
Abstract
Alcohol-induced liver disease (ALD) is a major health concern of alcohol abuse and a leading cause of liver-related morbidity and mortality. The pathogenesis of ALD is multifactorial and still ill characterized. One of the hallmarks of ALD common for both patients and experimental models is the alteration in the architecture and function of mitochondria. Due to their primordial role in energy production, metabolism and cell fate decisions, these changes in mitochondria caused by alcohol are considered an important contributory factor in ALD. A better understanding of the mechanisms underlying alcohol-mediated mitochondrial alterations may shed light on ALD pathogenesis and provide novel avenues for treatment. The purpose of the current review is to briefly update the latest developments in ALD research regarding morphological and functional mitochondrial regulation including mitochondrial dynamics and biogenesis, mitochondrial protein acetylation and evidence for an endoplasmic reticulum stress-mitochondrial cholesterol link of potential relevance for ALD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alcohol abuse is a leading cause of liver-related morbidity and mortality. Excessive chronic or binge alcohol consumption causes steatosis, which can progress to chronic alcoholic liver disease (ALD). Despite significant efforts, the pathogenesis of ALD is still poorly understood [1, 2]. Consequently, little improvement has been made in the treatment and management of ALD in the past decades. ALD encompasses a spectrum of liver alterations beginning with simple steatosis which can progress to alcoholic hepatitis and cirrhosis and that can ultimately culminate in hepatocellular carcinoma. There have been important advances in understanding the progression of the disease from its initial fatty liver stage to more advanced states. The transition from steatosis to alcoholic hepatitis reflects the induction of mechanisms triggered by alcohol metabolism such as decreased NAD+/NADH, perturbed methionine metabolism, induction of endoplasmic reticulum (ER) stress, oxidative stress, mitochondrial dysfunction and inflammasome activation that prime and sensitize the liver towards hepatocellular demise, TNF susceptibility, fibrosis and inflammation [3–9]. The superimposed participation of other players (e.g., innate immunity, epigenetics, microRNAs, and stem cells) accelerate the deterioration of liver function and ALD progression.
Mitochondria are key organelles that play a central role in energy generation from nutrient oxidation and in alcohol metabolism. Alterations in mitochondrial morphology and function are a hallmark of ALD. Early observations in alcoholic patients described the presence of giant mitochondria in hepatocytes, usually associated with a milder form of the disease [10, 11]. Moreover, alcohol-related liver disease with active drinking is associated with the expression of a mitochondrial DNA deletion fragment in human liver tissue [12, 13]. Moreover, studies in experimental models indicated a wide range of mitochondrial functional alterations [14]. Besides its primordial function in the generation of ATP via oxidative phosphorylation, mitochondria are dynamic organelles and participate in many other cellular functions and pathways, including the generation of metabolites via the tricarboxylic acid cycle, oxidative catabolism of amino acids, synthesis of ketone bodies, fatty acid breakdown in the β-oxidation pathway and generation of reactive oxygen and nitrogen species (ROS/RNS) [15–17]. Hence, it may be anticipated that mitochondrial alterations induced by alcohol may have a broad impact in cell’s function and thus stand as an important mechanism in ALD. Our goal is to provide an overview of recent mechanisms involved in the mitochondrial perturbations associated with alcohol consumption and their potential implications for ALD.
Mitochondrial Function in ALD
Mitochondria of patients with ALD exhibit structural abnormalities [10, 11]. The translation of these morphological alterations into functional impact has been assessed in ALD patients using a noninvasive breath test. The decarboxylation of ketoisocaproate is a specific mitochondrial reaction that results in the exhalation of CO2 from 2-keto-isocaproic acid [18]. Alcoholic patients exhibited reduced peak exhalation of 13CO2 from 2-keto[1-13C]isocaproic acid while aminopyrine breath test and galactose elimination capacity were not altered, indicating that the impaired mitochondrial ketoisocaproate decarboxylation is not a consequence of decreased functional hepatic mass. However, the mitochondrial impairment in alcoholic patients may be specific for decarboxylation reactions and does not appear to extend to other mitochondrial enzymes located in the inner (succinic dehydrogenase and cytochrome c oxidase) or outer mitochondrial membranes (monoamine oxidases) [19].
Pioneering studies in experimental models described mitochondrial damage as an early and characteristic feature of ALD [20]. Studies in rats fed alcohol orally by Lieber–DeCarli liquid diet reported decreases in mitochondrial respiration (state III) and lower respiratory control ratio (state III/state IV) in isolated liver mitochondria [21–23, 24•, 25, 26]. Furthermore, chronic alcohol intake impairs hepatic mitochondrial oxidative phosphorylation by suppressing the synthesis of protein subunits that are encoded by mitochondrial DNA, including subunits of the main respiratory complexes, NADH dehydrogenase (Complex I), cytochrome b–c1 (Complex III), and cytochrome oxidase (Complex IV), as well as the ATP synthase complex (Complex V) [25, 26]. In addition, alcohol metabolism decreases mitochondrial protein synthesis by impairing mitochondrial ribosomes [27]. Proteomic analyses revealed that chronic alcohol consumption alters the expression of several dozens mitochondrial proteins [26, 28]. Besides the direct impact of alcohol consumption on mitochondrial DNA and mitochondrial protein synthesis machinery, intramitochondrial proteins can be irreversibly oxidized by ROS/RNS as well as reactive lipid species such as 4-hydroxynonenal (4-HNE) generated in response to alcohol intake, contributing to the dysregulation of fatty acid metabolism and increased activation of the mitochondrial permeability transition pore [29–32].
In contrast to these effects in rats, feeding alcohol to mice results in increased mitochondrial respiration and higher level of complex I components in liver mitochondria [33, 34]. Interestingly, recent findings in mice fed alcohol orally in pair-fed liquid diet or given intragastrically reported increased state III respiration in isolated liver mitochondria, associated with enhanced levels of Complexes I, IV and V incorporated into the respiratory chain [35••]. Compared to oral alcohol intake, the magnitude of these changes was higher when alcohol was administered intragastrically. The increase in components of mitochondrial complexes following intragastric alcohol consumption was associated with increased PGC-1α protein expression but not of the mitochondrial transcription factor TFAM. Given its role as a master regulator of mitochondrial biogenesis, the increased PGC-1α levels observed in mice fed alcohol likely reflect enhanced mitochondrial biogenesis. However, the activity of PGC-1α is regulated not only at the transcriptional level but also post-translationally, primarily through phosphorylation and acetylation events, the latter regulated by the balance between acetylation mainly by the histone acetyltransferase GCN5 and deacetylation largely by the NAD+-dependent deacetylase SIRT1 [36, 37]. PGC-1α deacetylation by SIRT1 increases its transcriptional activity and has been shown to be essential in mitochondrial biogenesis induced by caloric restriction [38]. Although the status of SIRT1 in mice fed intragastric alcohol was not determined, previous findings reported that acetaldehyde impaired the transcription, translation and activity of SIRT1 in macrophages [39]. Thus, additional data are needed to further characterize the mechanisms of alcohol-induced mitochondrial biogenesis in mice fed alcohol.
The impact of the stimulation in mitochondrial respiration in mice fed alcohol may be of relevance to alcohol metabolism and injury [35••]. Mice fed alcohol orally or intragastrically exhibited increased levels of mitochondrial NAD+ and total NAD+–NADH. Alcohol metabolism by alcohol dehydrogenase (ADH) and the low Km acetaldehyde dehydrogenase (ALDH) is kinetically limited by NAD+, and thus the observed increase in NAD+ levels would be expected to enhance the activity of ADH and ALDH to accelerate alcohol and acetaldehyde oxidation, respectively. In principle, the increased mitochondrial respiration, in turn, would contribute to replenish NAD+ via enhanced NADH oxidation, resulting in an increased rate of alcohol oxidative metabolism. However, since alcohol can be metabolized by other mechanisms independent of ADH/ALDH (e.g. CYP2E1), further evidence is required for a direct link between increased mitochondrial respiration and NADH oxidation by Complex I and enhanced alcohol metabolism. Importantly, as mitochondria are the major consumers of molecular oxygen and ROS/RNS generators, the stimulation of mitochondrial respiration in mice fed alcohol may contribute to the characteristic hypermetabolic state and subsequent hypoxia and liver injury, both of which are preferentially seen in pericentral hepatocytes. Although the mechanisms that determine the species dependent increase (mice but not in rats) in mitochondrial respiration and respiratory complex assembly caused by alcohol intake are not fully understood, this stimulation correlated with alcohol-induced liver injury, being greater in mice than in rats. These findings hence imply that decreased mitochondrial respiratory capacity may protect against ALD. Further investigation is needed to establish the cause or consequence relationship between alterations in mitochondrial respiration and ALD progression. In line with what has been shown in nonalcoholic steatohepatitis [40], it may be of interest to examine the response of mice with intrinsic defects in hepatic mitochondrial respiration (e.g., liver-specific apoptosis inducing factor deletion) to alcohol feeding.
Mitochondrial ROS and Antioxidant Defenses
Consistent with the mitochondrial alterations, alcohol stimulates mitochondrial-derived ROS/RNS, which are likely critical mediators of important signs of ALD such as oxidative stress and cell injury. Moreover, as ALD is an inflammatory disease and recent evidence has shown that mitochondrial ROS induces the mitochondrial targeting of NRLP3 resulting in inflammasome activation [41], it is conceivable that alcohol-induced mitochondrial ROS may contribute to caspase 1 activation and IL-1β generation as described recently in ALD models [42•]. The oxidative metabolism of alcohol and acetaldehyde by ADH and ALDH produce an excess of reducing equivalents which enter the mitochondrial respiratory chain for oxidation. The more reduced electron transport chain then facilitates the transfer of an electron to molecular oxygen to produce superoxide anion [43, 44]. Moreover, decreased activity of the respiratory chain, resulting in accumulation of reduced respiratory carriers in Complexes I and III, stimulates superoxide anion generation. Besides this mechanism of mitochondrial ROS generation linked to alcohol oxidation via ADH/ALDH, the inducible CYP2E1 is an important player in ALD contributing to the hypoxia and superoxide anion generation in response to alcohol [45•]. Although targeted first to the ER by a signal recognition mechanism, CYP2E1 can undergo a post-translational modification for mitochondrial targeting via interaction with the membrane transporters TOM and TIM localized in the outer and inner mitochondrial membranes, respectively [46]. Alcohol feeding has been shown to induce the expression of CYP2E1 both in rat and mouse liver mitochondria, which is associated with increased steatosis, oxidant injury, and impaired mitochondrial antioxidant defense [47, 48]. Superoxide anion generation by alcohol can be the source of additional ROS/RNS species and oxidants, including peroxynitrite and hydrogen peroxide. The former is generated upon interaction of superoxide anion with nitric oxide to form the potent peroxynitrite, which has a short half-life and is responsible for many damaging modifications of tyrosine residues in mitochondrial target proteins. The dismutation of superoxide anion by Mn-SOD generates hydrogen peroxide, a potent oxidant. The degradation of mitochondrial hydrogen peroxide is carried out by the antioxidant enzymes GSH peroxidase 1 (GSHPx1), which requires reduced GSH for its activity, and peroxiredoxin-III (Prx-III) whose reactivation occurs by thioredoxin-2 (Trx2) [49]. In addition to Mn-SOD, hydrogen peroxide can be generated by other mechanisms within the mitochondria, including the action of p66Shc. p66Shc is a signaling adapter protein known to directly stimulate hydrogen peroxide generation by transferring electrons to cytochrome c [50]. In this regard, increased hepatic p66Shc expression has been reported in mice treated with alcohol in the drinking water and the deletion of p66Shc ameliorated alcohol-induced hepatic steatosis, oxidative stress and cell injury [51]. Intriguingly, these effects in the p66Shc null mice were accompanied by increased expression of MnSOD, raising the question of whether the protection in the null mice was due to the lack of direct generation of hydrogen peroxide by p66Shc or indirectly via increased antioxidant defenses.
The first line of defense against mitochondrial ROS generation is via MnSOD, and its deficiency leads to neonatal death in mice. However, the role of alcohol on MnSOD regulation is controversial, with findings indicating increased expression [52], no change [51, 53] or decreased expression [54]. Moreover, homozygous mutations in the MnSOD gene associated with increased mitochondrial localization has been shown to be a risk for severe ALD in humans [55]. Mitochondrial GSH, which is critical in the elimination of hydrogen peroxide, has been shown to be depleted by alcohol feeding both in rats or mice [56–59], with a preferential effect occurring in perivenous hepatocytes [57, 58]. The depletion of mitochondrial GSH by alcohol feeding is rapidly reversed upon alcohol withdrawal [60, 61] and occurs in mice fed alcohol intragastrically [53]. Furthermore, chronic alcohol intake has been reported to severely deplete mitochondrial GSH in alveolar type II cells, which is not restored by the GSH precursor N-acetylcysteine, (NAC) [62]. The inability of NAC to replenish alcohol-mediated mitochondrial GSH depletion has been observed in rat liver [58] and may be related to the defect in the transport of GSH into the mitochondrial matrix (GSH is not synthesized in mitochondria) caused by alcohol-mediated mitochondrial cholesterol accumulation [63•]. The impact of mitochondrial cholesterol loading in the depletion of mitochondrial GSH is not an exclusive feature of alcohol intake but has been reported in other contexts such as nonalcoholic steatohepatitis and anthrax-induced cell death [64, 65]. Given the differential role of alcohol on MnSOD and mitochondrial GSH regulation their interplay in ALD is of relevance. As recently exemplified in nonalcoholic steatohepatitis mitochondrial GSH determines the therapeutic potential of superoxide anion scavenging [66••]. Depletion of mitochondrial GSH worsened the consequences of superoxide anion scavenging by SOD mimetics or MnSOD overexpression due to increased hydrogen peroxide accumulation despite reduction in peroxynitrite formation. In line with this notion, it has been recently reported that MnSOD overexpression aggravates the deleterious effects of prolonged alcohol feeding on mitochondrial DNA in mice [67]. Therefore, in view of the critical balance between antioxidant enzymes to prevent oxidative stress, the combination of superoxide anion scavenging with mitochondrial GSH replenishment rather than improving only MnSOD may be important in human ALD [55].
ER Stress and Its Mitochondrial Link
ER plays a key role in Ca2+ homeostasis and is the primary site where synthesis of proteins and lipids take place in the cell. Sustained dysregulation in protein folding or altered lipid homeostasis in ER (e.g., increased PC/PE ratio) results in ER stress, which may ultimately lead to cell death if the stress is not resolved [68, 69]. To restore homeostasis the ER turns on the unfolded protein response (UPR). UPR is chiefly controlled by the binding immunoglobulin protein (BiP)/glucose-regulated protein 78 (GRP78) module, which regulates three specific transducers, inositol requiring (IRE) 1α, PKR-like ER kinase (PERK), and activating transcription factor (ATF) 6α that act in concert to increase ER content, expand the ER protein folding capacity, degrade misfolded proteins, and reduce the load of new proteins entering the ER. In addition, ER stress is known also to result in the activation of ER-resident transcription factors SREBPs, which undergo a controlled proteolytic processing in collaboration with Golgi proteases Sp1/Sp2 to induce the transcriptional activation of enzymes involved in lipid synthesis. SREBP-1c regulates the transcription of enzymes involved in fatty acid synthesis and esterification, contributing to enhanced triglyceride generation, while SREBP-2 controls key enzymes of de novo cholesterol synthesis in the mevalonate pathway [70–72]. Consequently, ER stress has been shown to regulate hepatic steatosis, hence playing a major role in metabolic liver diseases. Consistent with this scenario, alcohol feeding has been shown to induce ER stress, emerging as an important mechanism in ALD [73]. Indeed, liver specific deletion of GRP78 impairs global UPR exacerbating ER stress and alcohol-mediated liver injury [74]. A critical mechanism in alcohol-induced hepatic ER stress is the perturbation in one carbon metabolism and the increase in homocysteine [73, 74]. In favor of the homocysteine-ER stress link, nutritional therapy with betaine supplementation to alcohol fed mice decreased hyperhomocysteinemia preventing alcohol-mediated ER stress, hepatic steatosis and liver injury [74]. Interestingly, betaine treatment has also been shown to attenuate alcohol-induced alterations to the mitochondrial respiratory chain proteome [75]. However, whether this effect is due to the ability of betaine to prevent alcohol-induced ER stress remains to be investigated. Moreover, recent evidence addressing the inter-strain susceptibility of mice to ALD uncovered the correlation between hyperhomocysteinemia, one-carbon metabolism, and liver injury in sensitive mouse strain [76].
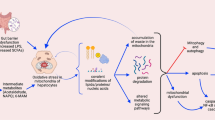
Recent evidence has suggested a ceramide-ER stress link that may change the perspective on the role of homocysteine as a direct ER stress trigger. In this regard, findings in mice deficient in cystathionine β-synthase have shown that the link between hyperhomocysteinemia and the subsequent glomerular injury is dependent on acid sphingomyelinase (ASMase)-mediated ceramide generation [77•]. Moreover, exogenous ASMase has been shown to directly induce ER stress in isolated hepatocytes by disrupting ER Ca2+ homeostasis, while the effects upon the addition of homocysteine in causing ER stress were modest and required suprapharmacological doses [63•]. Specific ASMase activation has been observed in association with hyperhomocysteinemia and decreased SAM/SAH ratio in mice fed the methionine and choline deficient diet [78], which is known to induce ER stress [79]. To further characterize the relationship between homocysteine, ASMase and ER stress in ALD, ASMase null mice were fed alcohol orally for 4 weeks. ASMase deletion abrogated alcohol-induced hepatic steatosis and ER stress [63•]. However, the level of hyperhomocysteinemia and the decrease in SAM/SAH caused by alcohol intake were similar in ASMase+/+ and ASMase−/− mice, indicating that the resistance of ASMase null mice to alcohol-induced ER stress is independent of hyperhomocysteinemia, with similar findings observed in ASMase null mice fed alcohol intragastrically. Intriguingly, in contrast to ASMase+/+ mice ASMase−/− mice were resistant to alcohol-induced mitochondrial cholesterol accumulation, an important event in hepatocyte apoptosis regulation due to mitochondrial GSH depletion [64, 80, 81]. Consequently, alcohol-induced susceptibility to liver injury caused by LPS and concanavalin-A was observed only in ASMase+/+ mice but not ASMase−/− mice. Consistent with the critical role of steroidogenic acute regulatory domain StARD1, the founding member of StARD protein family, in the trafficking of cholesterol to mitochondrial inner membrane [82], alcohol increased StARD1 expression in ASMase+/+ mice but not in ASMase−/−, indicating a correlation between alcohol-induced ER stress and StARD1 expression. Furthermore, the addition of tunicamycin to primary hepatocytes induced ER stress and StARD1 upregulation that was prevented by tauroursodeoxycholic acid [83], further suggesting that StARD1 is an ER stress target gene. As expected for SREBP-2 regulated genes, feeding a high-cholesterol diet downregulated hydroxymethylglutaryl CoA reductase but not StARD1, indicating that StARD1 upregulation is ER stress dependent. Thus, the link between ER stress and mitochondrial GSH depletion following alcohol intake involves ASMase-ER stress-StARD1 axis (Fig. 1). These results together with findings that ASMase expression increases in liver samples from patients with alcoholic hepatitis [63•] point to ASMase as a potential relevant target in the treatment of ALD by preventing alcohol-induced ER stress and subsequent downstream events.

Mechanisms linking alcohol metabolism to mitochondrial targeting. Ethanol metabolism induces ER stress via ASMase activation, and this event results in the transcriptional activation of StARD1, a mitochondrial cholesterol transporting polypeptide, resulting in mitochondrial cholesterol accumulation. In turn, this event contributes to mitochondrial GSH depletion. Ethanol metabolism and mitochondria are linked via the replenishment of NADH to NAD+ in the respiratory chain to ensure alcohol metabolism via ADH/ALDH. This in turn results in more reduced electron transport chain which facilitates the transfer of an electron to molecular oxygen to produce ROS. While ROS are recognized to contribute to oxidant cell death, their role in alcohol-regulated mitochondrial biogenesis and inflammation remain to be further established
Mitochondrial Morphology and Dynamics
Mitochondria exhibit a heterogeneous morphology and a dynamic nature. Mitochondrial dynamics involves movement of mitochondria along the cytoskeleton, regulation of mitochondrial architecture (morphology and distribution), and connectivity mediated by tethering and fusion/fission events [84]. The relevance of these factors in mitochondrial and cell physiology has been partially unraveled and the disruption of such processes results in mitochondrial heterogeneity and dysfunction [85, 86]. Therefore, a fine regulation of mitochondrial biogenesis and dynamics is necessary to obtain and maintain functional mitochondria. Mitochondrial morphology and dynamics in living cells depend on membrane fusion and fission events, which not only determine the presence of an integrated mitochondrial reticular network or the appearance of dispersed organelles but also allow for the exchange of components from the inner and outer membrane and matrix between individual mitochondria [87, 88]. Mitochondrial fusion is essential for normal cell function, and is regulated by mitofusins. Mitofusins 1 and 2 (Mfn1 and Mfn2) are integral proteins of the mitochondrial outer membrane, which interact in homo- or heterotypic fashion to tether neighbor mitochondria to initiate fusion [85–88]. In addition, optic atrophy 1 (Opa1), a mitochondrial inner membrane protein, is required to tether and fuse mitochondrial inner membrane and in the maintenance of respiratory function in hepatocytes and other cell types [89, 90]. On the other hand, mitochondrial fission is mediated by several proteins, such as fission 1 protein (Fis1), located in the outer mitochondrial membrane, mitochondrial fission factor (Mff), located in the outer mitochondrial membrane, and dynamin-related protein 1 (Drp1), which resides in the cytosol and translocates to the outer mitochondrial membrane during fission. Drp1 recruitment to the outer mitochondrial membrane and GTP hydrolysis are required for Drp1-mediated fission [84]. While many studies have examined the role of mitochondrial dynamics in apoptosis and, more recently, in nutrient supply and energy demand [91], the role of mitochondrial dynamics in alcohol-induced mitochondrial function and liver injury has been scarcely studied.
A consistent feature of alcohol intake is the alteration in hepatic mitochondrial morphology, as reported in alcoholic patients and ALD models [10, 11, 35••]. Using an organelle-targeted photoactivatable fluorescent protein approach and electron microscopy, Das et al. [92] examined the effect of oral chronic alcohol intake on mitochondrial morphology and dynamics. Mitochondrial fusion events, estimated by mtPAGFP and mtDsRed interexchange, decreased in hepatocytes from alcohol-fed rats. Electron microscopy analyses revealed the presence of matrix swelling in mitochondria from alcohol-fed rats. Moreover, hepatocarcinoma cells VL-17A, which overexpress ADH and CYP2E1 to metabolize alcohol, exhibited higher mitochondrial continuity and increased rate of mitochondrial dynamics and movement than primary rat hepatocytes. However, exposure of VL-17A cells to alcohol, significantly altered mitochondrial morphology and suppressed mitochondrial dynamics [92]. In line with these findings, recent observations have shown that hepatic mitochondria from mice fed alcohol intragastrically exhibited a heterogeneous morphological appearance manifested predominantly in the formation of longer tubular and thinner mitochondria compared with round or oval morphology of mitochondria from control mice [35••]. Moreover, intragastric alcohol feeding increased the frequency of very elongated mitochondria as well as the number of smaller round mitochondria. Whether these changes reflect increased fusion or decreased fission remains to be established as well as the impact of these morphological alterations in mitochondrial membrane potential and ROS generation. Interestingly, the increase in mitochondrial elongation caused by intragastric alcohol feeding resulted in enhanced mitochondrial respiratory capacity and biogenesis, as described above. The association of mitochondrial elongation due to fission inhibition and increased respiration coupled to ATP synthesis has been shown in states of increased energy demand and low nutrient supply, such as starvation, acute stress or G1/S cycle [93]. Moreover, mitochondrial elongation increases mitochondrial cristae number, which is associated with the dimerization of the ATP synthase and thus higher ATP synthesis activity [94••]. On the other hand, uncoupled respiration as found in conditions of nutrient excess is associated with mitochondrial fragmentation and fusion inhibition, strongly suggesting that mitochondrial dynamics regulate mitochondrial bioenergetic efficiency and capacity.
The mechanisms regulating mitochondrial dynamics in response to alcohol feeding and why this process is species dependent remain poorly understood. In this regard, whether alcohol-induced mitochondrial cholesterol loading (see above) plays a role in mitochondrial fusion/fission events needs further investigation. Interestingly, recent data reported that Opa-1 downregulation and cristae remodeling increased mitochondrial cholesterol trafficking in trophoblasts [95], raising the possibility that impairment in mitochondrial fusion may contribute, in part, to mitochondrial dysfunction by stimulating mitochondrial cholesterol accumulation, thus establishing a vicious circle of potential impact to ALD. In addition to mitochondrial cholesterol loading, accumulation of oxidized lipid aldehydes in mitochondria resulting from ROS-induced membrane damage may also regulate events involved in mitochondrial fusion. Further research in this area is needed to unravel the potential therapeutic value of targeting mitochondrial dynamics (promoting fusion or fission) in ALD.
Mitochondrial Protein Acetylation
Acetylation of proteins in lysine residues is an important regulatory post-translational mechanism of protein function that has a widespread impact in cell physiology, metabolism and longevity [96]. Protein acetylation relies on acetyl-CoA as the acetyl group donor and is controlled by deacetylation reactions. The principal enzymes involved in protein deacetylation are sirtuins. Sirtuins represent a small gene family with seven members in mammals, i.e. SIRT1–SIRT7, which share significant sequence homology and contain conserved catalytic and nicotinamide adenine dinucleotide (NAD+)-binding domains. All sirtuins require NAD+ for their enzymatic activity and catalyze deacetylation of target proteins by transferring the acetyl group to the ADP-ribose (ADPR) moiety of NAD+ generating a deacetylated protein, nicotinamide (NAM), and 2′-O-acetyl-ADP ribose [96]. Consequently, NAD+ activates the deacetylase activity of sirtuins, whereas NAM and the reduced form of NAD (NADH) inhibit their activity. Out of the 7 family members, only three sirtuins are known to reside in mitochondria, SIRT3–5. Based on mouse genetic studies, SIRT3 has been shown to be the major mitochondrial protein deacetylase [97]. Given the fact that about 20 % of mitochondrial proteins are acetylated, this outcome implies that SIRT3 is a key regulator of mitochondrial proteome landscape and function, and hence SIRT3 is known to regulate a wide range of cellular pathways such as oxidative phosphorylation, fatty acid metabolism, oxidative stress response and antioxidant defense, and alcohol metabolism [96, 98].
While the effect of alcohol intake on hepatic mitochondrial function has been widely studied, as described above, the contribution of mitochondrial protein acetylation in this context has been less examined. Recent reports indicated that feeding alcohol orally to mice and rats lead to mitochondrial protein hyperacetylation [99, 100•]. Consistent with these findings, intragastric alcohol feeding to mice also resulted in increased mitochondrial protein N-acetylation [35••], highlighting that mitochondrial protein hyperacetylation is likely a feature of alcohol metabolism independent of species and route of administration. Interestingly, while SIRT3 expression remained unchanged, alcohol feeding impaired SIRT3 activity [99]. Although SIRT3 is NAD+ dependent and decrease NAD+/NADH can inactivate SIRT3, the contribution of altered NAD+/NADH homeostasis to alcohol-induced SIRT3 inactivation remains controversial [35••, 99]. In addition to potential regulation of SIRT3 activity by NAD+, Fritz et al. [100•] provided a novel alternative mechanism to explain the alcohol-mediated SIRT 3 inactivation. SIRT3 carbonylation induced by alcohol-mediated 4-HNE allosterically inhibits SIRT3 activity. Importantly, in vitro studies using recombinant SIRT3 and physiological relevant 4HNE concentrations validated the carbonylation of SIRT3 at critical Cys280 residue, required for zinc binding. In silico analyses indicated that 4-HNE-induced SIRT-3 modification altered the conformation of the zinc-binding domain inducing minor changes within the active site, resulting in the allosteric inhibition of SIRT3 activity [101]. While these data revealed a novel mechanism of alcohol-mediated SIRT3 inactivation resulting in mitochondrial protein hyperacetylation, the impact on mitochondrial respiratory changes induced by alcohol remain unestablished. N-acetylation of mitochondrial proteins, including the respiratory complexes, negatively regulates their function [102], implying that hyperacetylation may be a potential mechanism whereby alcohol intake inhibits mitochondrial function. However, intragastric alcohol feeding to mice stimulates rather than inhibits mitochondrial respiratory chain function despite increased mitochondrial protein N-acetylation [35••]. Thus, further work is required to unravel whether modulation of SIRT3-mediated mitochondrial protein acetylation in lysine residues may affect mitochondrial function and hence regulate ALD. One interesting observation was the fact that alcohol-induced mitochondrial hyperacetylation was partially mediated by CYP2E1 [99]. Furthermore, comparison of the mitochondrial acetylome in wild-type and SIRT3 null mice revealed that alcohol-induced protein hyperacetylation targets similar proteins as the SIRT3 KO mice, affecting common biochemical pathways including fatty acid and lipid metabolism, electron-transport chain and amino acid biosynthesis, suggesting a central role for SIRT3 and altered protein acetylation in the pathogenesis of ALD [103]. Moreover, the mitochondrial antioxidant enzyme MnSOD is also regulated by SIRT3 via protein acetylation, thus suggesting that SIRT3 inactivation by alcohol intake may contribute to the superoxide anion generation as an additional mechanism of injury. Overall, while attractive as a potential target, the implication of modulating SIRT3 activity in ALD still needs further investigation.
Conclusions
Mitochondria not only contribute to alcohol metabolism via acetaldehyde oxidation and NAD+ replenishment by NADH oxidation in the respiratory chain but are also targets of the deleterious effects of alcohol. Over the years, there has been a wealth of evidence both in patients and experimental models of mitochondrial abnormalities at the morphological and functional levels caused by alcohol abuse. Perhaps with the exception of the recently shown increased mitochondrial respiration in mice, most studies in rats and alcoholic patients have shown impaired oxidative phosphorylation, damaged mitochondrial DNA and defective decarboxylation reactions, consistent and likely reflecting the alterations in architecture such as cristae organization and matrix swelling seen in both cases. While the underlying mechanisms for the species difference of alcohol stimulated mitochondrial respiration seen in mice but not in rats fed alcohol remain unknown, this contrast raises the question of whether mitochondrial alterations caused by alcohol intake are cause or consequence of the disease. The answer to this question requires further research using adequate experimental models of defective or enhanced mitochondrial respiratory capacity to examine the impact on alcohol-induced liver disease. Moreover, given the heterogeneous population of parenchymal cells within the acinus determined by anatomical features and oxygen availability, it remains unknown whether the increased mitochondrial respiration reported in mice fed alcohol may reflect the specific contribution of perivenous or periportal hepatocytes. Perhaps another process of potential relevance in sustaining defective mitochondria following alcohol intake may be impaired autophagy or more specifically mitophagy, although current available evidence in this area is limited. Thus, further work is needed to fully understand the mechanisms and impact of mitochondrial alterations in ALD to propose novel therapeutical avenues.
Abbreviations
- ALD:
-
Alcoholic liver disease
- ADH:
-
Alcohol dehydrogenase
- ALDH2:
-
Acetaldehyde dehydrogenase
- ASMase:
-
Acid sphingomyelinase
- Drp1:
-
Dynamic related protein 1
- ER stress:
-
Endoplasmic reticulum stress
- Mfn1/2:
-
Mitofusins 1/2
- Op1:
-
Optic atrophy 1
- ROS/RNS:
-
Reactive oxygen/nitrogen species
- StARD1:
-
Steroidogenic acute regulatory domain protein 1
- UPR:
-
Unfolded protein response
References
Recently published papers of particular interest have been highlighted as: • Of importance •• Of major importance
Lucey MR, Mathurin P, Morgan TR (2006) Alcoholic hepatitis. New Engl J Med 360:2758–2769
Gao B, Bataller R (2011) Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141:1572–1585
Chen X, Sebastian B, Tang H (2009) Taurine supplementation prevents ethanol-induced decrease in serum adiponectin and reduces hepatic steatosis in rats. Hepatology 49:1554–1562
Kaplowitz N, Than TA, Shinohara M, Ji C (2007) Endoplasmic reticulum stress and liver injury. Semin Liver Dis 27:367–377
Mato JM, Martinez-Chantar ML, Lu SC (2008) Methionine metabolism and liver disease. Annu Rev Nutr 28:273–293
Donohue DM Jr (2009) Autophagy and ethanol-induced liver injury. World J Gastroenterol 15:1178–1185
Wu D, Cederbaum AI (2009) Oxidative stress and alcoholic liver disease. Semin Liver Dis 29(2):141–154
Colell A, García-Ruiz C, Miranda M, Ardite E, Marí M, Morales A, Corrales F, Kaplowitz N, Fernández-Checa JC (1998) Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 115:1541–1551
Pastorino JG, Hoek JB (2000) Ethanol potentiates tumor necrosis factor-alpha cytotoxicity in hepatoma cells and primary rat hepatocytes by promoting induction of the mitochondrial permeability transition. Hepatology 31:1141–1152
Bruguera M, Bertran A, Bombi JA, Rodes J (1977) Giant mitochondria in hepatocytes: a diagnostic hint for alcoholic liver disease. Gastroenterology 73:1383–1887
Chedid A, Mendenhall CL, Tosch T, Chen T, Rabin L, Garcia-Pont P, Goldberg SJ, Kiernan T, Seeff LB, Sorrell M et al (1986) Significance of megamitochondria in alcoholic liver disease. Gastroenterology 90:1858–1864
Caldwell SH, Swerdlow RH, Khan EM, Iezzoni JC, Hespenheide EE, Parks JK, Parker WD Jr (1999) Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol 31:430–434
Fromenty B, Grimbert S, Mansouri A, Beaugrand M, Erlinger S, Rötig A, Pessayre D (1995) Hepatic mitochondrial DNA deletion in alcoholics: association with microvesicular steatosis. Gastroenterology 108:193–200
Hoek JB, Cahill A, Pastorino JG (2002) Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology 122:2049–2063
Kelly DP, Scarpulla RC (2004) Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18:357–368
Murgia M, Giorgi C, Pinton P, Rizzuto R (2009) Controlling metabolism and cell death: at the heart of mitochondrial calcium signalling. J Mol Cell Cardiol 46:781–788
Murphy MRP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Witschi A, Mossi S, Meyer B, Junker E, Lauterburg BH (1994) Mitochondrial function reflected by the decarboxylation of [13C]ketoisocaproate is impaired in alcoholics. Alcohol Clin Exp Res 18:951–955
Jenkins WJ, Peters TJ (1978) Mitochondrial enzyme activities in liver biopsies from patients with alcoholic liver disease. Gut 19:341–344
Rubin E, Beattie DS, Lieber CS (1970) Effects of ethanol on the biogenesis of mitochondrial membranes and associated mitochondrial functions. Lab Invest 23:620–627
Bernstein JD, Penniall R (1978) Effects of chronic ethanol treatment upon rat liver mitochondria. Biochem Pharmacol 27:2337–2342
Cederbaum AI, Lieber CS, Rubin E (1974) Effects of chronic ethanol treatment of mitochondrial functions damage to coupling site I. Arch Biochem Biophys 165:560–569
Spach PI, Cunningham CC (1987) Control of state 3 respiration in liver mitochondria from rats subjected to chronic ethanol consumption. Biochim Biophys Acta 894:460–467
• Chacko BK, Srivastava A, Johnson MS, Benavides GA, Chang MJ, Ye Y, Jhala N, Murphy MP, Kalyanaraman B, Darley-Usmar VM (2011) Mitochondria-targeted ubiquinone (MitoQ) decreases ethanol-dependent micro and macro hepatosteatosis. Hepatology 54:153–63. This study reports the effect of a mitochondrial-targeted antioxidant in the prevention of steatosis and hypoxia in alcohol fed rats
Cunningham CC, Coleman WB, Spach PI (1990) The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol 25:127–136
Venkatraman A, Landar A, Davis AJ, Chamlee L, Sanderson T, Kim H, Page G, Pompilius M, Ballinger S, Darley-Usmar V, Bailey SM (2004) Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J Biol Chem 279:22092–22101
Cahill A, Cunningham CC (2000) Effects of chronic ethanol feeding on the protein composition of mitochondrial ribosomes. Electrophoresis 21:3420–3426
Bailey SM, Robinson G, Pinner A, Chamlee L, Ulasova E, Pompilius M et al (2006) S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. Am J Physiol Gastrointest Liver Physiol 291:G857–G867
Lee JH, Yang ES, Park JW (2003) Inactivation of NADPH-dependent isocitrate dehydrogenase by peroxynitrite. Implications for cytotoxicity and alcohol-induced liver injury. J Biol Chem 278:51360–51371
Baraona E, Zeballos GA, Shoichet L, Mak KM, Lieber CS (2002) Ethanol consumption increases nitric oxide production in rats, and its peroxyni-trite-mediated toxicity is attenuated by polyenylphosphatidylcholine. Alcohol Clin Exp Res 26:883–889
King AL, Swain TM, Dickinson DA, Lesort MJ, Bailey SM (2010) Chronic ethanol consumption enhances sensitivity to Ca(2+)-mediated opening of the mitochondrial permeability transition pore and increases cyclophilin D in liver. Am J Physiol Gastrointest Liver Physiol 299:G954–G966
Pastorino JG, Marcineviciute A, Cahill A, Hoek JB (1999) Potentiation by chronic ethanol treatment of the mitochondrial permeability transition. Biochem Biophys Res Commun 265:405–409
Venkatraman A, Shiva S, Wigley A, Ulasova E, Chhieng D, Bailey SM, Darley-Usmar VM (2004) The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology 40:565–573
Zhang X, Tachibana S, Wang H, Hisada M, Williams GM, Gao B, Sun Z (2010) Interleukin-6 is an important mediator for mitochondrial DNA repair after alcoholic liver injury in mice. Hepatology 52:2137–2147
•• Han D, Ybanez MD, Johnson HS, et al (2012) Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: biogenesis, remodeling, and functional alterations. J Biol Chem 287:42165–42179. This study characterizes the increased mitochondrial respiratory capacity and biogenesis in liver of mice fed alcohol, which intriguingly correlate with increased liver injury
Jäger S, Handschin C, St-Pierre J, Spiegelman BM (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci USA 104:12017–12022
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434:113–118
Baur JA, Pearson KJ, Price NL et al (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444:337–342
Shen Z, Ajmo JM, Rogers CQ, Liang X, Le L, Murr MM, Peng Y, You M (2009) Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-α production in cultured macrophage cell lines. Am J Physiol Gastrointest Liver Physiol 296:G1047–G1053
Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD et al (2007) Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131:476–491
•• Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–224. This study demonstrates that mitochondrial oxidative stress and ROS generation result in the recruitment of NLRP3 and subsequent inflammation activation
• Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menas V, Barrieaud M, Min SY, Kurt-Jones EA, Szabo G (2012) IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 122:3476–3489. This study identifies the caspase 1 activation as an important mechanism of IL-1β generation and ALD
Bailey SM, Cunningham CC (2002) Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med 32:11–16
Wu D, Cederbaum AE (2009) Oxidative stress and alcoholic liver disease. Semin Liver Dis 29:141–154
• Wang X, Wu D, Yang L, Gan L, Cederbaum AI. CYP2E1 Potentiates Ethanol-induction of Hypoxia and HIF-1α in vivo. Free Rad Biol Med 2013 May 10. doi: 10.1016/j.freeradbiomed.2013.05.009. This paper demonstrates a critical role for CYP2E1 in the potentiation of alcohol on hypoxia and HIF-1α activation, and that HIF-1α inhibition prevents these effects
Knockaert L, Fromenty B, Robi MA (2011) Mechanisms of mitochondrial targeting of cytochrome P4502E1: physiopathological role in liver injury and obesity. FEBS J 278:4252–4260
Robin MA, Sauvage I, Grandperret T, Descatoire V, Pessayre D, Fromenty B (2005) Ethanol increases mitochondrial cytochrome P450 2E1 in mouse liver and rat hepatocytes. FEBS Lett 579:6895–6902
Bansal S, Liu CP, Sepuri NB et al (2010) Mitochondria-targeted cytochrome P450 2E1 induces oxidative damage and augments alcohol-mediated oxidative stress. J Biol Chem 285:24609–24619
Mari M, Colell A, Morales A et al (2009) Redox control of liver function in health and disease. Antioxid Redox Signal 12:1295–1331
Giorgio et al (2005) Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122:221–233
Koch OR, Fusco S, Ranieri SC, Maulucci G, Palozza P, Larocca LM, Cravero AA, Farre’ SM, De Spirito M, Galeotti T, Pani G (2008) Role of the life span determinant P66(shcA) in ethanol-induced liver damage. Lab Invest 88:750–760
Koch OR, De Leo ME, Borrello S et al (1994) Ethanol treatment up-regulates the expression of mitochondrial manganese superoxide dismutase in rat liver. Biochem Biophys Res Commun 201:1356–1365
Wheeler MD, Nakagami M, Bradford BU et al (2001) Overexpression of manganese superoxide dismutase prevents alcohol-induced liver injury in the rat. J Biol Chem 276:36664–36672
Nanji AA, Griniuviene B, Sadrzadeh SM et al (1995) Effect of type of dietary fat and ethanol on antioxidant enzyme mRNA induction in rat liver. J Lipid Res 36:736–744
Degoul F, Sutton A, Mansouri A et al (2001) Homozygosity for alanine in the mitochondrial targeting sequence of superoxide dismutase and risk for severe alcoholic liver disease. Gastroenterology 120:1468–1474
Hirano T, Kaplowitz N, Tsukamoto H, Kamimura S, Fernandez-Checa JC (1992) Hepatic mitochondrial glutathione depletion and progression of experimental alcoholic liver disease in rats. Hepatology 16:1423–1427
Garcia-Ruiz C, Morales A, Ballesta A et al (1994) Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J Clin Invest 94:193–201
Garcia-Ruiz C, Morales A, Colell A, Ballesta A, Rodes J, Kaplowitz N, Fernandez-Checa JC (1995) Feeding S-adenosyl-l-methionine attenuates both ethanol-induced depletion of mitochondrial glutathione and mitochondrial dysfunction in periportal and perivenous rat hepatocytes. Hepatology 21:207–214
Colell A, Coll O, García-Ruiz C, París R et al (2001) Tauroursodeoxycholic acid protects hepatocytes from ethanol-fed rats against tumor necrosis factor-induced cell death by replenishing mitochondrial glutathione. Hepatology 34:964–971
Zhao P, Kalhorn TF, Slattery JT (2002) Selective mitochondrial glutathione depletion by ethanol enhances acetaminophen toxicity in rat liver. Hepatology 36:326–335
Zhao P, Slattery JT (2002) Effects of ethanol dose and ethanol withdrawal on rat liver mitochondrial glutathione: implication of potentiated acetaminophen toxicity in alcoholics. Drug Metab Dispos 30:1413–1417
Guidot DM, Brown LAS (2000) Mitochondrial glutathione replacement restores surfactant synthesis and secretion in alveolar epithelial cells of ethanol-fed rats. Alcohol Clin Exp Res 24:1070–1076
• Fernandez A, Matias N, Fucho R, et al () ASMase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. J Hepatol. doi: 10.1016/j.jhep.2013.05.023 This study demonstrates that ASMase links alcohol feeding to ER stress, and that this event is critical in causing mitochondrial cholesterol loading
Josekutty J, Iqbal J, Iwawaki T et al (2013) Microsomal triglyceride transfer protein inhibition induces endoplasmic reticulum stress and increases gene transcription via Ire1α/cJun to enhance plasma ALT/AST. J Biol Chem 288:14372–14383
Ha SD, Park S, Han CY et al (2012) Cellular adaptation to anthrax lethal toxin-induced mitochondrial cholesterol enrichment, hyperpolarization, and reactive oxygen species generation through downregulating MLN64 in macrophages. Mol Cell Biol 32:4846–4860
•• von Montfort C, Matias N, Fernandez A, et al (2012) Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis. J Hepatol 57:852–859. This study illustrates the importance of a strict equilibrium between antioxidant enzymes in steatohepatitis
Mansouri A, Tarhuni A, Larosche I et al (2010) MnSOD overexpression prevents liver mitochondrial DNA depletion after an alcohol binge but worsens this effect after prolonged alcohol consumption in mice. Dig Dis 28:756–775
Malhi H, Kaufman RJ (2011) Endoplasmic reticulum stress in liver disease. J Hepatol 54:795–809
Fu S, Yang L, Li P, Hofmann O et al (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473:528–531
Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109:1125–1131
Browning JD, Horton JD (2004) Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 114:147–152
Goldstein JL, Brown MS (1990) Regulation of the mevalonate pathway. Nature 343:425–430
Ji C, Kaplowitz N (2006) ER stress: can the liver cope? J Hepatol 45(2):321–333
Ji C, Kaplowitz N (2003) Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 124(5):1488–1499
Kharbanda KK, Todero SL, King AL et al (2012) Betaine treatment attenuates chronic ethanol-induced hepatic steatosis and alterations to the mitochondrial respiratory chain proteome. Int J Hepatol 2012:962183
Tsuchiya M, Ji C, Kosyk O et al (2012) Interstrain differences in liver injury and one-carbon metabolism in alcohol-fed mice. Hepatology 56(1):130–139
• Boini KM, Xia M, Abais JM, et al (2012) Acid sphingomyelinase gene knockout ameliorates hyperhomocysteinemic glomerular injury in mice lacking cystathionine-b-synthase. PLoS One 7:e45020. This paper indicates that the deleterious effect of genetic hyperhomocysteinemia is dependent on the presence of acid sphingomyelinase
Caballero F, Fernández A, Matías N et al (2010) Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: impact on mitochondrial S-adenosyl-l-methionine and glutathione. J Biol Chem 285:18528–18536
Henkel AS, Dewey AM, Anderson KA et al (2012) Reducing endoplasmic reticulum stress does not improve steatohepatitis in mice fed a methionine- and choline-deficient diet. Am J Physiol Gastrointest Liver Physiol 303:G54–G59
Marí M, Caballero F, Colell A et al (2006) Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab 4:185–198
Mari M, Colell A, Morales A et al (2008) Mechanism of mitochondrial glutathione-dependent hepatocellular susceptibility to TNF despite NF-kappaB activation. Gastroenterology 134:1507–1520
Anuka E, Gal M, Stocco DM, Orly J (2013) Expression and roles of steroidogenic acute regulatory (StAR) protein in ‘non-classical’, extra-adrenal and extra-gonadal cells and tissues. Mol Cell Endocrinol 371:47–61
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E et al (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Liesa M, Palacín M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89(3):799–845
Zorzano A, Sebastián D, Segalés J, Palacín M (2009) The molecular machinery of mitochondrial fusion and fission: an opportunity for drug discovery? Curr Opin Drug Discov Devel 12(5):597–606
Chan DC (2006) Dissecting mitochondrial fusion. Dev Cell 11:592–594
Chen H, Chomyn A, Chan DC (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280(28):26185–26192
Shutt TE, McBride HM (2013) Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta 1833(2):417–424
Agier V, Oliviero P, Lainé J et al (2012) Defective mitochondrial fusion, altered respiratory function, and distorted cristae structure in skin fibroblasts with heterozygous OPA1 mutations. Biochim Biophys Acta 1822(10):1570–1580
Zhang Z, Wakabayashi N, Wakabayashi J et al (2011) The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell 22:2235–2245
Liesa M, Shirihai OS (2013) Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17:491–506
Das S, Hajnoczky N, Anthony AN et al (2012) Mitochondrial morphology and dynamics in hepatocytes from normal and ethanol-fed rats. Pfugers Arch 464:101–109
Mitra K, Wunder C, Roysam B et al (2009) A hyperfused mitochondrial state achieved at G1–S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA 106(29):11960–11965
•• Gomes, LC, Di Benedetto G, Scorrano,L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13:589–598. This study underscores the role of mitochondrial elongation on resistance to autophagy and increased ATP synthesis
Wasilewski M, Semenzato M, Rafelski SM et al (2012) Optic atrophy 1-dependent mitochondrial remodeling controls steroidogenesis in trophoblasts. Curr Biol 22:1228–1234
Huang JY, Hirschey MD, Shimazu T, Ho L, Verdin E (1804) Mitochondrial sirtuins. Biochim Biophys Acta 2010:1645–1651
Lombard DB, Alt FW, Cheng HL et al (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 27:8807–8814
Hirschey MD, Shimazu T, Goetzman E et al (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464:121–125
Picklo MJ (2008) Ethanol intoxication increases hepatic N-lysyl protein acetylation. Biochim Biophys Res Commun 376(3):615–619
• Fritz KS, Galligan JJ, Smathers RL, et al (2011) 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem Res Toxicol 24: 651–662. This study provides novel mechanisms of SIRT3 inactivation, which has widespread implications in oxidative stress including alcoholic liver disease
Verdin E, Hirschey MD, Finley LW, Haigis MC (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 35:669–675
Fritz KS, Galligan JJ, Hirschey MD et al (2012) Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J Proteom Res 11:1633–1643
Acknowledgments
This work was supported by Grant P50-AA-11999 from the Research Center for Liver and Pancreatic Diseases supported by National Institute on Alcohol Abuse and Alcoholism/National Institutes of Health; CIBERehd; Fundació la Marató de TV3; Fundación Mutua Madrileña; and Grants SAF2010-15760, SAF2011-23031, SAF2012-34831 (Plan Nacional de I+D), Spain.
Disclosure
Carmen García-Ruiz, Neil Kaplowitz, and José C. Fernandez-Checa declare that they have no conflict of interest.
Compliance With Ethics Guidelines
This article does not contain any studies with human subjects performed by any of the authors. With regard to the authors’ research cited in this paper, all institutional and national guidelines for the care and use of laboratory animals were followed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
García-Ruiz, C., Kaplowitz, N. & Fernandez-Checa, J.C. Role of Mitochondria in Alcoholic Liver Disease. Curr Pathobiol Rep 1, 159–168 (2013). https://doi.org/10.1007/s40139-013-0021-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-013-0021-z