law. This law was deduced from ball indentation experiments on a wide range of metals and it states that indentation diameter d after unloading is related to the applied load according to the following relation [40]:
where F is applied load and d is the resulting indentation size. A is a constant of proportionality. The exponent n is known as Meyer’s index, it is expected to be > 2 for RISE [41].
The values of A and n were determined from the slope of log F versus log d graph as listed in Table 3, indicating that the HDPE nanocomposites exhibited a reverse indentation size effect (RISE) [42].
A comparative study to the average Hv values versus F for HDPE/WO3 Bulk(A) composites revealed RISE behavior with a fluctuation of the Hv results of the different wt% of WO3 Bulk(A) with that of HDPE as shown in Fig. 8. Hv exhibited an increase up to 2 N for both fillers 10 wt% and 15 wt% and an increase up to 1 N for 25 wt% followed by a decrease compared to that of HDPE. Moreover, a decrease in Hv values is shown for 35 wt% at all loads compared to HDPE. An increase in wt% is accompanied by an increase of WO3 filler at the surface of the specimen which may affect the hardness values at loads less than 2 N. As described by the [43] study, the indentation depth is based on the relation between simultaneous piled-up dislocations forcing the indent's contact area and the dislocation network.

Hv versus F(N) for HDPE and HDPE/WO3 Bulk(A) composites
5.2.2 Tensile Test
Many studies revealed that the incorporation of nanoparticles into polymer matrix showed significantly more superior mechanical properties [44, 45]. The stress–strain curves of pure HDPE, HDPE/WO3 NPs(B), and HDPE/WO3 NPs(C) composites with different wt% measured at 25 °C and a constant strain rate (14 mm/min) are shown in Fig. 9a, b. The Young’s modulus, ultimate tensile strength, yield stress, and elongation at break were displayed as a function of WO3 wt% in Fig. 10, respectively with their values listed in Table 4. The standard deviations are also represented by error bars. HDPE exhibited a strain hardening region followed by a super ductile behavior with values of Young's modulus, UTS, yield stress, and elongation at break of 224.84 MPa, 24.76 MPa, 14.94 MPa, and 585.5%, respectively. The incorporation of both WO3 NPs to 25 wt% into the polymer matrix increased the values of Young’s modulus, UTS, and yield stress to 99%, 15%, and 35% for NPs (B) and 84%, 9%, and 19% for NPs(C), respectively compared to HDPE. Whereas, the elongation at break was declined to 94% and 82% for NPs(B) and NPs(C), respectively as compared to HDPE since the composites were brittle by increasing filler addition. Since WO3 is typically stiffer than HDPE, the filler particles inhibited the polymer matrix mobility [38, 46]. It is noteworthy that the stiffness increased with increasing wt% of the reinforcing particles up to 25 wt% as shown in Fig. 10 specially in the case of HDPE/WO3 NPs(B) composites where it reached its maximum which may be attributed to the apparent change in the degree of crystallinity that is higher in the NPs(B) than NPs(C) as reported from XRD data. This result is consistent with previous literature where the tensile elastic modulus increased with larger crystallinity [47]. However, a slight decrease in the modulus was noticed at 35 wt% for both WO3 NPs content which may be attributed to the formation of voids within the sample which initiated cracks and finally decreased the stiffness of the composites [38].

The stress–strain behavior of a HDPE and HDPE/WO3 NPs(B), and b HDPE and HDPE/WO3 NPs(C) composites at 14 mm/min

Effect of particles addition on young’s modulus, UTS, Yield stress, and % elongation at break of HDPE/WO3 nanocomposites
The strength is strongly dependent on the stress transfer between the particles and the matrix [45]. Besides, a slight decrease in the values of UTS and yield stress were recorded for the HDPE/ WO3 NPs(C) composites as revealed by Fig. 10. This may be due to the strong tendency of small particle size to agglomerate and the resulting clustered filler particles which were acting as stress-concentrators to reduce the tensile strength of the polymer matrix. Also, agglomerates were surrounded by HDPE matrix material and these agglomerates were like voids in the HDPE matrix. Thus, the strength of the polymer was diminished due to the presence of such voids [48, 49].
It is quite significant to notice that a further reduction of filler particles sizes was ineffective to enhance the composite strength as well as any further increase in filler content up to 35 wt% indicating that an optimum concentration of the filler should be taken into consideration to enhance the mechanical properties of the composite.
The elongation at break for composites decreased with increasing the filler wt% as prevailed by Fig. 10 which was mainly attributed to the flexibility decline caused by filler particles that perturbed the lamellae slippage, stretching, and orientation thus forming a barrier effect [50]. Moreover, the HDPE/WO3 NPs(C) composites showed higher strain at fracture compared to composites filled with WO3 NPs(B) at the same reinforcement content up to 25 wt%, which was associated with the increase in ductility of the composites. Also, the improvement in elongation at break reflects better adhesion of WO3 NPs(C) particles with the HDPE matrix [48, 49].
SEM and EDX observations are compatible with the mechanical properties of the synthesized composites. By increasing the amount of WO3 filler loading to 25 or 35 wt%, the mechanical properties of the composites were reduced due to the agglomeration of nanoparticles. With their big particle sizes, these agglomerated particles acted as defects and areas of stress concentrations which led to crack of the specimen and resulted in poor composite strength [45]. It can be concluded that the filler weight fraction and size significantly affect the mechanical properties of the composites.
The obtained results of the mechanical properties were comparable to other studies using HDPE incorporated with different metal oxide filler prepared via compression molding technique. The Stress–strain relationship of micro and nano CdO/HDPE composites with different particle content 10–40 wt% were studied by El-Khatib et al. [51]. They revealed that either micro CdO or nano CdO content leads to a substantial increase in the ultimate tensile strength and yield stress up to an optimum concentration of (30%). However, they tend to decrease if the concentration exceeds 30%. Moreover, the values of % elongation at break are dramatically decreased with increasing filler loading from 10 to 40 wt%. Alsayed et al. [52] studied the mechanical properties of HDPE composites filled with 0–40 wt% of micro and nano ZnO. Hv values showed a reverse indentation size effect (RISE) and the bulk composites revealed a fluctuation in Hv results. Also, a reduction in Hv values appeared starting from 30 wt% of ZnO nanofiller loading. On the other hand, It is quite significant that the 20 wt% of nano ZnO composite exhibited higher values of Young’s modulus and yield stress. Sepet et al. [53] studied the stress curve of unfilled and filled HDPE. They reported that 10 wt% of CaCO3 nanoparticles increased the tensile modulus of HDPE. At the same reinforcement ratio, the elongations at break of filled HDPE samples decreased substantially, as compared to the unfilled HDPE. The microhardness of 5%, 10%, and 15% weight percentage of MWCNT/MnO2 nanocomposite was measured by Hussain et al. [54] where they found that 10 wt% has higher microhardness in comparison to 5–15 wt%.
6 Conclusion
The different WO3 nano-sized and weight fractions showed a significant effect on the mechanical properties of HDPE. The crystalline structure of the synthesized composites showed a monoclinic phase for WO3 fillers and an orthorhombic structure for HDPE. The FTIR results confirmed the adsorption of the fillers on the surface polymer matrix. The morphological structure revealed a uniform dispersion of WO3 nanoparticles in the HDPE polymer matrix at low filler weight fractions. EDX analysis revealed the existence of W and C elements as main composed in the polymer matrix and showed an increase in the intensity of W peak for the WO3 NPs(C) composite indicating the large surface-to-volume ratio of the nanoparticles. The Vickers microhardness test showed that the samples exhibited a reverse indentation size effect (RISE) with increasing applied load through the application of Meyer’s law. Hv values for HDPE/WO3 NPs(C) composites were enhanced compared to HDPE/WO3 NPs (B) composites up to 25 wt%. The mechanical tensile properties of Young’s modulus, UTS, and yield stress revealed an increment in their values compared to HDPE with increasing the filler loading up to 25 wt% for both WO3 NPs, whereas HDPE/WO3 NPs(C) composites revealed higher values of elongation at break indicating a better adhesion of WO3 NPs(C) to the polymer matrix. It is deduced that the agglomeration of nano WO3 at high filler loading affected the properties of composites due to the filler adsorption on the surface of the polymer matrix as revealed by the FTIR results. Therefore, a fine dispersion of the nanofiller is needed by controlling some process parameters during compression molding technique such as: extending the agitation time in the mixer chamber to improve the homogeneity of the fillers in the polymer matrix, and reducing the water cooling rate of the shaped composite samples under the hot press to prevent the formation of cracks and voids.
References
P. Shukla, P. Saxena, Polymer nanocomposites in sensor applications: a review on present trends and future scope. Chin. J. Polym. Sci. (2021). https://doi.org/10.1007/s10118-021-2553-8
M. Nasirinezhad, S.R. Ghaffarian, M. Tohidian, Eco-friendly polyelectrolyte nanocomposite membranes based on chitosan and sulfonated chitin nanowhiskers for fuel cell applications. Iran. Polym. J. 30, 355–367 (2021)
S. Sherafatkhah Azari, A. Alizadeh, L. Roufegarinejad et al., Preparation and characterization of gelatin/β-glucan nanocomposite film incorporated with ZnO nanoparticles as an active food packaging system. J. Polym. Environ. 29, 1143–1152 (2021)
T. Hassan, A. Salam, A. Khan et al., Functional nanocomposites and their potential applications: a review. J. Polym. Res. 28, 36 (2021). https://doi.org/10.1007/s10965-021-02408-1
S.P. Aditya, W. Yixing, L. Xiaolin, I. Akshay, C. Wei et al., Investigating the effect of surface modification on the dispersion process of polymer nanocomposites. Nanocomposites 6, 111–124 (2020). https://doi.org/10.1080/20550324.2020.1809250
W. Longwen, C. Ziming, L. Longtu, W. Xiaohui, Breakdown strength and energy density enhancement in polymer-ceramic nanocomposites: Role of particle size distribution. Compos. Sci. Technol. 212, 108868 (2021). https://doi.org/10.1016/j.compscitech.2021.108868
G.I. Dzhardimalieva, I.E. Uflyand, Preparation of metal-polymer nanocomposites by chemical reduction of metal ions: functions of polymer matrices. J. Polym. Res. 25, 255 (2018). https://doi.org/10.1007/s10965-018-1646-8
S. Sánchez-Valdes, High-density polyethylene/recycled HDPE/nanoclay composites using an amine-alcohol modified polyethylene as a compatibilizer. Iran. Polym. J. 30, 297–305 (2021)
I.L.M. Costa, N.C. Zanini, D.R. Mulinari, Thermal and mechanical properties of HDPE reinforced with Al2O3 nanoparticles processed by thermokinectic mixer. J Inorg. Organomet. Polym. 31, 220–228 (2021). https://doi.org/10.1007/s10904-020-01709-0
J. Gupta, H. Shaik, K.N. Kumar, A review on the prominence of porosity in tungsten oxide thin films for electrochromism. Ionics (2021). https://doi.org/10.1007/s11581-021-04035-8
M. Ikram, M.M. Sajid, Y. Javed et al., Crystalline growth of tungsten trioxide (WO3) nanorods and their development as an electrochemical sensor for selective detection of vitamin C. J. Mater. Sci. Electron. 32, 6344–6357 (2021). https://doi.org/10.1007/s10854-021-05351-5
C.M. Wu, S. Naseem, M.H. Chou, J.H. Wang, Y.Q. Jian, Recent advances in tungsten-oxide-based materials and their applications. Front. Mater. 6, 49 (2019). https://doi.org/10.3389/fmats.2019.00049
Y. Shabdan, Y. Markhabayeva, N. Bakranov, N. Nuraje, Photoactive tungsten–oxide nanomaterials for water-splitting. Nanomaterials 10, 1871 (2020). https://doi.org/10.3390/nano10091871
Z. Zhijie Wei, Q. Zhou, W. Zeng, Hierarchical WO3–NiO microflower for high sensitivity detection of SF6 decomposition by product H2S. Nanotechnology 31, 215701 (2020). https://doi.org/10.1088/1361-6528/ab73bd
H. Quan, Y. Gao, W. Wang, Tungsten oxide-based visible light-driven photocatalysts: crystal and electronic structures and strategies for photocatalytic efficiency enhancement. Inorg. Chem. Front. 7, 817–838 (2020)
S. Kazuhiro, H. Hiroki, A. Takeo, Y. Masatoshi, G. Takahiro, S. Hideki, Highly active WO3 semiconductor photocatalyst prepared from amorphous peroxo-tungstic acid for the degradation of various organic compounds. Appl. Catal. B 94, 150–157 (2010)
J.R. Xavier, Effect of surface modified WO3 nanoparticle on the epoxy coatings for the adhesive and anticorrosion properties of mild steel. J. Appl. Polym. Sci. (2019). https://doi.org/10.1002/APP.48323
A. Henaish, A. Abouhaswa, Effect of WO3 nanoparticle doping on the physical properties of PVC polymer. Bull. Mater. Sci. 43, 149 (2020)
D. Toyen, A. Rittirong, W. Poltabtim et al., Flexible, lead-free, gamma-shielding materials based on natural rubber/metal oxide composites. Iran. Polym. J. 27, 33–41 (2018)
K. Rahmani, A. Sadooghi, M. Nokhberoosta, The effect of the double-action pressure on the physical, mechanical and tribology properties of Mg–WO3 nanocomposites. J. Mater. Res. Technol. 9, 1104–1118 (2020)
A.D. Mahdi, S.S. Irhayyim, S.F. Abduljabbar, Mechanical and wear behavior of Al7075 - graphite self-lubricating composite reinforced by nano-WO3 particles. Mater. Sci. Forum 1002, 151–160 (2020). https://doi.org/10.4028/www.scientific.net/MSF.1002.151
E. Besozzi, D. Dellasega, V. Russo, C. Conti, M. Passoni, M.G. Beghi, Thermomechanical properties of amorphous metallic tungsten-oxygen and tungsten-oxide. Mater. Design 165, 107565 (2018)
C.V. More, Z. Alsayed, M.S. Badawi, A.A. Thabet, P.P. Pawar, Polymeric composite materials for radiation shielding: a review. Environ. Chem. Lett. 19, 2057–2090 (2021). https://doi.org/10.1007/s10311-021-01189-9
N. Asari, L. Gholamzadeh, M. Aminian, Study the attenuation ability of the composites containing micro-size and nano-sized tungsten oxide and lead oxide as diagnostic X-ray shields. IJRSM 5, 15–22 (2017)
A.V. Machado, G. Botelho, M.M. Silva, I.C. Neves, A.M. Fonseca, Stability of nanocomposites of poly(ε-caprolactone) with tungsten trioxide. J. Polym. Res. 18, 1743–1749 (2011)
ASTM D638–14: Standard test method for tensile properties of plastics. ASTM International, (2014). https://doi.org/10.1520/D0638-14
A.H. Abdalsalam, E. Şakar, K.M. Kaky, M.H.A. Mhareb, B. Ceviz Şakar, M.I. Sayyed, A. Gürol, Investigation of gamma ray attenuation features of bismuth oxide nano powder reinforced high-density polyethylene matrix composites. Radiat. Phys. Chem. 168, 108537 (2019)
M. Joshi, B.S. Butola, G. Simon, N. Kukaleva, Rheological and viscoelastic behavior of HDPE/Octamethyl-POSS nanocomposites. Macromolecules 39, 1839–1849 (2006)
T. McNally, P. Pötschke, P. Halley, M. Murphy, D. Martin, S.E.J. Bell, G.P. Brennan, D. Bein, P. Lemoine, J.P. Quinn, Polyethylene multiwalled carbon nanotube composites. Polymer 46, 8222–8232 (2005)
V. Shakya, N.K. Pandey, S.K. Misra et al., Electrical and optical properties of ZnO–WO3 nanocomposite and its application as a solid-state humidity sensor. Bull. Mater. Sci. 40, 253–262 (2017)
S.R. Chandra, M. Hegde, C.N.R. Rao, H2S sensors based on tungsten oxide nanostructures. Sens. Actuators B Chem. 128, 488–493 (2008)
M.E. Mahmoud, A.M. El-Khatib, M.S. Badawi, A.R. Rashad, R.M. El-Sharkawy, A.A. Thabet, Fabrication, characterization and gamma rays shielding properties of nano and micro lead oxide-dispersed-high density polyethylene composites. Radiat. Phys. Chem. 145, 160–173 (2018)
R.C. Asensio et al., Analytical characterization of polymers used in conservation and restoration by ATR-FTIR spectroscopy. Anal. Bioanal. Chem. 395, 2081–2096 (2009)
G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, 3rd edn. (Wiley, Chichester, 2001)
M.R. Jung, F.D. Horgen, S.V. Orski, C.V. Rodriguez, K.L. Beers, G.H. Balazs, T.T. Jones, T.M. Work, K.C. Brignac, S.J. Royer, K.D. Hyrenbach, B.A. Jensen, J.M. Lynch, Validation of ATR FT-IR to identify polymers of plastic marine debris, including those ingested by marine organisms. Mar. Pollut. Bull. 127, 704–716 (2018)
B. Ingham, S.V. Chong, J.L. Tallon, Layered tungsten oxide-based organic− inorganic hybrid materials: an infrared and Raman study. J. Phys. Chem. B 109, 4936–4940 (2005)
N.Z. Azman, S.A. Siddiqui, I.M. Low, Characterization of micro-sized and nano-sized tungsten oxide-epoxy composites for radiation shielding of diagnostic X-rays. Mater. Sci. Eng. C Mater. Biol. Appl. 33, 4952–4957 (2013). https://doi.org/10.1016/j.msec.2013.08.023
M.R. Ambika, N. Nagaiah, K. Prashantha, Thermal resistance and mechanical stability of tungsten oxide filled polymer composite radiation shields. Int. J. Polym. Anal. Ch. 25, 431–443 (2020)
H. Li, R.C. Bradt, The effect of indentation-induced cracking on the apparent microhardness. J. Mater. Sci. 31, 1065–1070 (1996)
I.M. Hutchings, The contributions of David Tabor to the science of indentation hardness. J. Mater. Res. 24, 581–589 (2009)
S. Bhattacharya, R. Kundu, K. Bhattacharya, A. Poddar, D. Roy, Micromechanical hardness study and the effect of reverse indentation size on heat-treated silver doped zinc-molybdate glass nanocomposites. J. Alloys Compd. 770, 136–142 (2019)
K. Sangwal, On the reverse indentation size effect and microhardness measurement of solids. Mater. Chem. Phys. 63, 145–152 (2000)
M. Baitsch, K.C. Le, T.M. Tran, Dislocation structure during microindentation. Inter. J. Eng. Sci. 94, 195–211 (2015)
J. Cho, M.S. Joshi, C.T. Sun, Effect of inclusion size on mechanical properties of polymeric composites with micro and nano particles. Compos. Sci. Technol. 66, 1941–1952 (2006)
S.Y. Fu, X.Q. Feng, B. Lauke, Y.W. Mai, Effects of particle size, particle/matrix interface adhesion and particle loading on mechanical properties of particulate–polymer composites. Compos. B. Eng. 39, 933–961 (2008)
M. Saleh, Z. Al-Hajri, A. Popelka, S. Javaid Zaidi, Preparation and characterization of alumina HDPE composites. Materials (Basel) 13, 250 (2020). https://doi.org/10.3390/ma13010250
H. Mae, M. Omiya, K. Kishimoto, Effects of strain rate and relaxation rate on elastic modulus of semi-crystalline polymer. Trans. Jpn.Soc. Comput. Methods Eng. 7, 207–212 (2008)
E. Jenson Joseph, K. Panneerselvam, Investigation on the influence of tungsten particulate in mechanical and thermal properties of HD50MA180 high density polyethylene composites. Mater. Res. Express 7, 045306 (2020)
D. Tripathi, T.K. Dey, Thermal conductivity, coefficient of linear thermal expansion and mechanical properties of LDPE/Ni composites. Indian J. Phys. 87, 435–445 (2013)
B. Yuan, C. Bao, L. Song, N. Hong, K.M. Liew, Y. Hu, Preparation of functionalized graphene oxide/polypropylene nanocomposite with significantly improved thermal stability and studies on the crystallization behavior and mechanical properties. Chem. Eng. Technol. 237, 411–420 (2014)
A.M. El-Khatib, M.S. Hamada, M.T. Alabsy et al., Fast and thermal neutrons attenuation through micro-sized and nano-sized CdO reinforced HDPE composites. Radiat. Phys. Chem. 180, 109245 (2021)
Z. Alsayed, R. Awad, M.S. Badawi, Thermo-mechanical properties of high density polyethylene with zinc oxide as a filler. Iran. Polym. J. 29, 309–320 (2020). https://doi.org/10.1007/s13726-020-00796-7
H. Sepet, B. Aydemir, N. Tarakcioglu, Evaluation of mechanical and thermal properties and creep behavior of micro- and nano-CaCO3 particle-filled HDPE nano- and microcomposites produced in large scale. Polym. Bull. 77, 3677–3695 (2020). https://doi.org/10.1007/s00289-019-02922-9
M.Z. Hussain, S. Khan, R. Nagarajan, U. Khan, V. Vats, Fabrication and microhardness analysis of MWCNT/MnO2 nanocomposite. J. Mater. 2016, 10 (2016). https://doi.org/10.1155/2016/6070468
Acknowledgements
This work was done in the frame of the scientific collaboration between the Physics Department, Faculty of Science, Beirut Arab University (BAU), and the National Council for Scientific Research-Lebanon (CNRSL), Lebanese Atomic Energy Commission (LAEC). The main author would like to thank Dr. Malek Tabbal, Professor of Physics at the American University of Beirut (AUB), and the Lab’s Staff at KAS CRSL-AUB, for XRD measurements and their valuable support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Obeid, A., Roumie, M., Badawi, M.S. et al. Evaluation of the Effect of Different Nano-Size of WO3 on the Structural and Mechanical Properties of HDPE. J Inorg Organomet Polym 32, 1506–1519 (2022). https://doi.org/10.1007/s10904-021-02219-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-021-02219-3