
Abstract
Purpose
More than 50 different monogenic disorders causing inflammatory bowel disease (IBD) have been identified. Our goal was to characterize the clinical phenotype, genetic workup, and immunologic alterations in an Ashkenazi Jewish patient that presented during infancy with ulcerative colitis and unique clinical manifestations.
Methods
Immune workup and whole-exome sequencing were performed, along with Sanger sequencing for confirmation. Next-generation sequencing of the TCRB and IgH was conducted for immune repertoire analysis. Telomere length was evaluated by in-gel hybridization assay. Mass cytometry was performed on patient’s peripheral blood mononuclear cells, and compared with control subjects and patients with UC.
Results
The patient presented in infancy with failure to thrive and dysmorphic features, consistent with a diagnosis of dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Severe ulcerative colitis manifested in the first year of life and proceeded to the development of a primary immunodeficiency, presenting as Pneumocystis jiroveci pneumonia and hypogammaglobulinemia. Genetic studies identified a deleterious homozygous C.3791G>A missense mutation in the helicase regulator of telomere elongation 1 (RTEL1), leading to short telomeres in the index patient. Immune repertoire studies showed polyclonal T and B cell receptor distribution, while mass cytometry analysis demonstrated marked immunological alterations, including a predominance of naïve T cells, paucity of B cells, and a decrease in various innate immune subsets.
Conclusions
RTEL1 mutations are associated with significant alterations in immune landscape and can manifest with infantile-onset IBD. A high index of suspicion is required in Ashkenazi Jewish families where the carriage rate of the C.3791G>A variant is high.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the past decade, significant progress has been made in our understanding of how genetics plays a role in the pathogenesis of inflammatory bowel disease (IBD). Genome-wide association studies have identified > 240 specific single nucleotide polymorphisms (SNPs) that are relatively common and associate with increased risk for developing either Crohn’s disease (e.g., NOD2), ulcerative colitis (UC, e.g., HLA), or both (e.g., IL10) [1]. In contrast, rare and deleterious genetic variants can directly cause intestinal inflammation, and these disorders can be classified as monogenic IBD [2, 3]. With increased clinical awareness and interdisciplinary patient care, advances in sequencing technologies and lowering of the costs associated with these studies, more than 50 different monogenic disorders have been identified in recent years. Most of these rare disorders result from loss-of-function mutations in genes implicated in mucosal homeostasis, whether related to immune pathways, epithelial cell function, or both [2]. In many of these disorders, associated clinical features suggestive of severe immunodeficiency have been reported.
Dyskeratosis congenita (DC) belongs to a group of rare inherited disorders termed telomeropathies, which develop as a result of accelerated shortening or damage of telomeres [4]. Telomeres, located at the ends of the chromosomes, function to maintain chromosomal integrity and protect from DNA damage or loss, and abnormal fusion with other chromosomes [5]. Mutations in more than 10 genes encoding RNA or proteins required for telomere maintenance, with variable mode of inheritance, have been shown to cause telomeropathies, including dyskerin pseudouridine synthase 1 (DKC1), telomerase RNA component (TERC), TERF1 interacting nuclear factor 2 (TINF2), STN1, telomerase reverse transcriptase (TERT), and regulator of telomere elongation helicase 1 (RTEL1) [6, 7]. DC is a multi-systemic disease manifesting with a classic triad of dysplastic nails; lacy reticular hyperpigmented lesions, typically in the neck and chest; and oral leukoplakia [8]. In addition, these patients often develop bone marrow failure with increased risk of malignant transformation into leukemia, pulmonary fibrosis, and liver abnormalities [8]. Hoyeraal-Hreidarsson syndrome (HHS) is a severe form of DC, not limited to mutations in a single gene, and is characterized by intrauterine growth restriction (IUGR), microcephaly, and cerebellar hypoplasia [9].
One of the less appreciated phenotypes of DC is susceptibility to recurrent or atypical infections due to a combined immunodeficiency state. In addition, these patients can develop various gastrointestinal phenotypes, though intestinal inflammation has rarely been reported. We present the unusual clinical course as well as the genetic, molecular, and immunological workup of a patient who presented with infantile-onset colitis and was subsequently diagnosed with DC and HHS due to an RTEL1 mutation.
Methods
Genetic Studies
The study was approved by the local IRB committee at Sheba Medical Center. Informed written consent was obtained from the parents. Next-generation sequencing (NGS) was performed at the Dr. von Hauner Children’s Hospital NGS facility. Genomic DNA was isolated from whole blood, and whole-exome libraries were prepared with SureSelect XT Human All Exon V6 + UTR reagents (Agilent Technologies). Barcoded libraries were sequenced on a NextSeq 500 NGS platform (Illumina) to an average coverage depth of 100×. Bioinformatics analysis and subsequent filtering of the so-called variants led to the identification of the homozygous RTEL1 variant. Segregation of the sequence variant was confirmed by Sanger sequencing for the patient and informative family members.
Immune Workup
Cell surface markers of peripheral blood mononuclear cells (PBMCs) were determined by immunofluorescent staining using flow cytometry (Navios, Beckman Coulter, Brea, CA, USA) with antibodies purchased from Beckman Coulter. Lymphocyte proliferation assay was done in response to phytohemagglutinin and anti-CD3 (using tritiated thymidine incorporation). T cell receptor excision circles (TREC) analysis was performed using DNA extracted from the patient’s PBMCs. The amount of signal joint TREC copies per DNA content was determined by real-time quantitative PCR as previously described [10]. Representatives of specific TCR-variable b families were detected and quantified using patients’ PBMCs with flow cytometry according to the manufacturer’s instructions (Beta Mark TCR Vb repertoire kit, Beckman Coulter). Normal control values comprised of 58 healthy subjects were obtained from the kit.
NGS of the TCRB and IgH variable regions were performed to define the global TCR and B cell receptor (BCR) immune repertoire. Briefly, 2 μg genomic DNA was extracted from blood (Wizard kit, Promega, WI, USA) and used for TRB and IgH repertoire library generation. Amplified sequences were subjected to high-throughput sequencing using Illumina technology. Graphical presentation of the repertoire was presented using hierarchical tree maps using the TreeMap software (www.treemap.com).
In-gel Hybridization Assay
Genomic DNA was extracted from blood leukocytes following erythrocytes lysis by a standard proteinase K-phenol extraction, digested with HinfI, and analyzed by in-gel hybridization assay as described previously [11], except for the telomeric oligonucleotide probe, which was (AACCCT)3. Mean telomere restriction fragments (TRF) length was calculated using MaTeLo [12]. Following electrophoresis and gel drying, a telomeric probe was hybridized to native DNA in the gel to detect the single-stranded G-rich telomeric 3′ overhang (native). Then, the DNA was denatured and rehybridized to detect the overall length of the TRF (denatured). The native and denatured signals were quantified by ImageJ (http://imagej.nih.gov/ij/). The average length of the 3′ overhang was estimated by dividing the native by the denatured hybridization signals and then multiplying by the mean telomere length to correct for the effect of telomere length on the signal. The values were normalized to the control (C1).
Mass Cytometry (CyTOF) Studies
PBMCs from the RTEL1-deficient patient, four healthy controls and two patients with active UC, were stained with a panel of metal-tagged antibodies targeting markers of major immune cell lineages (Supplementary Table 1) per previously published protocol [13]. The samples were run on Helios2 mass cytometer (Fluidigm, San Francisco, CA, USA). FCS files were analyzed with premium Cytobank software and pre-gated on CD45+/viable/single/DNA+ events before initiating the analysis. Normalization beads were used and gated out of the analysis. The data was automatically clustered with the viSNE addon in the premium Cytobank using either all leukocytes (CD45+ cells) or all T lymphocytes (CD3+ cells), and cluster identity was manually labeled based on marker expressed in the individual clusters (Supplementary Table 2). Cluster percentage (% of CD45+ viable single events or CD3+CD45+ viable single events) was computed and plotted for comparison between patient groups using Prism8 software.
Results
Case Description
A female patient was referred at the age of 12 months for evaluation of failure to thrive, dysmorphic features, and chronic diarrhea, occasionally with blood, that started 4–6 months before. She was the third child to healthy non-consanguineous parents of Ashkenazi Jewish descent; both siblings were healthy. Pregnancy was noted for oligohydramnios and IUGR that was identified at the third trimester. She was born at 33 weeks weighing 1289 g (3 percentile) and spent one uneventful month in the neonatal admission ward. The patient was initially breast-fed and later transitioned to an amino acid–based formula due to non-specific feeding problems. There were no significant infections at that time.
At the age of 12 months physical exam revealed a small, microcephalic, and dysmorphic child. Her weight was 5.9 kg (-3.5 Z score), length was 66 cm (-3.1 Z score), and head circumference was 42 cm (-2.1 Z score). Dysmorphic features included bilateral epicanthal folds, large ears, and long philtrum. In addition, oral exam demonstrated a red demarcated tongue lesion, suspicious for oral leukoplakia (Fig. 1a).

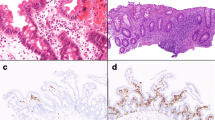
Clinical manifestations of the index patient. a Oral cavity examination showing leukoplakia. b Hematoxylin and eosin stain of rectal biopsy demonstrating chronic colitis with increased eosinophils in the lamina propria. c Atrophic cerebellum seen on brain MRI at the age of 14 months. b Severe rectal stricture demonstrated during colonoscopy at the age of 30 months
Initial blood tests showed normal complete blood counts, high inflammatory markers (CRP 30.5 mg/L, normal values < 5 mg/L), and low albumin levels (2.8 g/dL, normal 3.4–4.2 g/dL). Interestingly, although immunoglobulin levels were only slightly decreased, lymphocyte subset analysis at the age of 15 months demonstrated very low B cell percentages (1% CD20+ cells), while T cells were within normal limits (Table 1). Moreover, TREC quantification was within normal limits (583 copies/0.5 μg DNA, normal > 400), flow cytometry analysis of the TCR Vb repertoire was polyclonal, and the response to mitogenic stimulations was normal (Table 1). Further analysis of adaptive immune architecture was performed through NGS of the TCRB and IgH genetic segments. As can be witnessed in Fig. 2, T and B cell receptor repertoires were polyclonal, although lower number of B cell clones were appreciated, further demonstrating lower B cell numbers, compared with controls.

Polyclonal immune repertoire in the index patient. TreeMap images of a TCRB and b IgH repertoires for T and B cell receptors, respectively, from peripheral blood. In these analyses, each square represents a different clone, and the size correlates with its frequency
An upper endoscopy was normal at the age of 15 months. Stool calprotectin was modestly elevated (487 μg/g of stool). A colonoscopy at the same time showed moderate left-sided colitis (Mayo score 2) though biopsies demonstrated pancolitis with chronic inflammation throughout the entire colon, along with numerous eosinophils in sections from different regions (Fig. 1b). A brain MRI was also completed given microcephaly and demonstrated a small cerebellum which seemed atrophic (Fig. 1c).
Given the clinical, endoscopic, and histologic findings, the patient was diagnosed with UC. However, she failed to respond to sulfasalazine, antibiotics, and steroids. A Gtube was placed for management of feeding problems, but she was unable to tolerate bolus or continue amino acid–based formula feeds. As a result of ongoing diarrhea and severe feeding problems, administration of total parental nutrition was initiated. Despite evidence of intestinal inflammation, a decision was made not to treat her with anti-TNFα agents (typically used in steroid-resistant UC patients), based on the genetic diagnosis (see below).
At the age of 22 months, she developed severe Pneumocystis jiroveci pneumonia, which was the first manifestation of an immunodeficiency. At that time, she was not receiving immunosuppressive therapy, and blood tests demonstrated lymphopenia of ~ 500 cells/mm3 and hypogammaglobulinemia (IgG 98.5 mg/dL, IgM < 17.4 mg/dL, and IgA < 25 mg/dL). Following this infection, she started monthly replacement intravenous immune globulins, along with trimetophrim-sulfamethoxazole and fluconazone prophylaxis. A bone marrow biopsy around two and a half years of age confirmed the diagnosis of myelodysplasia, characterized by atypia of red cells and megakaryocytes and mild abnormal white cell maturation. Her condition continued to deteriorate, and a repeat endoscopic evaluation at the age of 32 months confirmed the presence of a high esophageal stricture in addition to a rectal stricture (Fig. 1d). The patient passed away before the age of 3 years due to adenovirus acute liver failure.
Identification of a Pathogenic RTEL1 Mutation Causing Short Telomers
Since the patient exhibited unique clinical features, whole-exome sequencing was performed at 18 months of age and identified a homozygote missense mutation in RTEL1 C.3791G>A (p.Arg1264His; VCV000042018.6) (CCDS63331) that was previously reported in patients with HHS [14, 15]. Sanger sequencing confirmed that the patient is homozygote for this mutation and the parents are carriers (Fig. 3a).

Identification of a homozygote RTEL1 mutation in the index patient leading to short telomers. a Sanger sequencing of the C.3791G>A variant in the patient and parents. b Genomic DNA was extracted from peripheral blood leukocytes, digested with HinfI restriction endonuclease and analyzed by in-gel hybridization with a C-rich telomeric probe to the native DNA (native panel) to detect the signal corresponding to the telomeric 3′ overhang. After exposure, the DNA was denatured in situ and rehybridized to the same probe to measure the total telomeric signal (denatured). Mean TRF in kilobyte is indicated below the lanes. c The average length of the 3′ overhang was estimated by dividing for each lane the native by the denatured hybridization signals, multiplying by the mean TRF to correct for the length-based bias in signal and then normalized to control 1. C1, control 1; C2, control 2; P, patient; M, mother; F, father
RTEL1 mutations have been associated with severely short telomeres, as well as diminished telomeric 3′ overhang [9, 11, 16]. Therefore, we examined telomere length and overhang length in leukocytes of the patient and compared them to age-matched controls and the parents. We used in-gel hybridization to genomic DNA digested with a frequent cutting restriction enzyme (HinfI), which does not cut the telomeric sequence but only adjacent to it. Leukocytes from the RTEL1-deficient patient showed severely short telomeres (mean TRF 4.6 kb, as compared with 6.7 and 7.1 kb of age-matched controls), with a significant portion that was below 2 kb (Fig. 3b). In addition, the patient, as well as the heterozygous parents, displayed short telomeric overhang compared with the non-carrier controls (Fig. 3c), as was shown previously in other RTEL1 mutations carriers [11, 17]. Altogether, the genetic data and telomere phenotype confirms the diagnosis of an RTEL1 mutation as the cause for telomere dysfunction and HHS.
Altered Immune Landscape in Peripheral Blood of the RTEL1-Deficient Patient
In order to characterize in detail the immune landscape associated with the RTEL1 mutation, we performed CyTOF analysis at the age of 15 months. The overall abundance of total T cells, CD4+, CD8+, and regulatory T cells (Tregs) were comparable between control subjects, patients with UC, and the RTEL1-deficient patient (Fig. 4a, b). In addition, the CD4/CD8 ratio was similarly preserved in the patient with RTEL1 mutation (data not shown). However, for both CD4+ and CD8+ T cells, we observed in the patient’s sample a predominance of naïve over memory T cells, which was also true for Tregs (Fig. 4c). In addition, as previously reported in association with RTEL1 deficiency [18], we observed paucity of all B cell subtypes (Fig. 4d). Interestingly, NK cells and ILCs were also significantly decreased in the patient (Fig. 4e), suggesting a defect likely at the early innate lymphoid progenitor (EILP) stage [19]. Additionally, we observed a nearly complete lack of myeloid cells, including monocytes and dendritic cells (Fig. 4e), again suggesting a defect at the progenitor state, likely at the myeloblast stage.

Altered immune landscape in the RTEL1-deficient patient. viSNE clustering of a leukocytes (CD45+) or b T cells of control, UC, and RETL1-deficient samples. Immune populations were identified using marker expression on the tSNE map and are color-coded according to the scheme on the right-hand side. Cumulative data of percent of c T cells, d B cells, and e innate immune cells in relationship to all leukocytes. Th, helper T cells; Tc, cytotoxic T cells; DCs, dendritic cells; NK, natural killer cells; ILCs, innate lymphoid cells; Tregs, regulatory T cells
Discussion
While the clinical phenotype of DC has been reported more than 50 years ago, the understanding that this disorder arise from telomere shortening and dysfunction was first appreciated in 1999 [20], and since then, numerous genes were identified as associated with telomeropathies [21]. Mutations in RTEL1 were initially identified in 2013 mostly in patients with features of HHS [14,15,16, 22, 23]. In the case we describe the initial presenting features were failure to thrive and infantile-onset UC, and as the patient grew, a multi-systemic disease evolved, including immunodeficiency and myelodysplasia.
The gastrointestinal phenotype in our patient was severe. Colitis presented at the age of several months and was unresponsive to standard therapy. The patient also did not tolerate enteral feeds and was dependent on parenteral nutrition. Finally, strictures of the esophagus and rectum were identified, highlighting the inflammatory-stricturing nature of this disorder. However, in contrast to patients with Crohn’s disease who can develop fibrostenotic complications as a result of chronic luminal inflammation, the strictures in DC likely result from a different etiology, as fibrosis can develop in these patients in other organs (e.g., liver, lungs) and often involves the esophagus, which was not shown to be inflamed by us and others.
There is scant data on intestinal inflammation in patients with RTEL1 mutations. Speckmann and colleagues reported that 5 of 6 patients with mutations in this gene developed chronic diarrhea [18], while in a different study, some patients developed enteropathy [14]. Nevertheless, these studies did not provide any endoscopic or histologic data, and it is unclear whether these patients developed intestinal inflammation per se. A different study summarized the gastrointestinal manifestations of 6 patients with telomere disorders, of whom 4 had mutations in DKC1, TERT, and TERC, and 2 exhibited typical DC/HHS phenotypes, but without an identifiable genetic cause [24]. Some of these patients developed enteropathy, and similar to the patient we report, one of them presented with pancolitis in infancy that was unresponsive to medical therapy and consequently was referred for colectomy [24]. Dysphagia due to a proximal esophageal web was also common, and according to a literature search performed by that group, it was highly characteristic of patients with telomeropathies (23/24 cases, 96%) [24]. A different presentation of GI bleeding due to vascular ectasia has been reported in patients with another telomeropathy, Coats plus syndrome, resulting from CTC1 [25] and STN1 [26] mutations. These observations highlight the diverse GI phenotypes in patients with telomere disorders.
Immune workup analysis by CyTOF demonstrated that consistent with published data [17, 18, 27], this patient had partial bone marrow failure at early myeloid and lymphocyte precursor stages, with a significant reduction of B, NK/ILC, and myeloid cells and preservation of T cells. However, even though the overall numbers of T cells were similar to control and IBD patients, functionally, these T cells were likely also significantly affected by the mutation as majority of T cells were of the naïve phenotype, with a marked reduction in memory T cell numbers. Interestingly, although this patient exhibited normal TREC values and polyclonal immune repertoire, she developed Pneumocystis jiroveci pneumonia, which also reflects abnormal T cell function. In addition, since all B cells were equally affected but the T cells were preserved, the underlying defect must have occurred after the differentiation from the common progenitor cell but in an early B cell precursor, perhaps at the proB or preB stage.
This patient failed to respond to standard anti-inflammatory medications typically administered in UC, including 5ASAs and steroids. We decided not to treat her with TNFα antagonists, due to the high risk of opportunistic infections in these patients [28], and especially in this patient who developed Pneumocystis jiroveci pneumonia, unrelated to immunosuppressive therapy. We also elected not to proceed with hematopoietic stem cell transplantation (HSCT), given increased morbidity and mortality associated with this procedure in patients with DC, and the high risk of long-term complications, likely not corrected by transplant, including liver, lung, and also GI diseases [29,30,31,32,33]. Androgens such as danazol were shown to improve bone marrow failure in patients with telomere disorders [34, 35]. However, while some of these studies showed that such therapy elongated telomers [34], others failed to demonstrate any effect [36]. Moreover, the effects of androgens on telomeres outside the bone marrow are unknown and therefore unclear whether they will influence other non-hematopoietic manifestations in patients with DC.
Telomere-associated proteins are critical for highly proliferative tissues, such as bone marrow, skin, and epithelium [5]. Woo and colleagues generated intestinal organoids from a patient with a DKC1 mutation and DC phenotype [37]. Compared with controls, DKC1-mutant organoids exhibited poor growth and differentiation, with a significant decrease in various epithelial markers, such as E-cadherin and β-catenin, and a marked reduction in Wnt signaling, resulting in attenuated intestinal stem cell self-renewal [37]. Decreased Wnt signaling was also observed in induced pluripotent stem cells from patients with DC [38]. Culture of DKC1-deficient intestinal organoids with Wnt agonists, including lithium, reversed their phenotype and enabled improved growth and differentiation [37]. More importantly, lithium administration to immunodeficient mice that received a xenograft model of DKC1-mutant intestinal organoids resulted in improved morphology and proliferation of the epithelium [37]. These studies highlight two important points: First, patients with telomeropathies have intrinsic epithelial defects, and therefore, while HSCT may correct the immunodeficiency state and bone marrow failure, it is unlikely to rescue the GI phenotypes. In fact, the phenotype in these patients may become more severe with age since telomere shortening and dysfunction is progressive. Second, lithium therapy may be considered in these patients with various GI disorders. Indeed, lithium was shown in mice to promote recovery following dextran sodium sulfate–induced colitis [39] and 2,4,6-trinitrobenzene sulfonic acid–induced colitis [40] in rats.
The patient we present, born to Ashkenazi Jewish non-consanguineous parents, harbored a homozygote missense mutation C.3791G>A in RTEL1, which has been reported previously in this population [14, 15]. Strikingly, Fedick and colleagues showed that 0.96% of the orthodox Ashkenazi Jews in New York were carriers of this variant, while in the general Ashkenazi Jewish population, the rate was 0.45%, compared with a carrier frequency of 0.01% in publicly available databases [41]. Nevertheless, DC and HHS are rare disorders, and the discrepancy between this observation and genetic data is unclear. As suggested by Fedick and colleagues [41], it is possible that these disorders are fatal early in life or in utero, or that the phenotype in some patients is mild. Regardless, these findings highlight the importance of screening for this variant in Ashkenazi Jewish subjects as part of carrier screening programs.
In conclusion, patients with RTEL1 mutations can develop severe colitis in the first years of life that is likely the result of aberrant telomere function in both immune and epithelial cells. Therefore, while HSCT might correct the bone marrow failure and immunodeficiency, it may not fully restore the GI phenotype. Additional studies are required to determine whether administration of Wnt agonists, such as lithium, potentially in gut-specific formulations, can alleviate the GI manifestations in these patients.
References
Graham DB, Xavier RJ. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature. 2020;578:527–39. https://doi.org/10.1038/s41586-020-2025-2.
Uhlig, H. H. et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 147, 990–1007. e1003 (2014).
Kelsen JR, Baldassano RN, Artis D, Sonnenberg GF. Maintaining intestinal health: the genetics and immunology of very early onset inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. 2015;1:462–76. https://doi.org/10.1016/j.jcmgh.2015.06.010.
Ballew BJ, Savage SA. Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol. 2013;6:327–37. https://doi.org/10.1586/ehm.13.23.
Roake CM, Artandi SE. Regulation of human telomerase in homeostasis and disease. Nat Rev Mol Cell Biol. 2020;21:384–97. https://doi.org/10.1038/s41580-020-0234-z.
Tangye SG, al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40:24–64. https://doi.org/10.1007/s10875-019-00737-x.
Savage, S. A. Beginning at the ends: telomeres and human disease. F1000Res 7, doi:https://doi.org/10.12688/f1000research.14068.1 (2018).
Fernandez Garcia MS, Teruya-Feldstein J. The diagnosis and treatment of dyskeratosis congenita: a review. J Blood Med. 2014;5:157–67. https://doi.org/10.2147/JBM.S47437.
Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol. 2015;170:457–71. https://doi.org/10.1111/bjh.13442.
Werner L, Lee YN, Rechavi E, Lev A, Yerushalmi B, Ling G, et al. Alterations in T and B cell receptor repertoires patterns in patients with IL10 signaling defects and history of infantile-onset IBD. Front Immunol. 2020;11:109. https://doi.org/10.3389/fimmu.2020.00109.
Lamm N, Ordan E, Shponkin R, Richler C, Aker M, Tzfati Y. Diminished telomeric 3′ overhangs are associated with telomere dysfunction in Hoyeraal-Hreidarsson syndrome. PLoS One. 2009;4:e5666. https://doi.org/10.1371/journal.pone.0005666.
Sagie S, Ellran E, Katzir H, Shaked R, Yehezkel S, Laevsky I, et al. Induced pluripotent stem cells as a model for telomeric abnormalities in ICF type I syndrome. Hum Mol Genet. 2014;23:3629–40. https://doi.org/10.1093/hmg/ddu071.
Konnikova L, Boschetti G, Rahman A, Mitsialis V, Lord J, Richmond C, et al. High-dimensional immune phenotyping and transcriptional analyses reveal robust recovery of viable human immune and epithelial cells from frozen gastrointestinal tissue. Mucosal Immunol. 2018;11:1684–93. https://doi.org/10.1038/s41385-018-0047-y.
Ballew BJ, Joseph V, de S, Sarek G, Vannier JB, Stracker T, et al. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet. 2013;9:e1003695. https://doi.org/10.1371/journal.pgen.1003695.
Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92:448–53. https://doi.org/10.1016/j.ajhg.2013.02.001.
Deng Z, Glousker G, Molczan A, Fox AJ, Lamm N, Dheekollu J, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci U S A. 2013;110:E3408–16. https://doi.org/10.1073/pnas.1300600110.
Marsh JCW, Gutierrez-Rodrigues F, Cooper J, Jiang J, Gandhi S, Kajigaya S, et al. Heterozygous RTEL1 variants in bone marrow failure and myeloid neoplasms. Blood Adv. 2018;2:36–48. https://doi.org/10.1182/bloodadvances.2017008110.
Speckmann C, et al. Clinical and molecular heterogeneity of RTEL1 deficiency. Front Immunol. 2017;8:449. https://doi.org/10.3389/fimmu.2017.00449.
Walker JA, et al. Polychromic reporter mice reveal unappreciated innate lymphoid cell progenitor heterogeneity and elusive ILC3 progenitors in bone marrow. Immunity. 2019;51:104–18 e107. https://doi.org/10.1016/j.immuni.2019.05.002.
Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–5. https://doi.org/10.1038/990141.
Bertuch AA. The molecular genetics of the telomere biology disorders. RNA Biol. 2016;13:696–706. https://doi.org/10.1080/15476286.2015.1094596.
Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in dyskeratosis congenita. Hum Genet. 2013;132:473–80. https://doi.org/10.1007/s00439-013-1265-8.
Le Guen T, et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013;22:3239–49. https://doi.org/10.1093/hmg/ddt178.
Jonassaint NL, Guo N, Califano JA, Montgomery EA, Armanios M. The gastrointestinal manifestations of telomere-mediated disease. Aging Cell. 2013;12:319–23. https://doi.org/10.1111/acel.12041.
Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet. 2012;44:338–42. https://doi.org/10.1038/ng.1084.
Simon AJ, Lev A, Zhang Y, Weiss B, Rylova A, Eyal E, et al. Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J Exp Med. 2016;213:1429–40. https://doi.org/10.1084/jem.20151618.
Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. 2018;131:717–32. https://doi.org/10.1182/blood-2017-09-806489.
Touzot F, Kermasson L, Jullien L, Moshous D, Ménard C, Ikincioğullari A, et al. Extended clinical and genetic spectrum associated with biallelic RTEL1 mutations. Blood Adv. 2016;1:36–46. https://doi.org/10.1182/bloodadvances.2016001313.
Fioredda F, Iacobelli S, Korthof ET, Knol C, van Biezen A, Bresters D, et al. Outcome of haematopoietic stem cell transplantation in dyskeratosis congenita. Br J Haematol. 2018;183:110–8. https://doi.org/10.1111/bjh.15495.
Tamura S, et al. Allogeneic hematopoietic cell transplantation for dyskeratosis congenita: a report of 3 cases. J Pediatr Hematol Oncol. 2017;39:e394–8. https://doi.org/10.1097/MPH.0000000000000844.
Nelson AS, Marsh RA, Myers KC, Davies SM, Jodele S, O’Brien TA, et al. A reduced-intensity conditioning regimen for patients with dyskeratosis congenita undergoing hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2016;22:884–8. https://doi.org/10.1016/j.bbmt.2016.01.026.
Sorge C, Pereboeva L, Westin E, Harris WT, Kelly DR, Goldman F. Pulmonary complications post hematopoietic stem cell transplant in dyskeratosis congenita: analysis of oxidative stress in lung fibroblasts. Bone Marrow Transplant. 2017;52:765–8. https://doi.org/10.1038/bmt.2016.353.
Chen, R. L., Lin, K. K. & Chen, L. Y. Complications for a Hoyeraal-Hreidarsson syndrome patient with a germline DKC1 A353V variant undergoing unrelated peripheral blood stem cell transplantation. Int J Mol Sci 20:3261, doi:https://doi.org/10.3390/ijms20133261 (2019).
Townsley DM, Dumitriu B, Liu D, Biancotto A, Weinstein B, Chen C, et al. Danazol treatment for telomere diseases. N Engl J Med. 2016;374:1922–31. https://doi.org/10.1056/NEJMoa1515319.
Catala A, et al. Androgen therapy in inherited bone marrow failure syndromes: analysis from the Canadian Inherited Marrow Failure Registry. Br J Haematol. 2020;189:976–81. https://doi.org/10.1111/bjh.16445.
Khincha PP, Bertuch AA, Gadalla SM, Giri N, Alter BP, Savage SA. Similar telomere attrition rates in androgen-treated and untreated patients with dyskeratosis congenita. Blood Adv. 2018;2:1243–9. https://doi.org/10.1182/bloodadvances.2018016964.
Woo DH, Chen Q, Yang TLB, Glineburg MR, Hoge C, Leu NA, et al. Enhancing a Wnt-telomere feedback loop restores intestinal stem cell function in a human organotypic model of dyskeratosis congenita. Cell Stem Cell. 2016;19:397–405. https://doi.org/10.1016/j.stem.2016.05.024.
Gu BW, Apicella M, Mills J, Fan JM, Reeves DA, French D, et al. Impaired telomere maintenance and decreased canonical WNT signaling but normal ribosome biogenesis in induced pluripotent stem cells from X-linked dyskeratosis congenita patients. PLoS One. 2015;10:e0127414. https://doi.org/10.1371/journal.pone.0127414.
Raup-Konsavage WM, Cooper TK, Yochum GS. A role for MYC in lithium-stimulated repair of the colonic epithelium after DSS-induced damage in mice. Dig Dis Sci. 2016;61:410–22. https://doi.org/10.1007/s10620-015-3852-0.
Daneshmand A, Mohammadi H, Rahimian R, Habibollahi P, Fakhfouri G, Talab SS, et al. Chronic lithium administration ameliorates 2,4,6-trinitrobenzene sulfonic acid-induced colitis in rats; potential role for adenosine triphosphate sensitive potassium channels. J Gastroenterol Hepatol. 2011;26:1174–81. https://doi.org/10.1111/j.1440-1746.2011.06719.x.
Fedick AM, Shi L, Jalas C, Treff NR, Ekstein J, Kornreich R, et al. Carrier screening of RTEL1 mutations in the Ashkenazi Jewish population. Clin Genet. 2015;88:177–81. https://doi.org/10.1111/cge.12459.
Acknowledgments
We would like to thank the patients and their families for participating in this study. We appreciate the support by the German Academic Exchange Service DAAD (CK, SS, and RS) as well as the Care-for-Rare Foundation.
Authorship Contribution
AZ, TS, BW, RS, and DSS contributed to sample acquisition. AZ, LW, LK, AA, TJ, MH, VM, SW, DK CK, SBS, YT, RS, and DSS contributed to data analysis. DSS designed the study, coordinated research studies, and wrote the manuscript.
Funding
DSS, SBS, and CK are supported by the Leona M. and Harry B. Helmsley Charitable Trust. SBS is also supported by the Wolpow Chair in IBD Research and Treatment and the Translational Research Program (Boston Children’s Hospital).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was approved by the local IRB committee at Sheba Medical Center. Informed written consent was obtained from the parents.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 17 kb)
Rights and permissions
About this article

Cite this article
Ziv, A., Werner, L., Konnikova, L. et al. An RTEL1 Mutation Links to Infantile-Onset Ulcerative Colitis and Severe Immunodeficiency. J Clin Immunol 40, 1010–1019 (2020). https://doi.org/10.1007/s10875-020-00829-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-020-00829-z