
Abstract
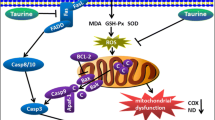
Chronic alcohol intoxication is associated with increased oxidative stress. However, the mechanisms by which ethanol triggers an increase in the production of reactive oxygen species (ROS) and the role of mitochondria in the development of oxidative stress has been insufficiently studied. The biochemical and proteomic data obtained in the present work suggest that one of the main causes of an increase in ROS generation is enhanced oxidation of glutamate in response to long-term alcohol exposure. In the course of glutamate oxidation, liver mitochondria from alcoholic rats generated more superoxide anion and H2O2 than in the presence of other substrates and more than control organelles. In mitochondria from alcoholic rats, rates of H2O2 production and NAD reduction in the presence of glutamate were almost twice higher than in the control. The proteomic study revealed a higher content of glutamate dehydrogenase in liver mitochondria of rats subjected to chronic alcohol exposure. Simultaneously, the content of mitochondrial catalase decreased compared to control. Each of these factors stimulates the production of ROS in addition to ROS generated by the respiratory chain complex I. The results are consistent with the conclusion that glutamate contributes to alcohol hepatotoxicity by enhancing oxidative stress in mitochondria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alcoholic liver disease is a worldwide health problem. The mechanisms that cause the progression of liver injury are complex. Although the pathogenesis of alcoholic liver disease involves changes in many systems and processes, a growing body of evidence indicates that the key elements are oxidative stress and mitochondrial dysfunction (Garcı’a-Ruiz et al. 2013; Louvet and Mathurin 2015; Mandrekar and Ambade 2012; Sid et al. 2013a; Teplova et al. 2010). Alcoholic intoxication causes not only substantial biochemical changes in the functioning of various mitochondrial enzymatic systems, such as the respiratory chain, the fatty acid oxidation pathway, and the urea cycle but also ultrastructural changes, mitochondrial DNA damage, and lipid accumulation (Auger et al. 2015; Cunningham and Bailey 2001; Grattagliano et al. 2011). Ethanol affects the mitochondrial functions, including respiration, ATP synthesis, and the production of reactive oxygen species (ROS) (Wu and Cederbaum 2009; Cederbaum et al. 2009; Mantena et al. 2008; Song et al. 2013; Sun et al. 2016; Yamashina et al. 2010). Recently, acute alcohol was shown to cause in vivo depolarization of hepatocellular mitochondria in a dose- and time-dependent fashion (Zhong et al. 2014). However, the reasons for the alcohol-induced increase in mitochondrial ROS production are not clear.
Recent data suggest that changes in the metabolism of pyridine nucleotides (PN) upon chronic alcohol intoxication contribute to the development of various liver disorders. The oxidation of alcohol by alcohol dehydrogenase and acetaldehyde dehydrogenase considerably changes cytosolic and mitochondrial NADH/NAD ratios due to NADH accumulation (Cahill et al. 2002; Manzo-Avalos and Saavedra-Molina 2010; Zhong et al. 2014). Mitochondrial acetaldehyde dehydrogenase ALDH2 converts the toxic ethanol metabolite acetaldehyde into acetate and generates NADH in the mitochondrial matrix. Hence, fast NADH oxidation through the respiratory chain and NAD regeneration are important for the minimization of the toxic effect of ethanol and its metabolites. However, the data on the effect of ethanol consumption on the mitochondrial NADH oxidation are contradictory. Thus, acute alcohol exposure was reported to lead to a substantial decrease in the mitochondrial transmembrane potential and an increase in hepatic oxygen consumption (a phenomenon named SIAM, swift increase in alcohol metabolism) (Zhong et al. 2014). Similar changes in mitochondrial metabolism were detected upon liver adaptation to chronic ethanol consumption (Han et al. 2017). According to other data, prolonged ethanol consumption may break this adaptation since it causes a decrease in the activity of mitochondrial respiratory enzymes (Marcinkeviciute et al. 2000; Zelickson et al. 2011). Thus, incomplete fatty acid oxidation inherent in chronic alcohol intoxication promotes the inactivation of numerous enzymes (about 20% of mitochondrial proteins) by acetylation; their reactivation also requires high concentrations of oxidized NAD, which is deficient under the conditions of ethanol consumption (Nassir and Ibdah 2014). Presumably, the shift of the NADH/NAD balance to a more reduced state facilitates ROS production by damaged respiratory complexes and nonmitochondrial redox systems (Bailey and Cunningham 1998; Das and Vasudevan 2007; Kukielka et al. 1994). In mitochondria, several sites (respiratory complexes I, III, and mtCYP2E1) were reported to be responsible for increased ROS production upon chronic ethanol consumption (Leung and Nieto 2013; Bailey et al. 1999). However, the relation of mitochondrial NADH and ROS production through possible alcohol-induced changes in the activity of NAD/NADH-dependent dehydrogenases has not been established. It has been shown earlier that ethanol consumption causes a remarkable increase in the levels of glutamate, aspartate, and free ammonia, a glutamate oxidation product, in the liver (Cascales et al. 1985). Ethanol- and acetaldehyde-dependent inhibition of hepatocellular ureagenesis caused by relative closure of voltage dependent anion channels (VDAC) in the outer membrane may underlie these increases in glutamate, aspartate and ammonia (Holmuhamedov et al. 2012). Based on these data, it can be hypothesized that glutamate dehydrogenase may enhance oxidative stress induced by alcohol.
The aim of the present study was to elucidate the participation of glutamate dehydrogenase in the ROS production and systemic oxidative stress in liver mitochondria during chronic alcohol intoxication. Experiments were performed to examine the effect of long-term chronic ethanol consumption by rats on NAD reduction and ROS production linked to oxidation of substrates of NAD-dependent dehydrogenases. It was found that chronic ethanol consumption caused an increase in the activity of mitochondrial glutamate dehydrogenase, which appeared to be accompanied by a strong increase in ROS generation.
Materials and methods
Materials
Alamethicin, superoxide dismutase, antimycin A, rotenone, and 3,7-dihydro-2-methyl-6-(4-methoxyphenyl)imidazol[1,2-a]pyrazine-3-one (MCLA) were from Fluka (Sigma-Aldrich Corporation); carbonyl cyanide p-(trifluoromethoxy)phenyl-hydrazone (FCCP), Amplex Red, NAD, NADH, menadione, and other reagents were from the Sigma-Aldrich Corporation (St. Louis, MO, USA).
Experimental model of chronic alcoholism
All manipulations with animals before the beginning of isolation of livers were performed in accordance with the Helsinki Declaration of 1975 (revised 1983) and national requirements for the care and use of laboratory animals. Manipulations were carried out by certified stuff of the Animal Department of the Institute of Theoretical and Experimental Biophysics (Russian Academy of Science) and approved by the Commission on Biomedical Ethics of ITEB RAS. We applied a widely used model of chronic rat alcogolization (Lang et al. 2004) as the closest to human alcoholism. Wistar male rats with a starting weight of 150 g were divided into two groups: a control group (n = 15), which received a food ration and drinking water, and an alcohol group (n = 15), in which animals were fed the same food and a 20% ethanol solution as the sole source of liquid for six to eight months. The addiction of the animals to alcohol was developed by gradually increasing its percent content in drinking water: 5% (days 1–3), 10% (days 4–6), 15% (days 7–10), and 20% (day 11– last day of experiment). During the study, the animals were kept in wire-mesh cages at a room temperature of 21–22 °C with a light/dark cycle of 12 h. Body weights were monitored weekly. Blood alcohol levels and liver weights were determined prior and after killing of animals, respectively, as physiological indicators of alcohol intoxication. The concentration of ethanol in blood serum was determined using enzymatic alcohol assay kit (Diagnostic Systems GmbH, Holzheim, Germany).
Liver samples were excised immediately after killing rats and homogenized in a 10-fold volume of 50 mM phosphate buffer solution, pH 7.4. The homogenates were centrifuged at 10000 x g for about 60 min, and the resulting supernatants were stored at −80 °C until measurements of malondialdehyde (MDA) and reduced thiols. Rat liver mitochondria were isolated simultaneously from control and alcoholic rats by a standard differential centrifugation (Teplova et al. 2006). The mitochondria were washed twice in mannitol buffer (220 mM mannitol, 70 mM sucrose, 10 mM HEPES-Tris, pH 7.4, potassium-free), resuspended in the same buffer to 60–80 mg protein/ml, and kept on ice. Protein levels in tissue and mitochondrial samples were determined by the Biuret method using BSA as a standard (Gornall et al. 1949).
Analysis of oxidative stress biomarkers
The level of liver free SH-groups was analyzed in liver homogenates by the method of Sedlak and Lindsay using the Ellman’s reagent (Sedlak and Lindsay 1968). Malondialdehyde (MDA) was used as a biomarker of the lipid peroxidation level. The rates of MDA formation by liver homogenates were measured using thiobarbituric acid (Ohkawa et al. 1979).
Measurement of oxygen uptake
Isolated mitochondria (1.5 mg protein/ml) were incubated at 25 °C in a medium containing 120 mM KCl, 2 mM KH2PO4, and 10 mM HEPES (pH adjusted to 7.3 with Trizma base) supplemented with substrates of complex I (5 mM pyruvate and 5 mM malate) or complex II (5 mM succinate) in the presence of 2 μM rotenone. Oxygen uptake was measured with a Clark-type electrode using Oroboros Oxygraph-2 k (Austria). In order to assess V3 and V4 respiration rates, 500 μM ADP was added to resting mitochondria (V2). The rate of uncoupled respiration (VDNP) was measured in the presence of 50 μM 2,4-dinitrophenol (DNP).
Determination of pyridine nucleotide redox state in isolated mitochondria
Changes in mitochondrial functions were determined as described earlier (Teplova et al. 2006). The pyridine nucleotide (PN) redox state was determined by recording fluorescence of PN on a Hitachi F 7000 fluorimeter with excitation at 340 nm and emission at 460 nm. All parameters were measured in a standard KCl- medium containing 120 mM KCl, 2 mM KH2PO4, and 10 mM HEPES (pH adjusted to 7.3 with a few grains of TRIZMA base) with 5 mM substrates.
The activity of NAD-dependent dehydrogenases was estimated from the reduction of exogenous NAD in permeabilized mitochondria by recording PN fluorescence on a Hitachi F 7000 fluorimeter with excitation at 340 nm and emission at 460 nm. The experiments were carried out in the mannitol-sucrose medium (220 mM mannitol, 70 mM sucrose, and 10 mM HEPES (pH adjusted to 7.3) in the presence of alamethicin, rotenone, and cyanide with different respiration substrates. The details are given in the figure legends.
Estimation of ROS production by isolated mitochondria
The production of superoxide anion in a rat liver mitochondria was measured in standard KCl- medium supplemented with 1 mM EGTA and respiration substrates using the highly sensitive chemiluminescent probe MCLA (20 μM), which can also detect singlet oxygen (Kambayashi and Ogino 2003). The kinetics of the SOD-sensitive MCLA-derived chemiluminescence (MDCL) was recorded in 96-well plates (Greiner Bio-One, Germany) using a multi-plate reader (Infinite 200 Tecan, Austria), as described earlier (Nikiforova et al. 2014). H2O2 released from isolated mitochondria was measured in standard KCl medium supplemented with 20 μM Amplex Red and horseradish peroxidase (HRP) (4 U/ml) (Starkov 2010). In order to measure H2O2 production in permeabilized RLM, alamethicin (10 μg/ml) was added 5 min prior to HRP. Where indicated, media also contained NAD, coenzyme A, substrates of NAD-dependent dehydrogenases, and inhibitors of respiratory chain. Resorufin formation was traced for 40 min using a multiplate fluorimeter (Infinite 200 Tecan, Austria) at excitation and emission wavelengths 530 and 595 nm. For the quantitative assessment of H2O2 production, fluorescence was calibrated by a bollus of H2O2 at the end of measurements.
Two-dimensional gel electrophoresis of mitochondrial proteins and mass spectrometry protein identification
Mitochondrial proteins from control and ethanol-fed rats were diluted in a rehydration solution containing 7 M urea, 2% triton X-100, 5% mercaptoethanol, and 2% ampholytes (pH 3.5–10). The polyacrylamide gel columns with the proteins separated by isoelectric focusing (IEF) were applied as a start zone for separation in the second direction. Two-dimensional IEF/SDS-PAGE was performed using a linear 7.5–0% gradient of acrylamide concentration in the presence of 0.1% SDS in a Helicon (Russia) vertical electrophoretic cell, as described earlier (Kovalyov et al. 1995). Protein visualization by Coomassie Blue R-250 and silver nitrate staining as well as the analysis of two-dimensional (2D) electrophoregrams were performed as described earlier (Kovalyov et al. 1995; Kovalev et al. 2006) with minor modifications (Kovalyova et al. 2009). Molecular masses of proteins in the electrophoretic fractions were determined using the kits of highly purified recombinant proteins SM0671 (10–170 kDa) (Fermentas, USA).
Protein spots with altered characteristics, found by the image analysis, were excised from gels and subjected to matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry. The tryptic hydrolysis of single protein fractions followed by an analysis of tryptic peptides using MALDI TOF MS and MS/MS were used for identification of the proteins as described earlier (Govorun et al. 2003; Kovalev et al. 2006; Shevchenko et al. 1996). Mass-spectra were obtained on a MALDI-TOF-mass-spectrometer Reflex III (Bruker, USA) with UV-laser (336 nm) in the regime of positive ions in the range of 500–8000 Da and calibrated in accordance with the known peaks of trypsin autolysis. Proteins were identified using the Mascot software, option Peptide Fingerprint (Matrix Science, USA) by matching experimental masses with the masses of proteins listed in the NCBI Рrotein and SwissProt/treMBL databases.
The densitometry of 2D electrophoregrams and/or their individual fragments (rectangles) was performed after scanning (Epson Expression 1680 scanner). The computer handling of the resultant densitometry images of protein fractions was performed using the Melanie Image Master software, versions 6 and 7 (Genebio, Switzerland).
Statistical analysis
The data given represent the means ± standard error of means (SEM) from 5 to 7 animals per treatment group or are the typical traces of four to five identical experiments with the use of different mitochondrial preparations. The statistical significance was estimated by the Student’s t-test with p < 0.05 as the criterion of significance.
Results
Signs of oxidative stress and mitochondrial dysfunction in the alcoholic liver
A quantitative estimation of oxidative stress biomarkers, such as MDA and free SH-groups in liver tissues, was performed. After seven months of alcohol consumption, rats showed a high content of alcohol in blood (165 ± 24 mg/dl), a smaller body weight (460 ± 25 g versus 570 ± 30 g in control), and a greater liver weight (14.3 ± 0.8 g versus 11.8 ± 0.6 g in control). The rate of MDA formation increased by 15% (755.8 ± 14.1 versus 603.7 ± 12.5 μmol/h/g of liver in control), and the concentration of free SH-groups decreased by 35% in alcoholic liver tissues (5.42 ± 0.24 versus 8.34 ± 0.35 μmol/g of liver in control). All changes were statistically significant. Earlier in the same model, we found that chronic alcohol intoxication increased the level of triglycerides in the liver from 9.6 ± 1.5 to 42.3 ± 5.2 μg/mg protein and in the blood serum from 102.1 ± 10.5 to 160.3 ± 21.0 mg/dL, which indicates an alcohol-induced disturbance of the catabolism of fats (Shubina et al. 2013).
In accordance with earlier observations (Marcinkeviciute et al. 2000), the respiratory activity (V2, V3, V4, and the rate of uncoupled respiration (VDNP)) was considerably depressed in alcoholic mitochondria in the presence of substrates, which feed both complex I (glutamate plus malate) and II (succinate in the presence of rotenone) (Figs. 1a and b). These data indicate an enhancement of oxidative stress, an increase in the intensity of peroxidation processes, and the development of mitochondrial dysfunction in the liver of rats that received alcohol for long time.

Oxygen consumption by control and alcoholic liver mitochondria, oxidizing glutamate plus malate and succinate in the presence of rotenone. Experimental details are given in Materials and methods. Rates of V2, V3, V4, and VDNP respiration in control mitochondria are taken as 100% and correspond to 8.5 ± 0.3, 42.8 ± 2.1, 9.5 ± 0.5, and 45 ± 3.2 ng-atom О∙min−1∙mg protein−1 in the presence of glutamate plus malate and 14.9 ± 0.7, 80.3 ± 4.1, 19.2 ± 0.9, and 135.4 ± 6.7 ng-atom О∙min−1∙mg protein−1 in the presence of succinate plus rotenone. All differences between control and alcoholic mitochondria are significant (p < 0.05)
Effect of long-term alcohol consumption on ROS production by liver mitochondria
To clarify the role of mitochondria in the development of oxidative stress in chronic alcohol intoxication, we compared the ability of liver mitochondria from control and alcoholic rats to generate ROS in the presence of different respiratory substrates, inhibitors of complex I and the redox cycler menadione. Long-term alcohol consumption caused an increase in H2O2 production by rat liver mitochondria (Fig. 2a). The higher rates were observed in the presence of glutamate plus malate than in the presence of succinate plus rotenone or without added substrate. The superoxide anion production in the presence of glutamate and malate was also higher in liver mitochondria of alcoholic animals (Fig. 2c–d). Menadione stimulated the superoxide production with a rate higher than in control mitochondria. Menadione bypasses the rotenone block in complex I and provides the transfer of electrons from NAD(P)H to cytochrome c and directly to molecular oxygen, leading to ROS generation (Criddle et al. 2006; Moore et al. 1986). The specific inhibitor of complex I rotenone, which acts at the quinone-binding site (Brand 2016), as well as the nonspecific inhibitor of complex I NEM, which, presumably, disturbs the flavine-binding site (Gavrikova and Vinogradov 1999; Gostimskaya et al. 2006 ), activated the superoxide production, which was also stronger in mitochondria of alcoholic rats. The most pronounced differences in superoxide anion generation were observed in the presence of the complex I inhibitors (rotenone, NEM) but not complex III inhibitor (antimycin A). These data suggest that complex I contributes to increased ROS production in liver mitochondria from alcoholic rats. Another reason for the faster ROS production in these mitochondria may be the contribution of NAD-dependent dehydrogenases.

Hydrogen peroxide and superoxide anion production by intact liver mitochondria from control and alcoholic rats in the presence of respiratory substrates and respiratory chain inhibitors. Panel a. Original traces of a representative experiment with alcoholic rat liver mitochondria (insert, AR – Amplex Red) and the rates of substrate-dependent H2O2 production are shown. Liver mitochondria from control and alcoholic rats (0.4 mg protein/ml) were added to standard medium, which contained 1 mM EGTA, 20 μM Amplex Red, 4 U/ml HRP and, where indicated, 5 mM glutamate plus 5 mM malate (GM), 5 mM succinate plus 2 μg/ml rotenone (SR). Asterisks show the significant difference between control and alcoholic mitochondria; double asterisks indicate significant difference between glutamate- and succinate-supplemented alcoholic mitochondria (n = 15). Panels b and c show representative traces of superoxide anion production simultaneously measured in liver mitochondria from control (panel b) and alcoholic rats (panel c). Mitochondria (0.3 mg protein/ml) were added to standard medium supplemented with 5 mM glutamate, 5 mM malate and 20 μM MCLA. Where indicated, medium also contained 20 μM NEM (+NEM), 20 μM menadione (+Men), and 1 μg/ml rotenone (+Rot). Data on traces in panels A–C are the means ± S.E.M. (n = 3). Panel d. Integrated MCLA-derived chemiluminescence (MDCL) measured for 25 min in the same samples and in antimycin A (1 μg/ml)-containing samples is presented. The SOD-insensitive part of MDCL was subtracted from total MDCL. Asterisks show the significant difference between control and alcoholic mitochondria
Effect of long-term alcohol consumption on reduced pyridine nucleotide levels in rat liver mitochondria
The level of endogenous NAD(P)H in liver mitochondria from alcoholic rats was about 15–25% higher than in mitochondria from control animals (Fig. 3a). Panels B, C and D in Fig. 3 show changes of NAD(P)H fluorescence on addition of rotenone, substrates of NAD-dependent dehydrogenases and menadione. Rotenone, which completely blocks NADH oxidation by complex I, increased the reduction of endogenous PN. Menadione induced complete oxidation of NAD(P)H in the presence of all substrates used: glutamate plus malate (B), glutamate (C), and malate (D) in control mitochondria. In contrast, in mitochondria of alcoholic rats in the presence of glutamate plus malate, menadione increased PN reduction (Fig. 3b). Glutamate alone but not malate alone was also capable of maintaining the NAD(P)H level in the presence of menadione (Figs. 3c and d). To oxidize NAD(P)H fully in mitochondria from alcoholic livers, menadione сoncentrations higher than required for control mitochondria were necessary. Thus, the glutamate oxidation made a major contribution to increased reduction of NAD(P)H in liver mitochondria of alcoholic rats.

Effect of substrates, rotenone and redox-cycling menadione on the redox state of PN in liver mitochondria from normal and alcoholic rats. Mitochondria (1 mg protein/ml) were added to standard medium without substrates (a, c, and d) or to medium supplemented with 5 mM glutamate plus 5 mM malate (b). Where indicated, 5 mM glutamate, 5 mM malate, 2 μg/ml rotenone, menadione and 0.5 μM FCCP were added. Traces are representative of at least four similar experiments
In subsequent experiments, the reduction of NAD in permeabilized liver mitochondria in the presence of different NAD-dependent substrates was analyzed (Fig. 4). Mitochondria were permeabilized by the freezing-thawing and the addition of pore-forming alamethicin. Under these conditions, NAD penetrated across mitochondrial membranes and was reduced by matrix dehydrogenases. Inserts A and B of Fig. 4 show the experimental design (A) and original traces of NAD reduction in the presence of glutamate in liver mitochondria from control and alcoholic rats (B). The main panel shows the initial rates of NAD reduction in the presence of malate, pyruvate, α-ketoglutarate, and glutamate. These data indicate that, in comparison with other substrates, glutamate provides the highest rate of NAD reduction in both mitochondrial samples. However, in mitochondria of alcoholic rats, this rate was almost twice higher than in control mitochondrial samples.

NAD reduction in permeabilized liver mitochondria from normal and alcoholic rats upon oxidation of NAD-dependent substrates. Liver mitochondria (1 mg/ml) from control and alcoholic rats were permeabilized by one freezing-thawing cycle and incubated in the presence of pore-forming alamethicin (10 μg/ml). Inserts A and B show the experimental design (a) and original traces of NAD reduction in the presence of 5 mM glutamate and 500 μM NAD in liver mitochondria from control and alcoholic rats (b). The main panel shows the calculated rates of NAD reduction. In the presence of pyruvate and α-ketoglutarate, the medium also contained 500 μM CoA. The values are means ± S.E.M. of four independent experiments
Contribution of NAD-dependent dehydrogenases to ROS production in alcoholic intoxication
The contribution of NAD-dependent dehydrogenases to ROS production was studied under the same conditions in mitochondria permeabilized by the freezing-thawing and alamethicin. In the presence of the substrates of glutamate dehydrogenase (NAD and glutamate), H2O2 production by permeabilized mitochondria from alcoholic rats was twice that of mitochondria of control rats (Fig. 5a). This difference persisted in the presence of rotenone and strongly increased in the presence of menadione. Figures 5b and c show that the H2O2 production in the presence of glutamate (+/− menadione) was much higher than in the presence of the substrates of other NAD-dependent dehydrogenases. These data indicate that in alcoholic intoxication the major part of mitochondrial ROS generation is due to the higher activity of glutamate dehydrogenase.

Hydrogen peroxide generation in permeabilized liver mitochondria from control and alcoholic rats in the presence of NAD-dependent substrates. Liver mitochondria (0.3 mg protein/ml) from control and alcoholic rats were added to standard medium supplemented with 20 μM Amplex Red, 4 U/ml HRP, 10 μg/ml alamethicin and, where indicated, substrates, 1 μg/ml rotenone and 25 μM menadione. Panel a. Rates of H2O2 production in the presence of substrates of glutamate dehydrogenase (5 mM glutamate and 0.5 mM NAD). Panel b Traces of Amplex Red fluorescence in the presence of the NAD-dependent substrates: 5 mM glutamate (Glut), 5 mM pyruvate (Pyr), 5 mM α-ketoglutarate (αKG), and 5 mM malate (Mal) without and with menadione. All samples were supplemented with 0.5 mM NAD. Samples containing pyruvate, α-ketoglutarate, and glutamate also included 0.5 mM CoA. Panel c. Menadione-stimulated H2O2 production in the presence of the substrates of different dehydrogenases in permeabilized mitochondria from alcoholic rats. Experimental conditions were as in panel b
Effect of chronic alcohol consumption on the mitochondrial proteome
The chronic alcohol consumption leads to a change in the expression of a number of mitochondrial proteins. Therefore, the higher glutamate dehydrogenase activity in alcoholic mitochondria may be caused by an increase in its content. In subsequent experiments, the effect of chronic alcohol consumption on the level of the glutamate dehydrogenase as well as antioxidant enzymes in the whole mitochondrial proteome was analyzed. Two-dimensional gel electrophoresis of liver mitochondria from control and alcoholic rats and analysis of changes in the mitochondrial proteome induced by chronic ethanol consumption revealed a strong increase in the amount of mitochondrial glutamate dehydrogenase (Fig. 6). In addition, catalase decreased. Details of mass spectrometric identification of protein fractions and characteristics of spots in 2D–gels are given in Table 1 (N 1–3). Three-dimensional densitometry of the zones of glutamate dehydrogenase and catalase in the 2D gel images confirmed the increases of glutamate dehydrogenase and decrease of catalase (Fig. 7).

Two-dimensional gel electrophoresis of liver mitochondria from control and alcoholic rats. Representative high resolution separation of mitochondrial proteins from a control and an alcoholic rats by two-dimensional IEF/SDS-PAGE. Ellipses depict the locations of glutamate dehydrogenase (GLUD1) and catalase (CAT)

Zones of glutamate dehydrogenase and catalase in 2D gel images (a, c) and 3D computer visualization of densitometry (b, d). Numbered arrows correspond to those in Table 1
As seen from Figs. 6 and 7, the content of glutamate dehydrogenase precursors increased in alcoholic mitochondria about threefold. Furthermore, proteomic studies revealed two fractions of catalase, acidic (2) and alkaline (3), the latter in normal rat liver mitochondria being significantly larger. In alcoholic rat liver mitochondria, the alkaline catalase fraction is distinctly reduced.
Overall, the data obtained are consistent with the conclusion that increased glutamate dehydrogenase and decreased catalase contributed to ROS overproduction in liver mitochondria of alcoholic rats.
Discussion
The results obtained indicate that chronic ethanol consumption caused an increase in the activity of mitochondrial glutamate dehydrogenase, which was related to an increase in protein expression of the mitochondrial glutamate dehydrogenase. In the presence of glutamate, mitochondria from alcoholic rats generated more superoxide anion and H2O2 than in the presence of other substrates and more than control mitochondria. ROS generation after alcohol was to a large measure associated with the high activity of glutamate dehydrogenase. In addition, a lower content of mitochondrial catalase was found compared to non-alcoholic rats. Both factors appeared to promote disorders in matrix systems of NADH reduction and oxidation, excessive ROS production, and systemic oxidative stress, which are the distinguishing features of chronic alcohol intoxication.
The alcohol-induced activation of glutamate dehydrogenase, in particular in the liver and leukocytes, has been reported earlier (Cascales et al. 1985; Kravos and Malesic 2010). The activity of mitochondrial glutamate dehydrogenase was shown to rise after both chronic ethanol and acetaldehyde treatment (Cascales et al. 1985). Interestingly, alcohol was also found to increase the glutamate in blood, and blood glutamate has been suggested as a biological marker of alcohol dependence (Tominaga et al. 1993; Walter et al. 2006). These data indicate a special role of glutamate in alcohol intoxication. It has been shown earlier that ethanol consumption causes a marked increase in the level of not only glutamate but also aspartate and free ammonia in the liver as well as a decrease in pyruvate and 2-oxoglutarate concentrations (Cascales et al. 1985). Ammonia and 2-oxoglutarate are the direct metabolites of glutamate oxidation, while aspartate can form in transamination reactions. Accumulation of these metabolites may be related to the more recently described inhibition of hepatocellular ureagenesis by ethanol and acetaldehyde (Holmuhamedov et al. 2012). Importantly, ammonia strongly activates H2O2 production in mitochondria, presumably, affecting the interaction of NAD(H) and lipoamide dehydrogenase (Grivennikova et al. 2008). These data agree well with our results showing a considerable contribution of glutamate oxidation to oxidative stress. Indeed, generation of ammonia by glutamate dehydrogenase can be responsible for the higher rates of ROS generation in the presence of glutamate. Consequently, glutamate can stimulate ROS production simultaneously by two pathways, namely, NAD reduction and ammonium formation. Against the background of the diminished activity of respiratory complex I (Bailey et al. 1999; Hoek et al. 2002; Manzo-Avalos and Saavedra-Molina 2010; Sun et al. 2016), the high glutamate dehydrogenase activity causes the overreduction of NAD, which, in turn, further stimulates ROS production.
The overproduction of NADH, also referred to as reductive stress (Gores et al. 1989), is inherent in many pathological and prepathological states. Along with alcohol intoxication, these are diabetes, ageing and others disorders (Gomes et al. 2013; Yan 2014). These states are as a rule associated with hypoxia or pseudohypoxia. These disorders are caused by the dysfunction of complex I and the increased NADH/NAD ratio. They are accompanied by the activation of fatty acid and triacylglycerol synthesis (He et al. 2013). Earlier, in our alcoholic model we found increased levels of triglycerides in the liver and blood serum, which confirms the disturbance of the catabolism of fats (Shubina et al. 2013). Mitochondrial dysfunction is assumed to be a main factor leading to obesity, a characteristic feature of chronic alcohol abuse. In this connection, glutamate itself induces the obesity syndrome with the attendant accumulation of triglyceride and cholesterol in blood, liver steatosis, and the formation of 4-hydroxynonenal (Sasaki et al. 2011). Besides neuroexcitotoxicity and obesity, the chronic administration of glutamate leads to oxidative stress in hepatic and cardiac tissues in experimental animals, which can be attenuated by antioxidants (Kumar and Bhandari 2013). Based on these data, the toxic action of glutamate in alcoholism may also promote liver steatosis.
Respiration regenerates NAD, a rate-limiting substrate in alcohol/acetaldehyde metabolism. Other mechanisms of NAD regeneration are also possible, among them the shunting of complex I (inhibited or damaged) by quinones. Quinones can bypass complex I and transfer electrons to cytochrome c, activate H+ pumps of complex IV, and thus create ΔΨm in deenergized mitochondria (Dedukhova et al. 1986; Dedukhova and Mokhova 1987; Fedotcheva et al. 2010). In turn, ΔΨm can maintain the NADH-dependent reduction of NADP in transhydrogenase reaction (Eytan et al. 1990). In agreement, menadione induced overreduction of NAD(P) in liver mitochondria from alcoholic rats in the presence of glutamate and rotenone, maintaining NADH in a reduced state. In order to decrease NADH in these mitochondria, menadione concentrations higher than in the control were necessary. Thus, glutamate contributes to alcohol hepatotoxicity, enhancing both oxidative and reductive stress in mitochondria.
Two-dimensional electrophoresis in combination with mass spectrometry is widely used for the identification of changes in protein expression (Uto et al. 2010). Proteomic changes in the liver have been investigated earlier on animal models of acute and chronic alcohol treatment. The studies by Cunningham and colleagues demonstrated that chronic alcohol consumption induces a decrease in the synthesis of 13 mitochondrial proteins that are components of complexes I, III, IV, and V (Coleman and Cunningham 1990; Coleman and Cunningham 1991). Alcohol exposure also changes the content of enzymes involved in the oxidation of fatty acids and oxidative phosphorylation (Venkatraman et al. 2004). Here, a proteomic approach was used to estimate alcohol-induced changes in the content of mitochondrial proteins, particularly glutamate dehydrogenase and antioxidant enzymes. The data obtained showed that alcohol increased the amount of the mitochondrial glutamate dehydrogenase and decreased the content of mitochondrial catalase.
Earlier it was shown that chronic alcohol consumption significantly decreased the level of mitochondrial complexes I, III, IV, and V, as well as the level of hepatic PGC1α, NRF1, and TFAM. It was assumed that the down regulation of transcription factors is the key mechanism underlying the chronic alcohol consumption-induced suppression of mitochondrial biogenesis and respiration (Sun et al. 2016). Nevertheless, in some cases, alcohol feeding caused an enhancement in glutamate/malate- and glycerol-3-phosphate-driven respiration and an increase in the expression of mitochondrial glycerol phosphate dehydrogenase (Han et al. 2017). It was suggested that liver mitochondria are remodeled in response to alcohol feeding. Our data show that the increase in GDH protein level and the decrease in the respiratory activity are a consequence of chronic alcohol consumption. Probably, the alcohol-induced ROS production can activate signaling pathways involved in the mitochondrial remodeling. Thus, it can induce the activation of AMP-activated protein kinase, which controls the mitochondrial functions under conditions of oxidative stress induced by alcohol (Sid et al. 2013b).
The decrease in the catalase level may be an adaptation to chronic ethanol exposure since catalase, in contrast to GSH-related systems, removes hydrogen peroxide without oxidation of NAD(P)H and hence is useless for the alleviation of the reductive stress. The literature data on the effect of alcohol on the activity of antioxidant enzymes in liver mitochondria (MnSOD, glutathione peroxidase, glutathione reductase) are controversial. For instance, the studies of Tang et al. (2012) and Zhang et al. (2015) showed that the activity of antioxidant enzymes decreases. In contrast, Zeng et al. (2008), Larosche et al. (2009), and Vidyashankar et al. (2012) showed no decrease and even found an increase in the activity. The activity of catalase, however, decreased in the majority of cases (Zeng et al. 2008; Vidyashankar et al. 2012).
Thus, the results obtained in the present work indicate that chronic ethanol consumption causes an increase in the activity of glutamate dehydrogenase, which is related to the increase in the amount of this enzyme in liver mitochondria. Together with the lower content of mitochondrial catalase, these factors promote disorders in matrix systems of NADH reduction and oxidation, excessive ROS production, and systemic oxidative stress, which are the distinguishing features of alcohol intoxication.
References
Auger C, Alhasawi A, Contavadoo M, Appanna VD (2015) Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front Cell Dev Biol 3:40. doi:10.3389/fcell.2015.00040 www.frontiersin.org
Bailey SM, Cunningham CC (1998) Acute and chronic ethanol increases reactive oxygen species generation and decreases viability in fresh, isolated rat hepatocytes. Hepatology 28:1318–1326
Bailey SM, Pietsch EC, Cunningham CC (1999) Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med 27:891–900
Brand MD (2016) Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 100:14–31
Cahill A, Cunningham CC, Adachi M, Ishil H, Bailey SM, Fromently B, Davies A (2002) Effect of alcohol and oxidative stress in liver pathology: the role of mitochondrion. Alcohol Clin Exper Res 26:907–915
Cascales C, Cascales M, Santos-Ruiz A (1985) Effect of chronic ethanol or acetaldehyde on hepatic alcohol and aldehyde dehydrogenases, aminotransferases and glutamate dehydrogenase. Rev Esp Fysiol 41:19–27
Cederbaum AI, Lu Y, Wu D (2009) Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol 83:519–548
Coleman WB, Cunningham CC (1990) Effects of chronic ethanol consumption on the synthesis of polypeptides encoded by the hepatic mitochondrial genome. Biochim Biophys Acta 1019:142–150
Coleman WB, Cunningham CC (1991) Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim Biophys Acta 1058:178–186
Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, Sutton R, Petersen OH (2006) Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem 281:40485–40492
Cunningham CC, Bailey SM (2001) Ethanol consumption and liver mitochondria function. Biol Signals Recept 10:271–282
Das SK, Vasudevan DM (2007) Alcohol-induced oxidative stress. Life Sci 81:177–187
Dedukhova VI, Kirillova GP, Mokhova EN, Rozovskaia IA, Skulachev VP (1986) Effect of menadione and vicasol on mitochondrial energy during inhibition of initiation sites of the respiration chain. Biochem Mosc 51:567–573
Dedukhova VI, Mokhova EN (1987) Therapeutic doses of menadione reduce the rotenone-induced inhibition of respiration and membrane potential generation in mitochondria. Biochem Mosc 52:1324–1334
Eytan GD, Carlenor E, Rydström J (1990) Energy-linked transhydrogenase. Effects of valinomycin and nigericin on the ATP-driven transhydrogenase reaction catalyzed by reconstituted transhydrogenase-ATPase vesicles. J Biol Chem 265:12949–12954
Fedotcheva NI, Teplova VV, Beloborodova NV (2010) The participation of phenolic acids of microbial origin in the dysfunction of mitochondria in sepsis. Biochemistry (Moscow) Suppl. Series A: Membrane and Cell Biology, 4: 50–55
Garcı’a-Ruiz C, Kaplowitz N, Fernandez-Checa JC (2013) Role of mitochondria in alcoholic liver disease. Curr Pathobiol Rep 1:159–168
Gavrikova EV, Vinogradov AD (1999) Active/de-active state transition of the mitochondrial complex I as revealed by specific sulfhydryl group labeling. FEBS Lett 455(1–2):36–40
Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA (2013) Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155:1624–1638. doi:10.1016/j.cell.2013.11.037
Govorun VM, Moshkovskii SA, Tikhonova OV, Goufman EI, Serebryakova MV, Momynaliev KT, Lokhov PG, Khryapova EV, Kudryavtseva LV, Smirnova OV, Toropygin IY, Maksimov BI, Archakov AI (2003) Comparative analysis of proteome maps of helicobacter pylori clinical isolates. Biochemistry (Mosc) 68:52–60
Gores GJ, Flarsheim CE, Dawson TL, Nieminen A-L, Herman B, Lemasters JJ (1989) Swelling, reductive stress and cell death during chemical hypoxia in hepatocytes. Am J Phys 257:C347–C354
Gornall AG, Bardawill CJ, David MM (1949) Determination of serum proteins by means of the biuret reaction. J Biol Chem 177:751–766
Gostimskaya IS, Cecchini G, Vinogradov AD (2006) Topography and chemical reactivity of the active-inactive transition-sensitive SH-group in the mitochondrial NADH:ubiquinone oxidoreductase (complex I). Biochim Biophys Acta 1757(9–10):1155–1161
Grattagliano I, Russmann S, Diogo C, Bonfrate L, Oliveira PJ, Wang DQ, Portincasa P (2011) Mitochondria in chronic liver disease. Curr. Drug targets 12: 879–893
Grivennikova VG, Cecchini G, Vinogradov AD (2008) Ammonium-dependent hydrogen peroxide production by mitochondria. FEBS Lett 582:2719–2724
Han D, Johnson HS, Rao MP, Martin G, Sancheti H, Silkwood KH, Decker CW, Nguyen KT, Casian JG, Cadenas E, Kaplowitz N (2017) Mitochondrial remodeling in the liver following chronic alcohol feeding to rats. Free Radic Biol Med 102:100–110. doi:10.1016/j.freeradbiomed.2016.11.020
He Q, Wang M, Petucci C, Gardell SJ, Han X (2013) Rotenone induces reductive stress and triacylglycerol deposition in C2C12 cells. Int J Biochem Cell Biol 45:2749–2755. doi:10.1016/j.biocel.2013.09.011
Hoek JB, Cahill A, Pastorino JG (2002) Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology 122:2049–2063
Holmuhamedov EL, Czerny C, Beeson CC, Lemasters JJ (2012) Ethanol suppresses ureagenesis in rat hepatocytes: role of acetaldehyde. J. Biol. Chem. 287: 7692–7670. PMCID: PMC3293599
Kambayashi Y, Ogino K (2003) Reestimation of cypridina luciferin analogs (MCLA) as a chemiluminescence probe to detect active oxygen species – cautionary note for use of MCLA. J Toxicol Sci 28:139–148
Kravos M, Malesic I (2010) Changes in leukocyte glutamate dehydrogenase activity in alcoholics upon break in alcohol consumption. Clin Biochem 43:272–277
Kukielka E, Dicker E, Cederbaum AI (1994) Increased production of reactive oxygen species by rat liver mitochondria after chronic ethanol treatment. Arch Biochem Biophys 309:377–386
Kumar P, Bhandari U (2013) Protective effect of Trigonella foenum-graecum Linn. On monosodium glutamate-induced dyslipidemia and oxidative stress in rats. Indian J Pharmacol 45:136–140
Kovalyov LI, Shishkin SS, Efimochkin AS, Kovalyova MA, Ershova ES, Egorov TA, Musalyamov AK (1995) The major protein expression profile and two-dimensional protein database of human heart. Electrophoresis 16:1160–1169
Kovalev LI, Kovaleva AA, Kovalev PL, Serebryakova MV, Moshkovskii SA, Shishkin SS (2006) Polymorphism of delta 3,5-delta 2,4-dienoyl-coenzyme a isomerase (the ECH1 gene product protein) in human striated muscle tissue. Biochemistry (Mosc) 71:554–560
Kovalyova MA, Kovalyov LI, Toropygin IY, Shigeev SV, Ivanov AV, Shishkin SS (2009) Proteomic analysis of human skeletal muscle (m. Vastus lateralis) proteins: identification of 89 gene expression products. Biochem Mosc 74:1239–1252. doi:10.1134/S0006297909110108
Lang CH, Frost RA, Svanberg E, Vary TC (2004) IGF-I/IGFBP-3 ameliorates alterations in protein synthesis, eIF4E availability, and myostatin in alcohol-fed rats. Am J Physiol Endocrinol Metab 286:E916–E926
Larosche I, Choumar A, Fromenty B, Lettéron P, Abbey-Toby A, Van Remmen H, Epstein CJ, Richardson A, Feldmann G, Pessayre D, Mansouri A (2009) Prolonged ethanol administration depletes mitochondrial DNA in MnSOD-overexpressing transgenic mice, but not in their wild type littermates. Toxicol Appl Pharmacol 234(3):326–338. doi:10.1016/j.taap.2008.11.004
Leung TM, Nieto N (2013) CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J Hepatol 58:395–398. doi:10.1016/j.jhep.2012.08.018
Louvet A, Mathurin P (2015) Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol 12:231–242
Mandrekar P, Ambade A (2012) Cellular signaling pathways in alcoholic liver disease. In: Shimizu I (ed) Trends in alcoholic liver disease research - Clinical and scientific aspects. InTech 91–112. doi:10.5772/27412. Available from: https://www.intechopen.com/books/trends-in-alcoholic-liver-disease-research-clinical-and-scientific-aspects/cellular-signaling-pathways-in-alcoholic-liver-disease
Mantena KS, King AL, Andringa KK, Eccleston HB, Bailey SM (2008) Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol and obesity induced fatty liver diseases. Free Radic Biol Med 44:1259–1272
Manzo-Avalos S, Saavedra-Molina A (2010) Cellular and mitochondrial effects of alcohol consumption. Int J Environ Res Public Health 7:4281–4304
Marcinkeviciute A, Mildaziene V, Crumm S, Demin O, Hoek JB, Kholodenko B (2000) Kinetics and control of oxidative phosphorylation in rat liver mitochondria after chronic ethanol feeding. Biochem J 349:519–526
Moore GA, O'Brien PJ, Orrenius S (1986) Menadione (2-methyl-1,4-naphthoquinone)-induced Ca2+ release from rat-liver mitochondria is caused by NAD(P)H oxidation. Xenobiotica 16:873–882
Nassir F, Ibdah JA (2014) Role of mitochondria in alcoholic liver disease. World J Gastroenterol 20:2136–2142
Nikiforova AB, Saris NE, Kruglov AG (2014) External mitochondrial NADH-dependent reductase of redox cyclers: VDAC1 or Cyb5R3? Free Radic. Biol Med 74:74–84
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95(2):351–358
Sasaki Y, Shimada T, Iizuka S, Suzuki W, Makihara H, Teraoka R, Tsuneyama K, Hokao R, Aburada M (2011) Effects of bezafibrate in nonalcoholic steatohepatitis model mice with monosodium glutamate-induced metabolic syndrome. Eur J Pharmacol 662:1–8
Sedlak J, Lindsay RH (1968) Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal Biochem 25:192–205
Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68:850–858
Shubina VS, Shatalin YuV, Teplova VV (2013) Oxidative stress in the pathogenesis of alcoholic encephalopathy. Proceedings’ book. International conference “receptors and signaling” (27-30 may Pushchino) 2: 626–630
Sid B, Verrax J, Calderon PB (2013a) Role of oxidative stress in the pathogenesis of alcohol-induced liver disease. Free Radic Res 47:894–904
Sid B, Verrax J, Calderon PB (2013b) Role of AMPK activation in oxidative cell damage: implications for alcohol-induced liver disease. Biochem Pharmacol 86(2):200–209
Song B-J, Abdelmegeed MA, Henderson LE, Yoo S-H, Wan J, Purohit V, Hardwick JP, Moon K-H (2013) Increased nitroxidative stress promotes mitochondrial dysfunction in alcoholic and nonalcoholic fatty liver disease Oxidative Medicine and Cellular Longevity Article ID 781050, doi:10.1155/2013/781050
Starkov AA (2010) Measurement of mitochondrial ROS production. Methods Mol Biol 648:245–255
Sun Q, Zhong W, Zhang W, Zhou Z (2016) Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: role of zinc deficiency. Am J Physiol Gastrointest Liver Physiol 310:G205–C214
Tang Y, Gao C, Xing M, Li Y, Zhu L, Wang D, Yang X, Liu L, Yao P (2012) Quercetin prevents ethanol-induced dyslipidemia and mitochondrial oxidative damage. Food Chem Toxicol 50(5):1194–1200. doi:10.1016/j.fct.2012.02.008
Teplova VV, Mikkola R, Tonshin AA, Saris NE, Salkinoja-Salonen MS (2006) The higher toxicity of cereulide relative to valinomycin is due to its higher affinity for potassium at physiological plasma concentration. Toxicol Appl Pharmacol 210:39–46
Teplova VV, Belosludtsev KN, Belosludtseva NV, Holmuhamedov EL (2010) Role of mitochondria in hepatotoxicity of ethanol. Biophysics (Russia) 55:1038–1047
Tominaga T, Suzuki H, Mizuno H, Kouno M, Suzuki M, Kato Y, Sato A, Okabe K, Miyashita M (1993) Clinical significance of measuring plasma concentrations of glutamine and glutamate in alcoholic liver diseases. Alcohol Alcohol Suppl 1A:103–109
Uto H, Kanumra S, Takami Y, Tsubouchi H (2010) Clinical proteomics for liver disease: a promising approach for discovery of novel biomarkers. Proteome Sci 8:70 http://www.proteomesci.com/content/8/1/70
Venkatraman A, Landar A, Davis AJ, Chamlee L, Sanderson T, Kim H, Page G, Pompilius M, Ballinger S, Darley-Usmar V, Bailey SM (2004) Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J Biol Chem 279:22092–22101
Vidyashankar S, Nandakumar KS, Patki PS (2012) Alcohol depletes coenzyme-Q(10) associated with increased TNF-alpha secretion to induce cytotoxicity in HepG2 cells. Toxicology 302(1):34–39. doi:10.1016/j.tox.2012.07.009
Walter H, Ramskogler-Skala K, Dvorak A, Gutierrez-Lobos K, Hartl D, Hertling I, Munda P, Thau K, Lesch OM, De Witte P (2006) Glutamic acid in withdrawal and weaning in patients classified according to Cloninger's and Lesch's typologies. Alcohol Alcohol 41:505–511
Wu D, Cederbaum AI (2009) Oxidative stress and alcoholic liver disease. Semin Liver Dis 29:141–154. doi:10.1055/s-0029-1214370
Yamashina S, Sato N, Kon K, Ikejima K, Watanabe S (2010) Role of mitochondria in liver pathophysiology, www.Drugdiscoverytoday.Com Elsevier ltd. doi:10.1016/j.ddmec.2010.05.00
Yan LJ (2014) Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res Article ID 137919, 11 pages. doi:10.1155/2014/13791
Zhang P, Qiang X, Zhang M, Ma D, Zhao Z, Zhou C, Liu X, Li R, Chen H, Zhang Y (2015) Demethyleneberberine, a natural mitochondria-targeted antioxidant, inhibits mitochondrial dysfunction, oxidative stress, and steatosis in alcoholic liver disease mouse model. J Pharmacol Exp Ther 352(1):139–147. doi:10.1124/jpet.114.219832
Zelickson BR, Benavides GA, Johnson MS, Chacko BK, Venkatraman A, Landar A, Betancourt AM, Bailey SM, Darley-Usmar VM (2011) Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochim Biophys Acta 1807:1573–1582
Zeng T, Zhang CL, Zhu ZP, Yu LH, Zhao XL, Xie KQ (2008) Diallyl trisulfide (DATS) effectively attenuated oxidative stress-mediated liver injury and hepatic mitochondrial dysfunction in acute ethanol-exposed mice. Toxicology 252(1–3):86–91. doi:10.1016/j.tox.2008.07.062
Zhong Z, Ramshesh VK, Rehman H, Liu Q, Theruvath TP, Krishnasamy Y, Lemasters JJ (2014) Acute ethanol causes hepatic mitochondrial depolarization in mice: role of ethanol metabolism. PLoS One 9, e91308. doi:10.1371/journal.pone.0091308. eCollection 2014
Acknowledgements
This work was supported by grant from the Ministry of Education and Science of the Russian Federation, Agreement № 14.Z50.31.0028 and grant from the Russian Foundation for Basic Research (project № 14-04-01664a).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Teplova, V.V., Kruglov, A.G., Kovalyov, L.I. et al. Glutamate contributes to alcohol hepatotoxicity by enhancing oxidative stress in mitochondria. J Bioenerg Biomembr 49, 253–264 (2017). https://doi.org/10.1007/s10863-017-9713-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10863-017-9713-0