
Abstract
Purpose
Pediatric inflammatory bowel disease (IBD) is a heterogeneous disorder caused by multiple factors. Although genetic and immunological analyses are required for a definitive diagnosis, no reports of a comprehensive genetic study of a Japanese population are available.
Methods
In total, 35 Japanese patients <16 years of age suffering from IBD, including 27 patients aged <6 years with very early-onset IBD, were enrolled in this multicenter study. Exome and targeted gene panel sequencing was performed for all patients. Mutations in genes responsible for primary immunodeficiency diseases (PID) and clinical and immunological parameters were evaluated according to disease type.
Results
We identified monogenic mutations in 5 of the 35 patients (14.3 %). We identified compound heterozygous and homozygous splice-site mutations in interleukin-10 receptor A (IL-10RA) in two patients, nonsense mutations in X-linked inhibitor of apoptosis protein (XIAP) in two patients, and a missense mutation in cytochrome b beta chain in one patient. Using assays for protein expression levels, IL-10 signaling, and cytokine production, we confirmed that the mutations resulted in loss of function. For each patient, genotype was significantly associated with clinical findings. We successfully treated a patient with a XIAP mutation by allogeneic cord blood hematopoietic stem cell transplantation, and his symptoms were ameliorated completely.
Conclusions
Targeted sequencing and immunological analysis are useful for screening monogenic disorders and selecting curative therapies in pediatric patients with IBD. The genes responsible for PID are frequently involved in pediatric IBD and play critical roles in normal immune homeostasis in the gastrointestinal tract.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammatory bowel disease (IBD), which includes ulcerative colitis (UC) and Crohn’s disease (CD), is a heterogeneous disorder caused by genetic background, host–microbe interactions, and environmental factors [1]. Pediatric patients with IBD, including patients with very early-onset IBD (VEO-IBD) under the age of 6 years at onset, have clinical features distinct from those of adult patients with IBD, and some show an unclassified pathology distinct from that of classical UC or CD [2]. Recent genome-wide association studies revealed that some patients with IBD have disease-causing mutations or single-nucleotide polymorphisms (SNPs) that increase the risk of IBD in adults [3]. The world-wide VEO-IBD consortium, created in 2014, has contributed to understanding the molecular basis of VEO-IBD and to developing personalized treatments for patients with these rare diseases. It has been reported that genes responsible for primary immunodeficiency diseases (PID), including Wiskott–Aldrich syndrome, dyskeratosis congenita, immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, interleukin-10 (IL-10) or IL-10 receptor (IL-10R) deficiency, X-linked inhibitor of apoptosis (XIAP) deficiency, chronic granulomatous disease (CGD), and common variable immunodeficiency including patients with ICOS gene mutation, are involved in the molecular pathogenesis of pediatric IBD [4–11]. Therefore, we hypothesized that pediatric patients with IBD in a Japanese population might also possess monogenic mutations or a disease-causing genetic background.
In Japanese patients with pediatric IBD, the disease may be caused by a genetic background distinct from that of patients in western countries. The cumulative incidence of pediatric IBD in patients under the age of 16 years [12–14] and the number of patients with a family history of IBD [15, 16] are lower than that in western countries. Mutations in nucleotide-binding oligomerization domain-containing protein 2 (NOD2) and unique SNP profiles that increased the risk of IBD have not been reported in adult Japanese patients with IBD [17, 18]. However, whole-exome sequencing and analysis of its links to clinical, pathological, and immunological evaluations have not been reported for Japanese patients with pediatric IBD.
In this study, we report the results of the first multicenter study for exome and targeted gene panel sequencing of pediatric patients with IBD in Japan. We identified monogenic mutations in 5 of 35 patients and analyzed whether genetic factors were associated with the pathogenesis of pediatric IBD.
Methods
Patients and Clinical Parameters
In total, 35 Japanese patients, from 33 families, under the age of 16 years, including 27 patients under the age of 6 years (VEO-IBD), who suffered from severe and refractory IBD with poor therapeutic response, were enrolled in this study from 2013 to 2015. A list of the 14 institutions that participated in this multicenter study is provided in the Supplementary Appendices. The following details were determined for each patient: genotype, sex, age at onset, stools at onset, histological classification, medical therapy, nutritional support, surgical therapy, and systemic complications.
Exome and Targeted Sequencing Analysis
Genomic DNA was obtained from peripheral blood using the PAXgene Blood DNA Kit (Qiagen, Valencia, CA). Exome analysis was performed using an Ion TargetSeq™ Exome kit and an Ion Proton System (Life Technologies, Carlsbad, CA) according to the manufacturer’s protocol. We selected nonsynonymous or splice-site mutations with a frequency of less than 1 %, as determined by the 1000 Genomes Project, the Exome Sequencing Project, Complete Genomics 46 (CG46), and the Human Genetic Variation Database (HGVD), and with a frequency of less than 5 % based on data obtained in-house for 64 healthy controls to cover all exons and examine whether the patients have mutations in candidates of novel responsible genes for VEO-IBD. To examine whether the patients had mutations in reported genes, we selected 55 genes reported to be responsible for PID and/or IBD for the targeted gene panel analysis, as listed in Table 1.
Mutation Analysis by Sanger Sequencing
All mutations identified by exome analysis were verified by direct Sanger sequencing. Polymerase chain reaction (PCR) using genomic DNA and subsequent Sanger sequencing of IL-10RA, XIAP, and CYBB were performed as previously described [4, 19]. The primers used are listed in Table S1. Total RNA was extracted from peripheral blood mononuclear cells (PBMCs) with an RNA Extraction Kit (Qiagen), and reverse transcription (RT)-PCR was performed with an RNA PCR kit, Ver. 3.0 (Takara, Otsu, Japan). The PCR products were subjected to sequencing using a BigDye Terminator v3.1 Cycle Sequencing kit and an ABI PRISM 310 Genetic Analyzer or a 3500xL Genetic Analyzer (Applied Biosystems, Carlsbad, CA).
Functional Assays for IL-10 Signaling
PBMCs were collected by Ficoll–Paque centrifugation and cryopreserved. For the signal transducer and activator of transcription 3 (STAT3) phosphorylation assays, PBMCs were cultured in RPMI 1640 medium supplemented with fetal bovine serum (FBS) at a density of 1 × 106 cells/ml at 37 °C, 5 % CO2 for 1 h. Cells were stimulated with human recombinant IL-10 (PeproTech Inc., Rocky Hill, NJ) at 37 °C, 5 % CO2 for 30 min, and lysed in sodium dodecyl sulfate sample buffer with 10 % mercaptoethanol. Lysed cells were sonicated twice for 15 s on ice, boiled for 5 min and subjected to electrophoresis, and then transferred to nitrocellulose membranes. Membranes were blocked with PVDF Blocking Reagent for Can Get Signal (Toyobo, Osaka, Japan) for 30 min. Western blot analysis was performed with anti-pSTAT3 rabbit monoclonal antibody (Tyr705) (Cell Signaling Technology, Danvers, MA) which recognized phosphorylated tyrosine residue 705, horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibody, and ECL Western Blotting Detection Reagents (GE Healthcare, Tokyo, Japan). Membranes were stripped with Restore Western Blot Stripping Buffer (Thermo Fisher Scientific Inc., Carlsbad, CA) for 15 min and probed with anti-STAT3 mouse monoclonal antibody and anti-actin monoclonal antibody (Sigma), as described previously [4].
For the cytokine production assay, PBMCs were cultured in RPMI 1640 medium supplemented with FBS at a density of 1 × 105 cells/ml at 37 °C, 5 % CO2 for 1 h. Lipopolysaccharide (LPS) (Sigma, St. Louis, MO) was added to the culture medium at a final concentration of 100 ng/ml in the presence or absence of 100 ng/ml of human recombinant IL-10, and cells were cultured at 37 °C, 5 % CO2 for 6 h. Culture supernatants were collected and proinflammatory cytokine levels were measured with a Human Inflammatory Cytokines Multi-Analyte ELISArray™ kit (Qiagen).
Flow Cytometry Analysis
Flow cytometry analysis of the XIAP protein in PBMCs permeabilized with 0.1 % Triton X-100 was performed using fluorescein isothiocyanate (FITC)-labeled anti-XIAP monoclonal antibody clone 48 (BD Biosciences, Franklin Lakes, NJ) and isotype-matched control IgG, and subjected to flow cytometry analysis.
A superoxide production assay in response to phorbol myristate acetate (PMA) stimulation was performed on peripheral blood from the patients and their parents using dihydrorhodamine (DHR)-123 staining and subjected to flow cytometry analysis. Percentage of superoxide production–positive cells was determined by the ratio of cell numbers in PMA stimulated cells producing superoxide more than upper limit of unstimulated cells. Stimulation index was determined by quantitating mean fluorescent intensity in PMA stimulated cells compared to unstimulated cells.
Serum Cytokine Profiles
Serum was obtained from peripheral blood of patients and healthy controls by centrifugation at 1500×g for 5 min. Serum cytokine levels were measured with a Human Inflammatory Cytokines Multi-Analyte ELISArray™ kit (Qiagen).
Statistical Analysis
The statistical significance of differences between groups was determined using the chi-square test; *p < 0.01 was considered to indicate significance.
Results
Patient Characteristics
Table 2 shows a summary of the genetic analysis and the clinical and pathological parameters for each of the 35 enrolled pediatric patients with IBD. In total, 23 male and 12 female patients were included, and all patients were Japanese, except patient (Pt) 4, who is half Japanese and half Chinese, and Pt 21, who is 7/8 Japanese and 1/8 Brazilian. No consanguineous families were included. Mean ± standard deviation (SD) for the age at onset of study group was 4.50 ± 4.64 years, and 27 patients were under the age of 6 years and 7 patients were under the age of 1 year at onset. Two pairs of siblings with IBD (Pts 31 and 33, 32 and 35) were included. In total, 17, 15, and 3 patients were diagnosed with UC, CD, and unclassified IBD or other types of IBD, respectively. All patients except Pts 1, 2, and 10 were treated with more than two immunosuppressive agents, 19 patients were treated with nutritional support, and 11 patients underwent surgical procedures.
Exome and Targeted Sequencing Analysis
In total, 5 of the 35 patients (14.3 %), including 4 patients with VEO-IBD, were diagnosed with monogenic disorders following exome and subsequent targeted sequencing analysis; 2, 2, and 1 patients were found to have IL10RA, XIAP, and CYBB mutations, respectively (Table 2). All mutations were verified by Sanger sequencing. No other new candidate genes responsible for VEO-IBD were identified in all genes examined by exome sequencing.
IL10RA Deficiency and Functional Assays
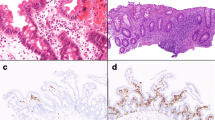
Pt 1 is a girl who showed initial symptoms of fever, vomiting, and bloody diarrhea at the age of 12 days and subsequently showed oral thrush, perianal abscess, and unique anal skin tags (Fig. 1a). Endoscopic examination showed stenosis and patchy erosion in the rectum (Fig. 1b). Pathological examination of biopsy specimens showed nonspecific active inflammation and granulation. Immunological screening data were normal, except elevated serum IL-10 levels (Fig. 5). Exome sequencing and Sanger sequencing revealed compound heterozygous mutations in IL10RA: a missense mutation at c.G350A (p.Arg117His) of maternal origin and a nonsense mutation at c.C634T (p.Arg212X) of paternal origin in the extracellular domain of the IL10RA protein (Fig. 1c). The former was reported to be a rare SNP (rs199989396) with an allele frequency of 1.7/1000 in the 1000 Genomes Project of Asian populations, and predicted impact score for the missense mutation by SIFT and Polyphen2 was 0.1 and 1 (both deleterious), respectively. She was treated with a surgical colostomy at the age of 2 months, due to severe stenosis of the sigmoid colon and rectum. Her symptoms have gradually improved and have no abdominal symptoms recently, except for a prolonged fever following viral infection. We confirmed that Pt 1 had no spontaneous reversion in IL10RA at the age of 6 years by Sanger sequencing analysis in PBMCs. She has been doing well for 6 years and has received no immunosuppressive therapy to date.

Genetic and clinicopathological analysis of patients 1 and 2 with IL10RA deficiency. a Unique manifestation of perianal lesion with abscess and skin tags, and b endoscopic evaluation showed stenosis and patchy erosion in the rectum as indicated by arrows in patient 1. c Mutations of IL10R gene in patient 1 and her parents. d Pathological evaluation of biopsy showed nonspecific active inflammation as indicated by arrows in patient 2. e Genomic (left panel) and cDNA (right panel) analysis of IL10RA mutations in patient 2 and his parents. Normal splicing donor site (black bar) and aberrant splicing donor site (red broken bar)
Pt 2 was a newborn boy from non-consanguineous parents who had initial symptoms of watery diarrhea, recurrent fever, skin candidiasis, failure to thrive, skin rash, and unique perianal lesions. Endoscopic examination showed stenosis, patchy erosion, and multiple ulcers in the rectum. Pathological examination of biopsy specimens showed nonspecific active inflammation (Fig. 1d). Immunological data were normal, with the exception of elevated serum IL-10 levels (Fig. 5). Exome sequencing revealed a homozygous mutation in IL10RA: c.G537A at the carboxy-terminal end of exon 4, which was of both paternal and maternal origin (Fig. 1e, left panel). The mutation was not reported in the 1000 Genomes Project or CG46; however, the allele frequency was reported to be 1.9/1000 in the HGVD. RT-PCR analysis confirmed that the mutation resulted in a splicing error and an 18-base pair deletion (c.520–537delGTGCCAGGAAACTTCACG, p.Val174_Thr179del) at the carboxy-terminal end of exon 4 in the extracellular domain of the IL10RA protein (Fig. 1e, right panel). He was treated with a colostomy at the age of 6 months, due to severe stenosis of the rectum. He died of multiple organ failure due to severe hypercytokinemia following infection with the influenza virus at the age of 11 months.
To confirm that these mutations in Pt 1 and Pt 2 were functionally significant, we examined STAT3 phosphorylation in PBMCs in response to recombinant human IL-10 stimulation. In contrast to the induction of STAT3 phosphorylation following IL-10 stimulation in normal controls, STAT3 phosphorylation was not observed in either patient (Fig. 2). We next examined cytokine production in response to LPS stimulation of PBMCs in the presence or absence of IL-10. Production of the proinflammatory cytokines IL-1β, IL-6, IL-8, and tumor necrosis factor-alpha (TNF-α), induced by LPS stimulation, was varied among Pts 1, 2, and normal controls. In contrast to normal controls, IL-10 did not have an inhibitory effect on proinflammatory cytokine production in PBMCs from either patient with an IL10RA deficiency. Minimal inhibitory effects of IL-10 on IL-1β and TNF-α secretion were observed in Pt 1 (Fig. S1).

Impaired STAT3 phosphorylation of patients 1 and 2 with IL10RA deficiency. Impaired tyrosine phosphorylation of STAT3 (Tyr705) in response to IL-10 stimulation in PBMCs. pSTAT3 tyrosine phosphorylated STAT3, M molecular weight marker, kDa kilodalton
XIAP Deficiency
Pt 3 is a boy who suffered from fever, bloody diarrhea, and abdominal pain at the age of 6 years. Endoscopic examination showed stenosis and ulcer formation in the sigmoid colon (Fig. 3a). Pathological examination of biopsy specimens showed nonspecific active inflammation (Fig. 3b). Whole-exome sequencing revealed a nonsense mutation in XIAP, c.C1141T (p.Arg381X), on the X chromosome, and we confirmed the mutation by Sanger sequencing (Fig. 3d). Blood samples from the parents were not available. Flow cytometry analysis of PBMCs from the patient confirmed that XIAP protein expression was significantly reduced compared to that in normal controls (Fig. 3e). He was treated with 5-aminosalicylic acid, prednisolone, and azathioprine, followed by infliximab, tacrolimus, and prednisolone. He was also treated with a surgical sigmoid colectomy at the age of 8 years; however, his symptoms were not sufficiently controlled. He received allogeneic cord blood hematopoietic stem cell transplantation (HSCT) following reduced intensity conditioning consisting of fludarabine, cyclophosphamide, and low-dose total body irradiation. He achieved full-donor chimerism in the bone marrow. His symptoms were completely ameliorated, the endoscopic findings at the site shown in Fig. 3a were improved (Fig. 3c), and XIAP protein expression became normal following allogeneic cord blood HSCT (Fig. 3e).

Genetic and clinicopathological analysis of patients 3 and 4 with XIAP deficiency. a Endoscopic evaluation at onset showed stenosis and ulcer formation in the sigmoid colon, b pathological evaluation of biopsy specimen at onset showed nonspecific active inflammation as indicated by arrows, and c improved endoscopic finding after allogeneic cord blood HSCT in patient 3. d XIAP mutation and e flow cytometric demonstration of XIAP protein expression levels before and after allogeneic cord blood HSCT in patient 3. f Endoscopic evaluation at onset showed multiple erosions and aphthoid lesions as indicated by arrows in the ileum, g mutations of XIAP gene in the patient and his mother, and h flow cytometric demonstration of reduced XIAP protein expression levels in patient 4. In Fig. 3e, h, red histograms showed isotype control and Y-axis denoted percent of maximum
Pt 4 is a boy who suffered from anorexia, weight loss, and general malaise at the age of 16 years, and computed tomography showed multiple swellings of the intestinal lymph nodes. Endoscopic examination showed multiple erosions and aphthoid lesions in the ileum (Fig. 3f). Pathological examination of biopsy specimens showed apoptosis in the mucosal layer and granulation. Exome sequencing revealed a nonsense mutation in XIAP, c.C847T (p.Gln283X); his mother is an asymptomatic carrier (Fig. 3g). No blood sample was available from his father. Flow cytometry analysis of PBMCs from the patient confirmed that XIAP protein expression was significantly reduced compared to that in normal controls (Fig. 3h). He has been treated with adalimumab and 5-aminosalicylic acid, and his symptoms are partly controlled to date. Allogeneic HSCT is under consideration as a curative therapy for IBD.
CYBB Deficiency
Pt 5 is a boy who suffered from bloody diarrhea at the age of 6 years. However, he has not suffered from severe bacterial and fungal infections, as are typically observed in patients with CGD. Endoscopic examination showed edema and multiple bleeding in the rectum (Fig. 4a), and pathological examination showed poor basal plasmacytosis but increased infiltration of neutrophils and pigmented vacuolated macrophages at disease onset (Fig. 4b). He then developed perianal abscess and ulcers, and formation of granuloma in the jejunum at the age of 14 years. Exome sequencing revealed a missense mutation in CYBB, c.T615A (p.Phe205Leu); his mother is an asymptomatic carrier (Fig. 4c). Predict impact score for the missense mutation by SIFT and Polyphen2 was 0 and 0 (both deleterious), respectively. Pt 5 showed significantly reduced (percentage of superoxide production–positive cells was 75 % and stimulation index was 4.17), but not total loss of superoxide production compared to that of his mother (94 % and 31.0) and of his father (93 % and 51.9) (Fig. 4d). His mother had minimally detectable negative, but not bimodal, peak of reduced superoxide production in the DHR analysis probably because of X-chromosome inactivation or skewing to normal allele. He was successfully treated with steroids and azathioprine. Based on the genetic diagnosis, we tapered azathioprine and added prophylactic use of oral sulfamethoxazole and trimethoprim antibiotics as a standard therapy for CGD.

Genetic and clinicopathological analysis of patient 5 with CYBB mutation. a Endoscopic evaluation showed edema and multiple bleeding in the rectum, and b pathological evaluation of biopsy specimen at onset showed increased infiltration of neutrophils (left panel) and pigmented vacuolated macrophages (right panel) as indicated by arrows in patient 5. c Mutations of CYBB gene in patient 5 and his parents. d DHR-123 histograms in unstimulated (blue, left panels) and PMA-stimulated (red, right panels) showed significantly reduced superoxide production in patient 5 compared to his parents
Serum Cytokine Profiles
It has been reported that serum IL-18 levels are significantly elevated in patients with XIAP deficiency who also showed hemophagocytic lymphohistiocytosis [20]. To identify new immunological parameters in patients with pediatric IBD, we measured serum proinflammatory cytokine levels. As shown in Fig. 5a, IL-8 levels were significantly elevated in both of the patients with XIAP deficiency who had IBD (785 ± 99 pg/ml) compared to those in healthy controls (mean = 12 ± 5.0 pg/ml) (p < 0.01). We could not examine immune response to bacterial muramyl dipeptide (MDP) in patients with XIAP deficiency due to limited blood samples. Serum IL-10 levels were significantly elevated in both patients with IL-10RA deficiency (105 ± 21 pg/ml) compared to those in healthy controls (mean = 10 ± 5.0 pg/ml) (p < 0.01), which was caused by compensation for impaired IL-10 signaling (Fig. 5b). Variable IL-8 and IL-10 levels were observed in other patients diagnosed with UC or CD without monogenic mutations. Levels of other cytokines did not differ significantly between Pts 1 and 5 and healthy controls.

Cytokine profiles in serum collected from patients with monogenic disorders. a IL-8 levels were significantly elevated in both of the patients with XIAP deficiency, and b IL-10 levels were significantly elevated in both of the patients with IL10RA deficiency, compared to those in healthy controls. XIAP deficiency (blue circles), IL10RA deficiency (red squares), CYBB deficiency (yellow triangle), and other patients diagnosed with UC or CD, and healthy controls (HC) (black Xs)
Variants of Genes Responsible for PID
Next, we examined whether the allele frequency of any SNP was significantly associated with an increased risk of IBD in 30 enrolled patients without monogenic mutations compared with in-house Japanese healthy controls. A SNP of TNFSF15 (rs4246905) only tended to increase the risk of VEO-IBD, although it was not statistically significant (Table S2).
Pt 28 had a heterozygous nonsense mutation in IL10RA, c.C634T (p.Arg212X) (Fig. S2a). A functional assay for proinflammatory cytokine production in response to LPS revealed that IL-10 had normal inhibitory effect on TNF-α secretion in Pt 28 similarly to healthy controls (Fig. S2b). Pt 9 had a heterozygous missense mutation in NCF4, c.G478A (p.V160M) (Fig. S2c). However, Pt 9 also possessed a wild-type allele of NCF4 and showed normal levels of superoxide production compared with healthy control. Therefore, we concluded that these heterozygous mutations were not disease-causing and were insufficient for the molecular diagnosis of the autosomal recessive IL10RA deficiency or the autosomal recessive form of CGD.
Discussion
In the present study, using exome sequencing, we identified 5 patients with monogenic disorders (IL10RA, XIAP, or CYBB deficiency), including 4 patients with VEO-IBD, among 35 Japanese patients with pediatric IBD, and confirmed that these mutations were functionally significant for the molecular pathogenesis of each patient. The frequency was concordant with a previous result that 4 out of 20 patients with VEO-IBD had mutations in some of 40 responsible genes, as determined by targeted gene panel sequencing [9].
IL-10 signaling plays an indispensable role in maintaining immune homeostasis and mediates a range of anti-inflammatory activities. IL-10 binds to a tetrameric receptor composed of two IL10RA and two IL10RB molecules, and IL-10-mediated signaling inhibits the production of proinflammatory cytokines [21]. Mutations in IL-10, IL10RA, and IL10RB have been reported in patients with VEO-IBD. The characteristic phenotypes are very early-onset, and at less than 3 months of age, some patients show perianal lesions with unique skin tags, fissures and abscess, eczematous skin rash, and susceptibility to infections. Endoscopic examination showed erosion and ulcers, as well as pathologically nonspecific inflammation, granuloma, infiltration of immune cells, and crypt abscess formation; these same symptoms were observed in our patients. Immunosuppressive therapy was not fully effective, and allogeneic HSCT, a potentially curative therapy, was also considered [4, 22–25]. Recently, Neven et al. reported an unexpected complication in five patients with mutations in IL10RA or IL10RB, who developed similar phenotypes of diffuse large B cell lymphoma caused by somatic second-hit mutations in tumor cells and loss of host immune surveillance against tumor cells [26].
The clinical severity differed between our two patients with IL10RA deficiency; Pt 1 improved spontaneously, whereas Pt 2 died of hypercytokinemia following influenza virus infection at the age of 11 months. Lu et al. reported a Chinese patient with the same homozygous IL10RA mutation as observed in Pt 2, who also showed severe clinical symptoms [27]. This mutated allele may be relatively frequent in east Asian countries. We confirmed that Pt 1 had no spontaneous reversion in IL10RA at the age of 6 years. Therefore, we speculated that the difference in clinical severity might have been caused by a partial inhibitory effect of IL-10 on production of proinflammatory cytokines, IL-1β and TNF-α, in response to LPS in Pt 1; however, this effect was not observed in Pt 2 (Fig. S1). Another possibility is that other compensatory mechanisms for IL-10 signaling defects may be involved in regulating immune homeostasis in Pt 1.
Mutation of XIAP is responsible for X-linked lymphoproliferative syndrome type II, and XIAP mediates signaling between NOD2 and nuclear factor-kappa B (NF-κB). NOD2 is stimulated by bacterial MDP and induces the production of IL-8 and monocyte chemotactic protein-1 via NF-κB. Therefore, loss of XIAP function results in impairment of the immune response via NF-κB and induces chronic inflammation [28]. Mutations in XIAP induce clinical manifestations similar to CD in the gastrointestinal tract [28–35]. Allogeneic HSCT is a curative therapy for the disease, and Pt 3, who received HSCT following reduced intensity conditioning, achieved full-donor chimerism in the bone marrow and improvement of gastrointestinal symptoms, as shown in Fig. 3c. Flow cytometric analysis for XIAP protein expression in PBMCs was useful for screening the patients with XIAP deficiency and for confirming the diagnosis and the improvement in XIAP protein expression levels after HSCT.
X-linked CGD is caused by mutations in CYBB, which encodes glycoprotein 91-phox protein as a component of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [36]. Loss of superoxide production in CGD patients induces IL-1β production and chronic inflammation in the gastrointestinal tract, which is recognized as CGD colitis [37]. Exome sequencing identified a missense mutation in CYBB that caused significant, but not complete, loss of NADPH oxidase activity in Pt 5. Pathological examination of CGD colitis shows infiltration of eosinophils and neutrophils, as well as pigmentation and inclusion bodies in macrophages [38], as was observed at disease onset in Pt 5. Granulation was observed at 8 years following disease onset in Pt 5. Therefore, a subtype of CGD with partial NADPH oxidase activity is included in the spectrum of pediatric IBD; these patients show a lack of susceptibility to bacterial and fungal infections, which is atypical for severe forms of CGD [39].
We identified heterozygous nonsense mutations of IL10RA in Pt 28, although this mutation was not functionally significant, as determined by the cytokine production assay. Kelsen et al. recently identified variants in PID genes that were more frequently observed in VEO-IBD patients than in healthy controls, including heterozygous mutations in IL10RA. In this paper, IL-10 was less efficient at reducing TNFα production from LPS-stimulated macrophages. Therefore, further analyses will be required to determine the exact effects of the heterozygous variant on the molecular pathogenesis of autosomal recessive forms of VEO-IBD [40].
The contribution of genetic background remains unclear in more than 80 % of the patients enrolled in this study. SNPs associated with an increased risk of pediatric IBD were not observed in the Japanese population enrolled in our study. Whole-genome sequencing may identify genetic factors in introns and transcriptional regulatory regions. In addition, we should consider whether a dysregulated immune response to microbiota and dysbiosis, as determined by metagenomic analysis, is involved in the pathogenesis of pediatric IBD.
An accurate molecular diagnosis is crucial to identify pediatric IBD patients with monogenic PID, such as IL-10 signaling defects, XIAP deficiency, CGD, IPEX syndrome, Wiskott–Aldrich syndrome, and common variable immunodeficiency. Our study suggests that comprehensive whole-exome sequencing is necessary for identifying or excluding monogenic PID and for determining the appropriate therapy, including the indication for allogeneic HSCT, in patients with refractory IBD and VEO-IBD.
Conclusions
Using whole-exome sequencing, we identified underlying PID gene mutations in pediatric patients with IBD in a Japanese population. Identifying links between genetic mutations and clinicopathological and immunological parameters helped us in understanding the pathogenesis and in selecting appropriate therapies for patients with IBD. Determining whether unidentified genes, a dysregulated immune response to intestinal microbiota, or dysbiosis is involved in the pathogenesis of these diseases may extend our understanding of normal immune regulation and host–microbe interactions in the gastrointestinal tract.
References
Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–78.
Benchimol EI, Mack DR, Nguyen GC, Snapper SB, Li W, Mojaverian N, et al. Incidence, outcome, and health services burden of very early onset inflammatory bowel disease. Gastroenterology. 2014;147:803–13.
Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24.
Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–45.
Glocker EO, Frede N, Perro M, Sebire N, Elawad M, Shah N, et al. Infant colitis--it’s in the genes. Lancet. 2010;376:1272.
Blaydon DC, Biancheri P, Di WL, Plaqnol V, Cabral RM, Brooke MA, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. 2011;365:1502–8.
Uhlig HH. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut. 2013;62:1795–805.
Cutler DJ, Zwick ME, Okou DT, Prahalad S, Walters T, Guthery SL, et al. Dissecting allele architecture of early onset IBD using high-density genotyping. PLoS ONE. 2015;10:e0128074.
Kammermeier J, Drury S, James CT, Dziubak R, Ocaka L, Elawad M, et al. Targeted gene panel sequencing in children with very early onset inflammatory bowel disease-evaluation and prospective analysis. J Med Genet. 2014;51:748–55.
Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. 2014;147:990–1007.
Christodoulou K, Wiskin AE, Gibson J, Tapper W, Willis C, Afzal NA, et al. Next generation exome sequencing of pediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut. 2013;62:977–84.
Ishige T, Tomomasa T, Takebayashi T, Asakura K, Watanabe M, Suzuki T, et al. Inflammatory bowel disease in children: epidemiological analysis of the nationwide IBD registry in Japan. J Gastroenterol. 2010;45:911–7.
Maisawa S, Sasaki M, Ida S, Uchida K, Kagimoto S, Shimizu T, et al. Characteristics of inflammatory bowel disease with an onset before eight years of age: a multicenter epidemiological survey in Japan. J Gastroenterol Hepatol. 2013;28:499–504.
Asakura K, Nishiwaki Y, Inoue N, Hibi T, Watanabe M, Takebayashi T. Prevalence of ulcerative colitis and Crohn’s disease in Japan. J Gastroenterol. 2009;44:659–65.
Kuwahara E, Asakura K, Nishiwaki Y, Inoue N, Watanabe M, Hibi T, et al. Effects of family history on inflammatory bowel disease characteristics in Japanese patients. J Gastroenterol. 2012;47:961–8.
Russell RK, Satsangi J. IBD: a family affair. Best Pract Res Clin Gastroenterol. 2004;18:525–39.
Inoue N, Tamura K, Kinouchi Y, Fukuda Y, Takahashi S, Ogura Y, et al. Lack of common NOD2 variants in Japanese patients with Crohn’s disease. Gastroenterology. 2002;123:86–91.
Yamazaki K, Umeno J, Takahashi A, Hirano A, Johnson TA, Kumasaka N, et al. A genome-wide association study identifies 2 susceptibility loci for Crohn’s disease in a Japanese population. Gastroenterology. 2013;144:781–8.
Tsai MF, Lin YJ, Cheng YC, Lee KH, Huang CC, Chen YT, et al. Primer Z: streamlined primer design for promoters, exons and human SNPs. Nucleic Acids Res. 2007;35:W63–5.
Wada T, Kanegane H, Ohta K, Katoh F, Imamura T, Nakazawa Y, et al. Sustained elevation of serum interleukin-18 and its association with hemophagocytic lymphohistiocytosis in XIAP deficiency. Cytokine. 2014;65:74–8.
Donnelly RP, Dickensheets H, Finbloom DS. The interleukin-10 signal transduction pathway and regulation of gene expression in mononuclear phagocytes. J Interferon Cytokine Res. 1999;19:563–73.
Begue B, Verdier J, Rieux-Laucat F, Goulet O, Morali A, Canioni D, et al. Defective IL-10 signaling defining a subgroup of patients with inflammatory bowel disease. Am J Gastroenterol. 2011;106:1544–55.
Shim JO, Hwang S, Yang HR, Moon JS, Chang JY, Ko JS, et al. Interleukin-10 receptor mutations in children with neonatal-onset Crohn’s disease and intractable ulcerating enterocolitis. Eur J Gastroenterol Hepatol. 2013;25:1235–40.
Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143:347–55.
Pigneur B, Escher J, Elawad M, Lima R, Buderus S, Kierkus J, et al. Phenotypic characterization of very early-onset IBD due to mutations in the IL-10, IL-10 receptor alpha or beta gene: a survey of the genius working group. Inflamm Bowel Dis. 2013;19:2820–8.
Neven B, Mamessier E, Bruneau J, Kaltenbach S, Kotlarz D, Suarez F, et al. A Mendelian predisposition to B-cell lymphoma caused by IL-10R deficiency. Blood. 2013;122:3713–22.
Lu D, Xu Y, Chen Y, Zeng P, Chen H, Zeng H. Interleukin-10 receptor mutations in children with neonatal onset inflammatory bowel disease: genetic diagnosis and pathogenesis. Chin J Pediatr. 2015;53:348–54.
Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci U S A. 2009;106:14524–9.
Latour S, Aguilar C. XIAP deficiency syndrome in humans. Semin Cell Dev Biol. 2015;39:115–23.
Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–62.
Aguilar C, Lenoir C, Lambert N, Bèque B, Brousse N, Canioni D, et al. Characterization of Crohn disease in X-linked inhibitor of apoptosis-deficient male patients and female symptomatic carriers. J Allergy Clin Immunol. 2014;134:1131–41.
Zeissig Y, Petersen BS, Milutinovic S, Bosse E, Mayr G, Peuker K, et al. XIAP variants in male Crohn’s disease. Gut. 2015;64:66–76.
Yang X, Kanegane H, Nishida N, Imamura T, Hamamoto K, Miyashita R, et al. Clinical and genetic characteristics of XIAP deficiency in Japan. J Clin Immunol. 2012;32:411–20.
Speckmann C, Lehmberg K, Albert MH, Damgaard RB, Fretsch M, Gyrd-Hansen M, et al. X-linked inhibitor of apoptosis (XIAP) deficiency: the spectrum of presenting manifestations beyond hemophagocytic lymphohistiocytosis. Clin Immunol. 2013;149:133–41.
Speckmann C, Ehl S. XIAP deficiency is a Mendelian cause of late-onset IBD. Gut. 2014;63:1031–2.
Royer-Pokora B, Kunkel LM, Monaco AP, Goff SC, Newburger PE, Baehner RL, et al. Cloning the gene for an inherited human disorder-chronic granulomatous disease-on the basis of its chromosomal location. Nature. 1986;322:32–8.
van de Veerdonk FL, Dinarello CA. Deficient autophagy unravels the ROS paradox in chronic granulomatous disease. Autophagy. 2014;10:1141–2.
Dhillon SS, Fattouh R, Elkadri A, Xu W, Murchie R, Walters T, et al. Variants in nicotinamide adenine dinucleotide phosphate oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology. 2014;147:680–9.
Schappi MG, Klein NJ, Lindley KJ, Rampling D, Smith VV, Goldblatt D, et al. The nature of colitis in chronic granulomatous disease. J Pediatr Gastroenterol Nutr. 2003;36:623–31.
Kelsen JR, Dawany N, Moran CJ, Petersen BS, Sarmady M, Sasson A, et al. Exome sequencing analysis reveals variants in primary immunodeficiency genes in patients with very early onset inflammatory bowel disease. Gastroenterology. 2015;149:1415–24.
Acknowledgments
The authors thank all of our patients and their parents for participating in this study. We thank Drs. H Kumagai, M Sasaki, T Saitoh, T Nanbu, N Abe, and J Suzuki for providing patients’ samples and clinical data. We also thank M Uchiyama, N Ichinoi, H Kudoh, and R Satoh for technical assistance.
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (26461562), a grant from the Japanese Ministry of Health, Labour and Welfare (H26-037) and a grant from the Japan Agency for Medical Research and Development (J150001095) to YS.
Authorship Contributions
TS and YS designed and performed the research, analyzed data, and wrote the manuscript. AH and HK performed the research. HK, TK, TI, YN, and DA provided patients’ samples and clinical data. MT performed pathological evaluations. AK and SK provided scientific advice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors declare that they have no conflict of interest and have nothing to disclose.
Ethical Approval
This study was approved by the Ethics Committee of the Tohoku University Graduate School of Medicine on 30th September 2013. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendment or comparable ethical standards.
Informed Consent
Written informed consent was obtained from all individual participants or their guardians included in this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 277 kb)
Rights and permissions
About this article

Cite this article
Suzuki, T., Sasahara, Y., Kikuchi, A. et al. Targeted Sequencing and Immunological Analysis Reveal the Involvement of Primary Immunodeficiency Genes in Pediatric IBD: a Japanese Multicenter Study. J Clin Immunol 37, 67–79 (2017). https://doi.org/10.1007/s10875-016-0339-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-016-0339-5