
Abstract
Dyskeratosis congenita (DC) is an inherited bone marrow failure and cancer predisposition syndrome caused by aberrant telomere biology. The classic triad of dysplastic nails, abnormal skin pigmentation, and oral leukoplakia is diagnostic of DC, but substantial clinical heterogeneity exists; the clinically severe variant Hoyeraal Hreidarsson syndrome (HH) also includes cerebellar hypoplasia, severe immunodeficiency, enteropathy, and intrauterine growth retardation. Germline mutations in telomere biology genes account for approximately one-half of known DC families. Using exome sequencing, we identified mutations in RTEL1, a helicase with critical telomeric functions, in two families with HH. In the first family, two siblings with HH and very short telomeres inherited a premature stop codon from their mother who has short telomeres. The proband from the second family has HH and inherited a premature stop codon in RTEL1 from his father and a missense mutation from his mother, who also has short telomeres. In addition, inheritance of only the missense mutation led to very short telomeres in the proband’s brother. Targeted sequencing identified a different RTEL1 missense mutation in one additional DC proband who has bone marrow failure and short telomeres. Both missense mutations affect the helicase domain of RTEL1, and three in silico prediction algorithms suggest that they are likely deleterious. The nonsense mutations both cause truncation of the RTEL1 protein, resulting in loss of the PIP box; this may abrogate an important protein–protein interaction. These findings implicate a new telomere biology gene, RTEL1, in the etiology of DC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dyskeratosis congenita (DC) is an inherited bone marrow failure syndrome caused by germline defects in telomere biology genes (Savage and Bertuch 2010). The classic triad of dysplastic nails, skin pigmentation, and oral leukoplakia is diagnostic, but substantial clinical heterogeneity exists; patients may also have pulmonary fibrosis, liver disease, esophageal, urethral, or lacrimal duct stenosis, developmental delay, and/or other complications. Individuals with DC are at very high risk of bone marrow failure (BMF), myelodysplastic syndrome, and cancer (Alter et al. 2010).
Hoyeraal Hreidarsson syndrome (HH) is a clinically severe variant of DC. In addition to features of DC, patients with HH have cerebellar hypoplasia, severe immunodeficiency, enteropathy, and intrauterine growth retardation. The clinical consequences of DC manifest at variable ages and in different patterns, even within the same family. Independent of the classic triad, lymphocyte telomere lengths less than the first percentile for age are diagnostic of DC (Alter et al. 2012).
The inheritance of DC is variable, with germline mutations reported in X-linked ([XL] DKC1), autosomal dominant ([AD] TERC, TERT, or TINF2), and autosomal recessive ([AR] TERT, CTC1, NOP10, NHP2, or WRAP53) patterns (Nelson and Bertuch 2012; Walne et al. 2012) accounting for approximately one-half of classic DC cases. The reported AR mutations in TERT are homozygous missense mutations, whereas WRAP53 and CTC1 AR inheritance is due to compound heterozygous mutations. In addition to the different modes of DC inheritance, genetic anticipation has been reported in TERC, TERT, and TINF2 pedigrees (Armanios et al. 2005; Savage and Bertuch 2010; Vulliamy and Dokal 2008); this phenomenon is marked by increasing severity in the clinical phenotype and shorter telomeres with each successive generation.
In order to advance understanding of the genetic etiology of DC and related telomere biology disorders, we conducted exome sequencing on two families with DC. Novel variants in biologically and genetically plausible genes were evaluated by targeted sequencing. This led to the discovery of germline mutations in the regulator of telomere elongation helicase 1, RTEL1, as a new cause of both AR and AD DC.
Results
We performed whole exome sequencing (WES) on two families with children affected by the clinically severe DC subtype, Hoyeraal Hreidarsson syndrome (HH) (Table 1). The probands were clinically tested and negative for mutations in DKC1, TERC, TERT, TINF2, NOP10, NHP2, and WRAP53. We specifically assessed exome sequencing coverage of the known DC genes, including CTC1, mutations in which were recently discovered to cause DC (Keller et al. 2012). Variants identified in WES were evaluated in AD, AR, and XL inheritance models (“Materials and methods”, Online Resource 2). WES variants of interest were validated to rule out false positive findings using an alternative sequencing technology (“Materials and methods”). In both families, the most biologically plausible gene containing novel or extremely rare variants was regulation of telomere elongation helicase 1 (RTEL1, OMIM #608833). RTEL1 is an evolutionarily conserved DNA helicase that is important in telomeric replication and stability (Uringa et al. 2012). Depletion of murine Rtel1 from mouse embryonic stem (ES) cells results in loss of telomeric sequence and chromosomal abnormalities upon differentiation (Ding et al. 2004).
Family NCI-164 includes two brothers with HH, a healthy mother with short telomeres, and a healthy father with normal telomeres (Table 1; Fig. 1). An RTEL1 variant, g.20:62324600C>T (p.Arg1010X, NM_032957), resulting in a premature stop codon in exon 30 was present in both the affected siblings and their mother, indicating AD inheritance (Table 2; Fig. 2). This mutation has been reported twice in the Exome Sequencing Project (ESP) database with a minor allele frequency (MAF) of 0.015 %, but is not present in 1,000 Genomes, Kaviar, or dbSNP, implying that the prevalence of this mutation in the general population is much lower than 0.015 %. This truncation results in the loss of the PCNA (proliferating cell nuclear antigen) interacting protein (PIP) motif (Fig. 2). It is likely that genetic anticipation contributes to the clinical status of the children, since the mother is currently healthy. This pattern of genetic anticipation, including the presence of clinically silent mutation carriers, has been seen previously in DC (Armanios et al. 2005; Savage and Bertuch 2010; Vulliamy et al. 2004), and has been used to inform DC-related gene discovery (Savage et al. 2008).

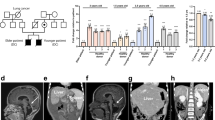
Lymphocyte telomere lengths in families with RTEL1 mutations. Lymphocyte telomere lengths for DC or DC-like patients and unaffected relatives were measured by flow cytometry with fluorescent in situ hybridization (Alter et al. 2012). In family NCI-238, telomere lengths for the two siblings are shown; however, genotype data were unavailable for the sister and parents. In both the pedigrees and the telomere length graphs, the proband is indicated with an arrow

Schematic of RTEL1 genomic structure and conserved domains. a RTEL1 is comprised of 35 exons spanning nearly 40,000 bases of genomic sequence on chromosome 20q13.33. Exons 20 through 35 have been expanded (blue boxes). In a and b, the positions of the mutations in DC patients are labeled relative to transcript NP_116575. b Comparison of amino acid conservation of RTEL1 homologs ("Materials and methods"). Higher percent identity at a given amino acid position is indicated by a deeper purple color
Family NCI-180 includes a male with HH whose mother and brother, while currently healthy, both have very short telomeres (Table 1; Fig. 1). The healthy father has normal telomeres. The proband, brother, and mother share two novel variants in RTEL1: g.20:62319931G>T (p.Glu615Asp, NM_032957), a likely deleterious mutation in a highly conserved residue in a helicase domain, and g.20:62322230A>C (p.Gln853Pro, NM_032957), which is likely benign (Table 2; Fig. 2). In addition, the proband and his father are heterozygous for a mutation g.20:62324564C>T (p.Arg998X, NM_032957) in exon 30 that results in deletion of the PIP motif (Fig. 2). The presence of two likely deleterious mutations in the proband and the correspondingly severe phenotypes indicates compound heterozygous AR inheritance (Fig. 1). The brother’s inheritance of only the missense mutation has so far resulted in no obvious clinical manifestations of disease. However, his telomeres are significantly below the first percentile for his age and he has hypocellular bone marrow with a cytogenetic clone. Consequently, he will be monitored for development of DC-related complications. Mutations in a DC-associated gene causing both AD and AR (or compound heterozygous) inheritance of DC is not unprecedented. For example, individuals with AD TERT mutations may not have medical problems until middle age, whereas AR TERT mutations can cause HH with manifestations in infancy (Marrone et al. 2007; Nelson and Bertuch 2012).
We then performed targeted sequencing of all exons in RTEL1 in 10 DC and 14 DC-like families who are negative for mutations in known DC-associated genes (“Materials and methods”, Online Resources 3 and 4). DC-like individuals have short telomeres and features similar to, but not diagnostic of DC (Savage and Bertuch 2010). We identified likely deleterious mutations in RTEL1 in one additional, DC-like proband.
A heterozygous mutation g.20:62320468G>A (p.Ala645Thr, NM_032957) in exon 22, which encodes part of the Helicase_C_2 region, was identified in a DC-like patient, NCI-238-1, who has short telomeres and BMF (Tables 1, 2; Fig. 1). This residue is highly conserved, and this mutation is predicted to be deleterious. His family members were not available for sequencing; however, his sister has nail dysplasia and her telomeres are at the first percentile for age.
Targeted sequencing of RTEL1 also identified common single nucleotide polymorphisms (SNPs) in patients and their relatives. Three additional nonsynonymous SNPs were present in three unrelated patients, but are either present in other databases (MAF > 1 %), predicted to be benign, or not in an evolutionarily conserved region (Online Resource 3).
Discussion
RTEL1 encodes an essential, evolutionarily conserved DNA helicase that is important for DNA replication and telomere elongation. By employing WES followed by targeted sequencing, we discovered mutations in RTEL1 in three DC families, indicating that dysfunctional RTEL1 is a biologically plausible cause of DC, a disorder of aberrant telomere biology. Clinical data show that RTEL1 mutations may be associated with very severe clinical symptoms; this is exemplified by the fact that two of the three probands with RTEL1 mutations have been diagnosed with HH, the clinically severe variant of DC. As seen in families with TINF2 and TERT mutations, genetic anticipation appears to be evident. However, we cannot rule out the presence of other disease-modifying factors. Correlations between clinical manifestations of DC and genetic mutations are complicated by the presence of disease heterogeneity, incomplete penetrance, and genetic anticipation. However, telomere length is an accurate diagnostic indicator (Alter et al. 2012), and we have found that in eight of nine patients, RTEL1 mutations correlate with telomeres at or below the first percentile. The only exception is the father in family NCI-180, whose telomeres are near the tenth percentile for his age.
RTEL1 is an essential protein in mice, and depletion from murine ES cells results in loss of telomeric sequence and chromosomal abnormalities upon differentiation (Ding et al. 2004), indicating that it is required to maintain both telomeric and genomic stability. In mice, RTEL1 is widely expressed in proliferating cells, including lymphocytes (Ding et al. 2004). These data support the model that human RTEL1 influences telomere length and that perturbation of RTEL1 results in a disorder marked by bone marrow failure and elevated risk of leukemia. More recently, mouse RTEL1 has been shown to disassemble T-loops, thereby promoting telomeric replication (Vannier et al. 2012). In the absence of functional RTEL1, T-loops are excised by the SLX4 nuclease complex, resulting in dramatic changes in telomere length. The discovery of RTEL1 dysfunction in DC marks a potentially novel mechanism of disease-associated telomere shortening; other DC genes mediate telomerase activity, localization, or biogenesis, while RTEL1-associated telomere length change appears to be telomerase-independent.
The likely deleterious mutations discovered in our DC families may have significant effects on domains of RTEL1 that are critical for proper protein function. The point mutations are located in conserved helicase domains. The truncations result in loss of the C-terminus, which seems unlikely to affect the helicase activity. A putative PIP motif in this region may mediate interactions between RTEL1 and PCNA, a sliding clamp that functions in DNA replication and repair and which localizes to stalled replication forks in telomeric sequence (Verdun and Karlseder 2006). RTEL1 may interact with PCNA to facilitate replication of telomeres, or to mediate T-loop stability (Vannier et al. 2012; Wang et al. 2004). However, an interaction between RTEL1 and PCNA remains speculative, and there may be important domains other than the PIP motif in the truncated region of RTEL1. Ongoing functional characterization of the mutations reported here will elucidate the impact of these mutations on telomere maintenance and human disease.
Mutations in DNA helicases are associated with other human disorders, including Bloom’s syndrome, Werner’s syndrome, Rothmund–Thomson syndrome, and Fanconi anemia, all of which affect genomic stability and result in predisposition to cancer (Ellis et al. 1995; Kitao et al. 1999; Levitus et al. 2005; Yu et al. 1996). The role of the RTEL1 helicase in human disease is just now being explored, but there is a clear precedent for dysfunctional DNA helicases leading to diseases of chromosomal instability and increased cancer risk. Recently, non-coding SNPs in RTEL1 have been found to be associated with susceptibility to high-grade glioma (Egan et al. 2011; Shete et al. 2009; Wrensch et al. 2009). DC is a cancer predisposition syndrome; affected individuals are at an 11-fold increased risk of cancer compared with the general population. Notably, the risk of tongue squamous cell cancer is increased by 1,000-fold and the risk of AML is increased by 195-fold (Alter et al. 2009). These findings suggest that the RTEL1 locus may influence cancer susceptibility, possibly via alterations in telomere biology. Similarly, variations in the TERT-CLPTM1L locus have been implicated in modulating risk of a wide variety of cancers (Rafnar et al. 2009). TERT encodes the reverse transcriptase component of the enzyme telomerase and germline mutations can cause DC and related telomere biology disorders. Taken together, these data suggest that preserving genomic integrity through appropriate telomere maintenance is critical for preventing oncogenesis.
Overall, in our cohort of 57 classic DC and 19 DC-like families, RTEL1 accounts for approximately 4 % of this complex telomere biology disorder. Ongoing gene discovery studies are required to more thoroughly understand the genetic etiology of DC and related disorders, and to further define the molecular consequences of germline RTEL1 mutations.
Materials and methods
Patients and families
Families with DC and their relatives are participants in an IRB-approved longitudinal cohort study at the National Cancer Institute (NCI) entitled “Etiologic Investigation of Cancer Susceptibility in Inherited Bone Marrow Failure Syndromes” (www.marrowfailure.cancer.gov, NCI 02-C-0052, ClinicalTrials.gov Identifier: NCT00027274). Patients and their family members complete detailed family history and medical history questionnaires. We conduct detailed medical record review, comprehensive questionnaires, and thorough clinical evaluations of affected individuals and their relatives at the NIH Clinical Center (Alter et al. 2010). To date, 57 families with DC have enrolled, including 86 affected and 212 unaffected relatives. Individuals with short telomeres and features similar to, but not diagnostic of DC, who lack a DC-associated mutation, are classified as DC-like. Nineteen families are classified as DC-like and consist of 24 affected individuals and 48 relatives.
All DC and DC-like probands had mutation testing of DKC1 (if male), TINF2, TERT, TERC, and WRAP53. NOP10 and NHP2 were sequenced in the DC probands only. None of the patients reported in this study had a germline mutation in one of these genes. DNA was extracted from whole blood using standard methods. Telomere length was measured by flow cytometry with fluorescent in situ hybridization (flow FISH) in leukocytes of all patients and family members reported (Baerlocher et al. 2006).
Exome sequencing
Whole exome sequencing for families NCI-164 and NCI-180 was performed at the NCI’s Cancer Genomics Research Laboratory. Adapter-ligated genomic DNA libraries were prepared with the TruSeq DNA Sample Preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol, and then amplified by ligation-mediated PCR, purified with the QIAquick PCR Purification kit (Qiagen, Valencia, CA, USA), and evaluated electrophoretically. Exome enrichment was performed with NimbleGen’s SeqCap EZ Human Exome Library v2.0, targeting 44.1 Mb of exonic sequence (Roche NimbleGen, Inc., Madison, WI, USA). Sample libraries were hybridized with the EZ Exome Probe Library, and then DNA was washed and recovered as described in the NimbleGen SeqCap EZ Library SR protocol. The exome-enriched libraries were amplified by ligation-mediated PCR, purified, and evaluated as above. The resulting post-capture enriched multiplexed sequencing libraries were used in cluster formation on an Illumina cBOT and paired-end sequencing was performed using an Illumina HiSeq following Illumina-provided protocols for 2 × 100-cycle sequencing. Exomes were sequenced to sufficient depth to achieve a minimum threshold of 80 % of coding sequence covered with at least 15 reads (see Online Resource 1), based on UCSC hg19 “known gene” transcripts (http://genome.ucsd.edu/). This minimum threshold resulted in an average coding sequence coverage of 160 reads.
Exome analysis and variant prioritization
The human reference genome and the “known gene” transcript annotation were downloaded from the UCSC database (http://genome.ucsc.edu/), version hg19 (corresponding to Genome Reference Consortium assembly GRCh37). Reads were aligned to the hg19 reference genome using the Novoalign software version 2.07.14 (http://www.novocraft.com). Duplicate reads based on paired ends aligning to the same start locations due to either optical or PCR artifacts were marked and dropped from further analysis using the MarkDuplicated module of the Picard software version 1.67 (http://picard.sourceforge.net/) using default parameters. Alignments for each individual were refined using a local realignment strategy around known and novel sites of insertion and deletion polymorphisms using the RealignerTargetCreator and IndelRealigner modules from the Genome Analysis Toolkit (GATK, http://www.broadinstitute.org/gatk/) (DePristo et al. 2011). Variant discovery and genotype calling of multi-allelic substitutions, insertions and deletions were performed on all individuals simultaneously using the UnifiedGenotyper module from GATK with the minimum call quality parameter set to 30. Annotation, fitting genetic models, and filtering of each variant locus were performed using a custom locally developed software pipeline using data from the UCSC GoldenPath database (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/), the ESP6500 dataset from the Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/) (accessed August 2012), the Institute of Systems Biology KAVIAR (Known VARiants) database (http://db.systemsbiology.net/kaviar/) (Glusman et al. 2011), the National Center for Biotechnology Information dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/) (Sherry et al. 2001) build 137, and the 1,000 Genomes (http://www.1000genomes.org/) (1000 Genomes Project Consortium 2010). Variants were also annotated for their presence in an in-house database consisting of 366 whole exomes that were sequenced in parallel with our DC families. Variants within each family were filtered and categorized as indicated in Online Resource 2.
Candidate variant validation
Primers for sequencing were designed using Primer3 software (http://jura.wi.mit.edu/rozen/papers/rozen-and-skaletsky-2000-primer3.pdf). The BLAT feature on the UCSC Genome Browser (ucsc.genome.edu) and NetPrimer software (http://www.premierbiosoft.com/netprimer/index.html ) were used to evaluate sequence specificity and oligo folding irregularities. Primers were provided by IDT Technologies (Coralville, Iowa, USA). See Online Resource 4 for primer sequences. All samples were amplified using KAPA2 RobustHotstart Readymix (2X) (Kapa Biosystems, Johannesburg, South Africa) and the following cycling conditions: 3 min at 95°, followed by 30 cycles of 15 s at 95°, 15 s at 60°, 15 s at 72°, followed by 10 min at 72°. Amplicons were purified using Agencourt’s Ampure XP beads, then libraries were constructed and barcoded using the Ion Xpress Plus Fragment Library Kit (Life Technologies, Carlsbad, CA, USA). DNA tagged beads were generated for sequencing using Life Technologies’ OneTouch and run on an Ion 316 chip on the Ion PGM Sequencer (Life Technologies). The default TMAP aligner and variant caller were used to generate a variant list per sample.
In silico analysis
PolyPhen-2 (Adzhubei et al. 2010) (http://genetics.bwh.harvard.edu/pph2), SIFT (Kumar et al. 2009) (http://sift.jcvi.org), and Condel (Gonzalez-Perez and Lopez-Bigas 2011) (http://bg.upf.edu/condel/home) were used to predict the severity of RTEL1 amino acid substitutions. Multiple sequence alignments were generated for homologous RTEL1 protein sequences using M-Coffee (Wallace et al. 2006) and T-Coffee (Notredame et al. 2000) (http://www.tcoffee.org) to evaluate conservation. Alignments were generated with NCBI Reference Sequence proteins NP_116575 (Homo sapiens), NP_001124929 (Pongo abelii), NP_001091044 (Bos taurus), NP_001160137 (Mus musculus), and NP_001013328 (Danio rerio). Jalview (http://www.jalview.org) (Waterhouse et al. 2009) was used to visualize and format the alignments. ProPhylER (Binkley et al. 2010) (http://www.prophyler.org) was also employed to examine the evolutionary constraint on each affected amino acid.
References
1000 Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073. doi:10.1038/nature09534
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. doi:10.1038/nmeth0410-248
Alter BP, Giri N, Savage SA, Rosenberg PS (2009) Cancer in dyskeratosis congenita. Blood 113:6549–6557. doi:10.1182/blood-2008-12-192880
Alter BP, Giri N, Savage SA, Peters JA, Loud JT, Leathwood L, Carr AG, Greene MH, Rosenberg PS (2010) Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol 150:179–188. doi:10.1111/j.1365-2141.2010.08212.x
Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA (2012) Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica 97:353–359. doi:10.3324/haematol.2011.055269
Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA, Eshleman JR, Cohen AR, Chakravarti A, Hamosh A, Greider CW (2005) Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci USA 102:15960–15964. doi:10.1073/pnas.0508124102
Baerlocher GM, Vulto I, de Jong G, Lansdorp PM (2006) Flow cytometry and FISH to measure the average length of telomeres (flow FISH). Nat Protoc 1:2365–2376. doi:10.1038/nprot.2006.263
Binkley J, Karra K, Kirby A, Hosobuchi M, Stone EA, Sidow A (2010) ProPhylER: a curated online resource for protein function and structure based on evolutionary constraint analyses. Genome Res 20:142–154. doi:10.1101/gr.097121.109
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. doi:10.1038/ng.806
Ding H, Schertzer M, Wu X, Gertsenstein M, Selig S, Kammori M, Pourvali R, Poon S, Vulto I, Chavez E, Tam PP, Nagy A, Lansdorp PM (2004) Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell 117:873–886. doi:10.1016/j.cell.2004.05.026
Egan KM, Thompson RC, Nabors LB, Olson JJ, Brat DJ, Larocca RV, Brem S, Moots PL, Madden MH, Browning JE, Ann Chen Y (2011) Cancer susceptibility variants and the risk of adult glioma in a US case–control study. J Neurooncol 104:535–542. doi:10.1007/s11060-010-0506-0
Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J (1995) The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 83:655–666
Glusman G, Caballero J, Mauldin DE, Hood L, Roach JC (2011) Kaviar: an accessible system for testing SNV novelty. Bioinformatics 27:3216–3217. doi:10.1093/bioinformatics/btr540
Gonzalez-Perez A, Lopez-Bigas N (2011) Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 88:440–449. doi:10.1016/j.ajhg.2011.03.004
Keller RB, Gagne KE, Usmani GN, Asdourian GK, Williams DA, Hofmann I, Agarwal S (2012) CTC1 Mutations in a patient with dyskeratosis congenita. Pediatr Blood Cancer 59:311–314. doi:10.1002/pbc.24193
Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y (1999) Mutations in RECQL4 cause a subset of cases of Rothmund–Thomson syndrome. Nat Genet 22:82–84. doi:10.1038/8788
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081. doi:10.1038/nprot.2009.86
Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H (2005) The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J. Nat Genet 37:934–935. doi:10.1038/ng1625
Marrone A, Walne A, Tamary H, Masunari Y, Kirwan M, Beswick R, Vulliamy T, Dokal I (2007) Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood 110:4198–4205. doi:10.1182/blood-2006-12-062851
Nelson ND, Bertuch AA (2012) Dyskeratosis congenita as a disorder of telomere maintenance. Mutat Res 730:43–51. doi:10.1016/j.mrfmmm.2011.06.008
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217. doi:10.1006/jmbi.2000.4042
Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, Sigurdsson A, Jakobsdottir M, Helgadottir H, Thorlacius S, Aben KK, Blondal T, Thorgeirsson TE, Thorleifsson G, Kristjansson K, Thorisdottir K, Ragnarsson R, Sigurgeirsson B, Skuladottir H, Gudbjartsson T, Isaksson HJ, Einarsson GV, Benediktsdottir KR, Agnarsson BA, Olafsson K, Salvarsdottir A, Bjarnason H, Asgeirsdottir M, Kristinsson KT, Matthiasdottir S, Sveinsdottir SG, Polidoro S, Hoiom V, Botella-Estrada R, Hemminki K, Rudnai P, Bishop DT, Campagna M, Kellen E, Zeegers MP, de Verdier P, Ferrer A, Isla D, Vidal MJ, Andres R, Saez B, Juberias P, Banzo J, Navarrete S, Tres A, Kan D, Lindblom A, Gurzau E, Koppova K, de Vegt F, Schalken JA, van der Heijden HF, Smit HJ, Termeer RA, Oosterwijk E, van Hooij O, Nagore E, Porru S, Steineck G, Hansson J, Buntinx F, Catalona WJ, Matullo G, Vineis P, Kiltie AE, Mayordomo JI, Kumar R, Kiemeney LA, Frigge ML, Jonsson T, Saemundsson H, Barkardottir RB, Jonsson E, Jonsson S, Olafsson JH, Gulcher JR, Masson G, Gudbjartsson DF, Kong A, Thorsteinsdottir U, Stefansson K (2009) Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet 41:221–227. doi:10.1038/ng.296
Savage SA, Bertuch AA (2010) The genetics and clinical manifestations of telomere biology disorders. Genet Med 12:753–764. doi:10.1097GIM.0b013e3181f415b5
Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP (2008) TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet 82:501–509. doi:10.1016/j.ajhg.2007.10.004
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311
Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, Simon M, Marie Y, Boisselier B, Delattre JY, Hoang-Xuan K, El Hallani S, Idbaih A, Zelenika D, Andersson U, Henriksson R, Bergenheim AT, Feychting M, Lonn S, Ahlbom A, Schramm J, Linnebank M, Hemminki K, Kumar R, Hepworth SJ, Price A, Armstrong G, Liu Y, Gu X, Yu R, Lau C, Schoemaker M, Muir K, Swerdlow A, Lathrop M, Bondy M, Houlston RS (2009) Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet 41:899–904. doi:10.1038/ng.407
Uringa EJ, Lisaingo K, Pickett HA, Brind’amour J, Rohde JH, Zelensky A, Essers J, Lansdorp PM (2012) RTEL1 contributes to DNA replication and repair and telomere maintenance. Mol Biol Cell 23:2782–2792. doi:10.1091/mbc.E12-03-0179
Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ (2012) RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149:795–806. doi:10.1016/j.cell.2012.03.030
Verdun RE, Karlseder J (2006) The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 127:709–720. doi:10.1016/j.cell.2006.09.034
Vulliamy TJ, Dokal I (2008) Dyskeratosis congenita: the diverse clinical presentation of mutations in the telomerase complex. Biochimie 90:122–130. doi:10.1016/j.biochi.2007.07.017
Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I (2004) Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet 36:447–449. doi:10.1038/ng1346
Wallace IM, O’Sullivan O, Higgins DG, Notredame C (2006) M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res 34:1692–1699. doi:10.1093/nar/gkl091
Walne A, Bhagat T, Kirwan M, Gitaux C, Desguerre I, Leonard N, Nogales E, Vulliamy T, Dokal I (2012) Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica. doi:10.3324/haematol.2012.071068
Wang RC, Smogorzewska A, de Lange T (2004) Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 119:355–368. doi:10.1016/j.cell.2004.10.011
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi:10.1093/bioinformatics/btp033
Wrensch M, Jenkins RB, Chang JS, Yeh RF, Xiao Y, Decker PA, Ballman KV, Berger M, Buckner JC, Chang S, Giannini C, Halder C, Kollmeyer TM, Kosel ML, LaChance DH, McCoy L, O’Neill BP, Patoka J, Pico AR, Prados M, Quesenberry C, Rice T, Rynearson AL, Smirnov I, Tihan T, Wiemels J, Yang P, Wiencke JK (2009) Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet 41:905–908. doi:10.1038/ng.408
Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD (1996) Positional cloning of the Werner’s syndrome gene. Science 272:258–262
Acknowledgments
We thank all of the study participants for their valuable contributions. Lisa Leathwood, RN and Maureen Risch, RN, Westat, Inc., provided excellent study support.
This study was funded by the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health and by contracts N02-CP-91026, N02-CP-11019, and HHSN261200655001C with Westat, Inc.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2013_1265_MOESM1_ESM.pdf
Supplementary material 1 (PDF 13 kb)Online Resource 1: Exome coverage statistics. Exome coverage, off-target and on-target bases are indicated for two DC families
439_2013_1265_MOESM2_ESM.pdf
Supplementary material 2 (PDF 15 kb)Online Resource 2: Exome variant filtering strategy. Variants were sequentially filtered by the criteria described
439_2013_1265_MOESM3_ESM.pdf
Supplementary material 3 (PDF 29 kb)Online Resource 3: Variants found in RTEL1 by targeted sequencing. Targeted sequencing of RTEL1 exons revealed variants that were common (MAF > 1 %), synonymous, or predicted to be benign
439_2013_1265_MOESM4_ESM.pdf
Supplementary material 4 (PDF 15 kb)Online Resource 4: Primers for the RTEL1 locus used in IonTorrent sequencing. These primers were used for targeted sequencing of the RTEL1 exons
Rights and permissions
About this article
Cite this article
Ballew, B.J., Yeager, M., Jacobs, K. et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet 132, 473–480 (2013). https://doi.org/10.1007/s00439-013-1265-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-013-1265-8