
Abstract
Specific isotopic labeling of methyl groups in proteins has greatly extended the applicability of solution NMR spectroscopy. Simultaneous labeling of the methyl groups of several different amino acid types can offer a larger number of useful probes that can be used for structural characterisations of challenging proteins. Herein, we propose an improved AILV methyl-labeling protocol in which L and V are stereo-specifically labeled. We show that 2-ketobutyrate cannot be combined with Ala and 2-acetolactate (for the stereo-specific labeling of L and V) as this results in co-incorporation incompatibility and isotopic scrambling. Thus, we developed a robust and cost-effective enzymatic synthesis of the isoleucine precursor, 2-hydroxy-2-(1′-[2H2], 2′-[13C])ethyl-3-keto-4-[2H3]butanoic acid, as well as an incorporation protocol that eliminates metabolic leakage. We show that application of this labeling scheme to a large 82 kDa protein permits the detection of long-range 1H–1H NOE cross-peaks between methyl probes separated by up to 10 Å.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is unequivocally recognized that strategies for the specific isotopic labeling of methyl groups in proteins have substantially extended the applicability of solution NMR spectroscopy. Indeed, these advances have permitted solution NMR studies of supra-molecular complexes (>100 kDa), which were previously inaccessible to this technique (Plevin and Boisbouvier 2012; Rosenzweig and Kay 2014; Ruschak et al. 2010b). While such protein systems remain challenging for NMR structure determination, there are a growing number of elegant NMR studies of dynamics (Religa et al. 2010; Sprangers et al. 2007; Audin et al. 2013), interactions (Amero et al. 2011; Sprangers and Kay 2007) and function (Ruschak et al. 2010b; Shi and Kay 2014) of such systems; all of which have benefited from specific methyl group labeling technology. The first report of methyl selective labeling concerned the δ1-methyl group of isoleucine (I) (Gardner and Kay 1997). In this protocol, 2-keto, 3,3-[2H2],4-[13C]-butyrate was the sole source of protons added to perdeuterated culture media, to generate a [U-2H], Iδ1-[13CH3] labeled overexpressed protein. Subsequently, numerous protocols and an array of precursors have been developed to label the remaining methyl-containing amino acids, including: leucine (L) and valine (V) labeling with either 2-keto-isovalerate (Goto et al. 1999; Gross et al. 2003; Lichtenecker et al. 2004; Tugarinov and Kay 2004); 2-ketoisocaproate (Lichtenecker et al. 2013) or acetolactate (Gans et al. 2010; Mas et al. 2013; Miyanoiri et al. 2013) for either stereospecific or non-stereospecific labeling of L and V; alanine (A) (Isaacson et al. 2007; Ayala et al. 2009); the γ2 methyl group of isoleucine using 2-hydroxy-2-ethyl-3-keto-4-butanoic acid (Ayala et al. 2012), also known as 2-aceto-hydroxy butanoic acid (Ruschak et al. 2010a); methionine (M) (Fischer et al. 2007; Gelis et al. 2007); and recently threonine (T) (Velyvis et al. 2012).
Working with [13CH3]-labeled methyl groups in a perdeuterated background is a prerequisite for detecting high-quality NMR spectra of large proteins. A side effect of this strategy is the considerable loss of structural information due to the low number of remaining protonated probes. Methyl-containing residues represent 30–40 % of the amino acids in proteins and they are particularly abundant in the hydrophobic cores of folded proteins. Therefore, simultaneous labeling of the methyl groups of several amino acids represents an obvious route for increasing the number of NMR-visible sites from which useful structural restraints can be obtained. Various combinatorial methyl labeling strategies have been reported, including ILV (Gross et al. 2003; Lichtenecker et al. 2004; Tugarinov et al. 2005) and MILV(Gelis et al. 2007). For the ILV scheme, most studies reported the use of 2-ketoisovalerate and 2-ketobutyrate to label LV and the Iδ1 methyl groups, respectively. Because of the high abundance of alanine in proteins, this residue was later added to the ILV ensemble to further increase the measureable number of distance constraints (Godoy-Ruiz et al. 2010). However, 2-ketoisovalerate leads to racemic labeling of the LV methyl groups such that each prochiral methyl group is protonated at 50 %. Consequently, the intensity of inter-methyl group NOEs will be reduced by a factor of 4. Furthermore, the labeling of all methyl groups of leucine and valine generates two NMR-visible sites per residue and thus renders the analysis of the NMR 3D and 4D matrices more complex and time consuming.
In this work, we propose an alternative AILV labeling scheme, in which L and V are stereo-specifically labeled. Additionally, we demonstrate that combining diverse precursors for the simultaneous labeling of AILV can lead to “cross-talk” in the metabolic pathways. We verify that adding 2-ketobutyrate in combination with alanine and 2-acetolactate results in co-incorporation incompatibility and isotopic scrambling, which affects the quality of the prepared samples. Indeed, E. coli enzymatic machinery preferentially processes isoleucine precursors over 2-acetolactate. Therefore, these metabolites cannot be simultaneously added to the bacterial culture. Furthermore, 2-ketobutyrate cannot be used in combination with alanine as this leads to isotopic scrambling at Iδ2 positions. Herein, we suggest a modified protocol to enhance 2-acetolactate incorporation in the presence of isoleucine precursors. We also present a robust and cost-effective enzymatic synthesis of an alternative candidate for the Iδ1 labeling: 2-hydroxy-2-(1′-[2H2], 2′-[13C]) ethyl-3-keto-4-[2H3]butanoic acid. This precursor can be successfully combined with [13CH3]-alanine to obtain Iδ1 and Aβ methyl probes with no detectable isotopic scrambling. Finally, we demonstrate that our proposed AILV labeling scheme is a useful tool for the detection of long-range 1H–1H NOEs between methyl probes separated by up to 10 Å in large proteins.
Materials and methods
Preparation of aceto-hydroxy-acid synthase II (AHAS II)
The overexpression and purification of AHAS II (also known as acetolactate synthase ALS II) followed the protocol previously described (Vyazmensky et al. 1996). E. coli BL21(DE3) cells carrying the plasmid containing the AHAS II gene were grown at 37 °C in Luria Broth (LB) media. When the O.D. (600 nm) reached 0.5–0.7, AHAS II expression was induced by the addition of IPTG to a final concentration of 0.4 mM. Expression was performed for 12 h at 20 °C. The cells were harvested by centrifugation at 5,000g for 15 min at 4 °C, resuspended in 10 mL of 0.1 M TRIS–HCl, pH 7.5, and centrifuged at 4,000g for 15 min at 4 °C. The cells were resuspended in 10 mL of buffer A (50 mM TRIS, pH 8, 0.5 M KCl, 10 mM imidazole and 20 µM FAD). The cells were disrupted by sonication for 2 min and the insoluble materials were removed by centrifugation at 45,000g for 45 min at 4 °C. The supernatant was passed over a Ni–NTA column pre-equilibrated with buffer A. After washing with 5 equivalent volumes of buffer A, the protein was eluted using buffer B (50 mM TRIS, pH 8, 0.5 M KCl, 400 mM imidazole and 20 μM FAD). The fractions containing AHAS II were pooled, concentrated (55 mg/L), dialyzed against pure water and lyophilized. The activity of AHAS II was determined by measuring the decrease in absorbance at 333 nm of pyruvate.
Synthesis of 2-hydroxy-2- (1′-[2H2], 2′-[13C])ethyl-3-keto-4-[2H3]butanoic acid
The synthesis of 2-hydroxy-2-(1′-[2H2], 2′-[13C]) ethyl-3-keto-4-[2H3]butanoic acid was performed following the protocol previously described by D. Chipman (Engel et al. 2004). The reaction was initiated by adding an aliquot of AHAS II at 6 μM (420 ng/ml) to an equimolar (33 mM) mixture of deuterated pyruvate (perdeuterated by incubating unlabeled pyruvate in 2H2O at pH 10.7 for 72 h) and 3,3-[2H2],4-[13C]-2-ketobutyrate (Isotec) in 3 mL of 2H2O buffer containing 50 mM potassium phosphate, pH 7.8, 10 mM MgCl2, 1 mM thiamine diphosphate, and 20 μM FAD. This reaction was followed by 1D NMR.
Protein expression and purification
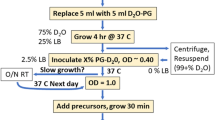
E. coli BL21(DE3) cells carrying the plasmids for ubiquitin or Malate synthase G (MSG) were progressively adapted to M9/2H2O media containing 1 g/L 15N2H4Cl (CortecNet) and 2 g/L d-glucose-d7 (Isotec) in three stages over 24 h. In the final culture, the bacteria were grown at 37 °C in M9 media prepared with 99.85 % 2H2O (Eurisotop). When the O.D. at 600 nm reached 0.7, a solution containing the labeled precursors was added.
-
For the production of [U-2H], I-[13CH3]δ1, L-[13CH3]proS, V-[13CH3]proS MSG sample:
2-[13CH3], 4-[2H3] acetolactate (NMR-Bio) at 300 mg/L was added 1 h prior to induction, 40 min later (i.e. 20 min prior to induction), 3,3-[2H2],4-[13C]-2-ketobutyrate (Isotec) was added to a final concentration of 60 mg/L. The incorporation of 13CH3 isotopomers was 50 % lower when both precursors are added simultaneously.
-
For the production of a [U-2H], A-[13CH3]β, I-[13CH3]δ1 ubiquitin sample:
A mixture of 60 mg/L 3,3-[2H2],4-[13C]-2-ketobutyrate (Isotec), 700 mg/L 2-[2H], 3-[13C]alanine (NMR-Bio) and 200 mg/L U-[2H] 2-ketoisovalerate (Isotec) was added 1 h prior to induction, according to Godoy-Ruiz et al. (2010). 13CH3 scrambling in the Iγ2 position was observed using this protocol.
-
For the production of a scrambling-free [U-2H], A-[13CH3]β, I-[13CH3]δ1 ubiquitin sample:
A mixture of 60 mg/L 2-hydroxy-2-(1′-[2H2], 2′-[13C])ethyl-3-keto-4-[2H3]butanoic acid, 700 mg/L 2-[2H], 3-[13C]alanine (NMR-Bio) and 200 mg/L U-[2H] 2-ketoisovalerate (Isotec) was added 1 h prior to induction.
-
For the production of a scrambling-free [U-2H], A-[13CH3]β, I-[13CH3]δ1, L-[13CH3]proS, V-[13CH3]proS MSG sample:
2-[13CH3], 4-[2H3] acetolactate (NMR-Bio) at 300 mg/L was added 1 h prior to induction. Forty minutes later (20 min prior to induction), 60 mg/L 2-hydroxy-2-(1′-[2H2], 2′-[13C])ethyl-3-keto-4-[2H3]butanoic acid and 700 mg/L 2-[2H], 3-[13C]alanine (NMR-Bio) were added. Protein expression was induced by a final IPTG concentration of 1 mM.
In order to quantify incorporation of precursors in I, L, and V residues, [U-2H], 200 mg/L U-[13C]-methionine was added 1 h before induction, to each ubiquitine cultures. The expression was performed overnight at 20 °C for MSG and 3 h at 37 °C for ubiquitin before harvesting. Ubiquitin was purified by Ni–NTA (Qiagen) chromatography in a single step. MSG was purified initially by Chelating Sepharose chromatography (GE Healthcare) followed by gel filtration chromatography (Superdex 200 pg; GE Healthcare). The typical final yields after purification were 40 and 80 mg/L of methyl-specific protonated ubiquitin and MSG, respectively.
NMR spectroscopy
The typical concentrations of ubiquitin and MSG in the NMR samples were 2 and 1 mM, respectively, in a 100 % 2H2O buffer containing either 20 mM Tris and 20 mM NaCl at pH 7.4 (ubiquitin) or 25 mM MES, 20 mM MgCl2 and 5 mM DTT at pH 7.1 (MSG). The 2D (1H,13C) NMR spectra of ubiquitin and MSG were recorded at 37 °C on a Varian (Agilent) DirectDrive spectrometer operating at a proton frequency of 800 MHz equipped with a cryogenic triple resonance probehead. The 3D HMQC-NOESY experiment was recorded for 82 h with a 1 mM [U-2H], A-[13CH3]β, I-[13CH3]δ1, L-[13CH3]proS, V-[13CH3]proS MSG sample and a NOE mixing time of 500 ms. The experiment was recorded with 4 scans per increment and maximum acquisition times of 20 ms in both the 13C and 1H indirect dimensions. All data were processed and analyzed using nmrPipe/nmrDraw (Delaglio et al. 1995) and CCPN software (Vranken et al. 2005).
Results and discussion
Co-incorporation of precursors for isoleucine and stereospecific leucine and valine labelling
ILV combinatorial labeling has been used as a tool for the study of several biological systems (Gross et al. 2003; Lichtenecker et al. 2004; Tugarinov and Kay 2004). Most of the previous reports have used 2-ketobutyrate and 2-ketoisovalerate for the labeling of Iδ1 and the non-stereospecific labeling of the prochiral methyl groups of L and V, respectively. Depending on the size of the system studied, reducing the number of resonances and resonance overlap can be crucial for interpretation of spectra. The use of 2-acetolactate offers a robust solution for crowded spectra through stereospecific labeling of only one of the prochiral methyl groups of leucine and valine. In addition to spectral resolution enhancement, stereospecific labeling can increase the intensity of long-range NOEs by a factor of 4 compared to the labeling pattern obtained with 2-ketoisovalerate precursor (Gans et al. 2010). To test the Iδ1(LV)proS combination, a culture of ubiquitin was grown as described in “Materials and methods” section. A 2D (1H,13C) spectrum of this sample of ubiquitin is shown in Fig. 1a. Interestingly, signals corresponding to (LV)proS methyl groups were significantly less intense than those of Iδ1 methyl groups. Integration of these signals shows a substantial reduction in isotopic incorporation at (LV)proS sites (Fig. 1c). Under our culture conditions, isotopic incorporation at (LV)proS sites was estimated to be 50–60 % of that at Iδ1 sites. Low incorporation rate severely affects the detection of NMR data. Indeed, the intensity of the detectable NOE correlations between the Iδ1 and (LV)proS residues is estimated to be diminished by a factor of up to 2. For NOEs involving LproS and VproS methyl groups, the intensity reduction is expected to be 3 to 4 fold. Notably, this co-incorporation incompatibility is not observed with the 2-ketoisovalerate precursor. Thus, the co-incorporation of 2-ketobutyrate and 2-acetolactate had to be investigated.

The co-incorporation of Iδ1 and (LV)proS precursors in a ubiquitin sample: 2D HSQC NMR spectra were recorded at 37 °C in 2H2O buffer (20 mM Tris, pH 7.4, and 20 mM NaCl) on a 800 MHz NMR spectrometer equipped with a cryogenic probe. The [U-2H], A-[13CH3]β, I-[13CH3]δ1-ubiquitin sample was prepared using 2-[13CH3], 4-[2H3]acetolactate and 3,3-[2H2], 4-[13C]-2-ketobutyrate. a Both precursors were added simultaneously to the culture 1 h before induction (3 h). b Labeled acetolactate was added to the culture 1 h before induction (3 h), while the labeled 2-ketobutyrate was added 20 min before induction. c Quantification of the I-δ1 and (LV)-pro-S signals. For incorporation level quantification of precursors in I, L, and V residues, [U-2H], 200 mg/L U-[13C]-methionine was added to the culture 1 h before induction. Met-ε methyl groups were used as an internal reference. The experimental I-δ1/M-ε and LV-pro-S/Met-ε ratios were compared to those obtained for conditions with complete incorporation of each precursor added alone
Enhancement of the co-incorporation level of 2-acetolactate and the isoleucine precursor
Thorough analysis of the enzymatic machinery involved in the processing of both 2-ketobutyrate and 2-acetolactate (Fig. 2) indicates that the ketol-acid reductoisomerase (EC.1.1.1.86), known as KARI, more efficiently processes the isoleucine precursors than those of leucine and valine. KARI presents a 5- to 8-fold higher activity with 2-hydroxy-2-ethyl-3-ketobutanoic acid (isoleucine precursor) than with 2-acetolactate (Dumas et al. 2001). This information could explain the more efficient incorporation of 2-ketobutyrate compared to 2-acetolactate (Fig. 1). It may also explain why the incorporation of 2-ketoisovalerate, whose enzymatic processing occurs after KARI step, is not affected in presence of 2-ketobutyrate. Considering this information, we hypothesized that adding 2-ketobutyrate to the culture after 2-acetolactate would promote enzymatic processing of the LV precursor. To test this hypothesis, a ubiquitin culture was prepared in which the two precursors were added at separate times: 2-acetolactate was added 1 h before induction, while 2-ketobutyrate was added 40 min later (i.e. 20 min before induction). Quantification of the resulting isotopic-labeling patterns clearly shows that this 2-step approach enhances the incorporation of the 2-acetolactate precursors to more than 90 %, without significantly affecting the level of Iδ1 labeling (Fig. 1c).

Biosynthetic pathway of the AILV residues. The pathway for the scrambling of 13CH3 group from alanine into isoleucine, leucine and valine is indicated in red. The carbons from 2-ketobutyrate are depicted in blue. Each biosynthetic intermediate has been named according to the Kyoto Encyclopedia of Genes and Genomes (KEGG). The enzymes responsible for catalyzing each reaction are indicated by EC number. EC 2.2.1.6: aceto-hydroxy-acid synthase; EC 1.1.1.86: ketol-acid reductoisomerase (for simplification, we indicated only the enzymes referenced in the text). Dashed arrows indicate multistep reactions. Further information on the Ala, Ile, Leu and Val metabolic pathway can be found online: http://www.genome.jp/kegg/
Isotopic scrambling at the Iγ2 position in the standard alanine and isoleucine (AβIδ1) labeling scheme
To achieve a AβIδ1 (LV)proS labeling pattern, 2-[2H], 3-[13C]alanine must be used in conjunction with 2-[13CH3], 4-[2H3]acetolactate and 3,3-[2H2], 4-[13C]-2-ketobutyrate. However, the simultaneous availability of 2-ketobutyrate and labeled alanine in the cell is expected to generate an isotopic leak to the Iγ2 methyl group position (Ayala et al. 2009). In E. coli, the first common step in the biosynthesis of the branched-chain amino acids (I, L and V) is catalyzed by three aceto-hydroxy-acid synthases (AHAS: EC 2.2.1.6) (Bar-Ilan et al. 2001; Umbarger 1996). These enzymes catalyse two condensation reactions involving pyruvate: (1) reaction of pyruvate with 2-ketobutyrate to produce the I precursor 2-hydroxy-2-ethyl 3-ketobutanoic acid; or (2) reaction of two molecules of pyruvate producing the L/V precursor 2-acetolactate (see Fig. 2). The origin of the Iγ2 methyl group is the pyruvate. The addition of an excess of 2-[2H], 3-[13C]alanine to the culture medium causes the cellular pyruvate pool to become partially labeled due to the transamination activity of various enzymes (Ayala et al. 2009), such as alanine-valine transaminase (AvtA), YfbQ and YfbZ. Deamination of 2-[2H], 3-[13C]alanine produces 3-[13C]-pyruvate, which is subsequently combined with the added 3,3-[2H2], 4-[13C]-2-ketobutyrate, leading to the appearance of a fraction of isoleucine that is labeled at both the δ1 and γ2 methyl groups. To demonstrate this phenomenon, “standard” AβIδ1 labeling was applied to ubiquitin (see Fig. 3). As expected, signals corresponding to the Iγ2 methyl groups were also observed, demonstrating the predicted scrambling pathway. Integration of the crosspeaks revealed that these signals represent approximately 2–5 % of the Iδ1 methyl group content. For this calculation, we considered solely the signals of the CH3 isotopomer because the 13CH 22 H and 13CH2H2 isotopomers represent <5 % of the Iγ2 13CH3 signals. Similar artifacts were also detected when MSG (82 kDa) was overexpressed using the same protocol (spectra available online as electronic supplementary material).

Isotope scrambling of A-β methyl groups into I-γ2 positions using combination of 2-ketobutyrate and alanine. 2D HSQC NMR spectra were recorded at 37 °C in 2H2O buffer (20 mM Tris, 20 mM NaCl pH 7.4) on an NMR spectrometer operating at a proton frequency of 600 MHz. [U-2H], A-[13CH3]β, I-[13CH3]δ1ubiquitin was prepared using 3,3-[2H2],4-[13C]-2-ketobutyrate, 2-[2H], 3-[13C]alanine and U-[2H] 2-ketoisovalerate. Spectrum was plotted at 10 % of the average maximal intensity of I-δ1 signals. I-γ2 part of the spectra (dashed box) was plotted at 1 % of the average maximal intensity of I-δ1 signals. Signals for β alanine and δ1 methyl carbons of isoleucine were observed in 13C-HSQC spectra, but also peaks corresponding to the resonance γ2 methyl carbons of isoleucine. The level of isotopic scrambling in γ2 methyl carbons of isoleucine was estimated to be approximately 2–5 %
Suppression of isotopic scrambling at the Iγ2 position in AβIδ1 labelling
Low level scrambling to Iγ2 methyl groups can be neglected for many types of NMR applications (e.g., interaction, dynamics and assignment using triple resonance experiments). However, for structural studies based on the detection and analysis of NOEs, these spurious correlations could cause erroneous assignments and consequently incorrect distance restraints. The intensity of an NOE between two sites is inversely proportional to sixth power of the inter-site distance. In the case of isoleucine, where the distance between the δ1 and γ2 methyl groups is on the order of 3 Å, the expected NOE intensity between a fully-labeled δ1 site and a fractionally-labeled γ2 site would correspond to NOE between two fully-labeled sites distant by c.a. 6 Å. Given that it is possible to detect inter-methyl NOEs for distances as long as 12 Å (Sounier et al. 2007), fractional labeling of γ2 methyl sites due to isotopic scrambling can introduce misleading artefacts in 3D 13C edited NOESY experiments.
To avoid scrambling to Iγ2 methyl groups it would be necessary to use an alternative I precursor to 2-ketobutyrate. Analysis of the relevant metabolic pathways shows that 2-hydroxy-2-ethyl-3-ketobutanoic acid (see Fig. 2) may be an alternative option. This molecule has already been used for Iγ2 labeling (Ayala et al. 2012; Ruschak et al. 2010a). In these studies, this precursor was produced chemically and the protocol for its synthesis is well described. However, utilization of the same compound for Iδ1 labeling would require an alternative synthesis strategy that uses 1-[2H2], 2-[13C]-ethyl iodide. However, this compound is unavailable commercially. Therefore, we sought to develop an alternative synthesis that could be easily performed in any biochemistry laboratory. We evaluated whether AHAS II could be used to synthesize the required precursor by catalysing the condensation of pyruvate with 2-ketobutyrate. Considering this approach, 2-hydroxy-2- (1′-[2H2], 2′-[13C])ethyl-3-keto-4-[2H3]butanoic acid was produced using AHAS II from an equimolar mixture of deuterated pyruvate and 3,3-[2H2],4-[13C]-2-ketobutyrate, according to the protocol described in Materials & Methods section. The 1H NMR spectra of the initial and final compounds from the synthetic reaction are presented in Fig. 4. The reaction reached completion in 2 h, as shown by the disappearance of the signals from 2-ketobutyrate. The 2-hydroxy-2-(1′-[2H2], 2′-[13C])ethyl-3-keto-4-[2H3]butanoic acid appears as a doublet at 0.8 ppm. A fully deuterated 2-acetolactate (not visible by NMR) is also synthesized as a side-product during the reaction. This compound is produced at approximately 10 % of the 2-hydroxy-2-ethyl-3-ketobutanoic acid, as determined in comparable reactions performed with protonated and non-labeled reactants (data not shown).

1D NMR spectra representing the enzymatic synthesis of the isoleucine precursor. 2-hydroxy-2-(1′-[2H2], 2′-[13C])ethyl-3-keto-4-[2H3]butanoic acid (3) was synthesized by condensation of 3,3-[2H2],4-[13C]-2-ketobutyrate (1) with [U-2H]-pyruvate (2) using AHAS II as described in “Materials and methods” section. In black: 1D spectrum of the reaction solution before condensation (deuterated pyruvate is not visible). In red: 1D spectrum after condensation. Single asterisk indicates the minor gem-diol form of 2-ketobutyrate (CH3–CH2–C(OH)2–COOH). Double asterisks indicates buffer signal
The ability of the synthesized precursor to suppress undesired enrichment of Iγ2 methyl groups was tested. Ubiquitin was expressed using the produced compound without any purification (100 mg/L) in conjunction with an excess of labeled alanine (700 mg/L) and [U-2H]-2-ketoisovalerate (200 mg/L). NMR analysis of the resulting ubiquitin sample shows that the Iγ2 methyl group signals were efficiently removed without affecting those of the Iδ1 methyl groups (Fig. 5). We then sought to determine the efficiency of precursor incorporation. A previous protocol for synthesizing 2-hydroxy-ethyl-3-ketobutanoic acid produced a racemic mixture of R and S forms, with only the S form being utilized by the cell (Ayala et al. 2012). The enzymatic synthesis scheme outlined here produces only the S form. Consequently, only half the amount of 2-hydroxy-2- (1′-[2H2], 2′-[13C])ethyl-3-keto-4-[2H3]butanoic acid should be required to achieved maximal Iδ1 incorporation compared to the chemically synthesized precursor. To test this hypothesis, increasing amounts of both compounds were added to different ubiquitin cultures. The resulting obtained incorporation curves are shown in Fig. 6. 95 % incorporation at Iδ1 sites was achieved using 80 mg/mL of the enzymatically synthesized compound compared to 100 mg/L of the chemically synthesized compound (Fig. 6). In addition, the enzymatic synthesis described offers a considerable amount of flexibility in the final labeling pattern. For example, simply by modifying the initial reactants, this approach can be used to produce precursors for both Iδ1 and Iγ2 labeling.

Scrambling-Free Combinatorial labeling of Alanine and Isoleucine-δ1. The 2D HSQC NMR spectrum were recorded at 37 °C in a 2H2O buffer (20 mM Tris, pH 7.4, and 20 mM NaCl) on an NMR spectrometer operating at a proton frequency of 600 MHz. The [U-2H], [U-12C], I-[13C 1H3]δ1 ubiquitin was prepared using 2-hydroxy-2-(1′-[2H2], 2′-[13C])ethyl-3-keto-4-[2H3]butanoic acid and 2-[2H], 3-[13C]alanine. The spectrum is displayed and was quantified as described in Fig. 3. The incorporation level of the 13CH3 groups in the δ1 position of isoleucine and the β position of Ala was estimated to be higher than 95 %, based on the integration of the NMR signals observed in a two-dimensional 2D (1H–13C) spectrum of the labeled proteins. Only the signals for the δ1 isoleucine methyl carbons were observed in the 13C-HSQC spectra, indicating that the 13C1H3 groups of (S)-2-hydroxy-2-(1′-[2H2], 2′-[13C])ethyl-3-keto-4-[2H3]butanoic acid were not incorporated into the metabolic pathways of the other amino acid precursors, respectively

Incorporation level of the chemically and enzymatically synthesized precursors in overexpressed ubiquitin as a function of the amount of exogenous labeled precursor added. The ubiquitin samples were prepared in E. coli cultures in M9 minimal media in the presence of increasing amounts of either racemic chemically synthesized precursor (full blue circles) or enzymatically synthesized precursor (open red squares) along with 200 mg/L U-[13C]-methionine. An isoleucine side-chain incorporation level of up to 95 % was achieved by adding 100 and 80 mg of the chemically and enzymatically synthesized compounds, respectively, per liter of M9/2H2O culture medium
AβIδ1(LV)proS application on MSG for the detection of long-range NOEs
Protein structure determination by NMR relies on the extraction of a large set of meaningful structural restraints, such as NOEs, dihedral angle restraints and residual dipolar couplings (RDCs). However, while accurate local structural information can be easily achieved, information on overall folding is seriously compromised, particularly for elongated or modular biological systems. Therefore, long-range 1H–1H NOEs are valuable for insight into the global shape of the protein. NOE correlations for 1H–1H distances of 8–12 Å have been previously reported in a perdeuterated protein where a restricted number of methyl groups were selectively protonated (Sounier et al. 2007). While advantageous to improve spectra quality, reducing proton concentration also decreases the number of detectable NOEs and therefore the overall numbers of distance restraints obtainable.
In this study, we were interested in confirming whether these long-range NOEs remain detectable in large perdeuterated systems where a higher number of protonated methyl probes has been introduced. Taking advantage of the optimised AβIδ1(LV)proS labeling protocol described above, a [U-2H], A-[13CH3]β, I-[13CH3]δ1, (LV)-[13CH3]proS labeled sample of MSG (82 kDa) was prepared. The 2D (1H,13C) HSQC spectrum recorded on this sample is shown in Fig. 7, confirming the high quality of the sample prepared and the excellent fidelity of the labeling strategy. Analysis of a 3D (1H,1H,13C) HMQC-NOESY spectrum indicated that 96 % of the expected NOEs between the methyl pairs separated by 2.5–8.5 Å were detected (theoretical distances were predicted from the 1D8C PDB co-ordinates, Howard et al. 2000). Moreover, ~50 % of the NOEs arising from methyl groups separated by 9.5–10.5 Å could also be detected. Examples of 2D (1H,1H) strips showing long-range NOEs are presented in Fig. 8. Thus, despite higher proton density (14 %) than the sample used in previous analyses, a large number of long-range NOEs are still detectable. Likewise a higher number of distance restraints could be determined: out of 2,294 expected NOEs within a distance range of 2.5–10.5 Å, 1,714 were experimentally observed. However, the contribution of spin diffusion can not be neglected as relatively long mixing times (500 ms) was used to extract NOEs from a 50 ns tumbling protein. Full-relaxation-matrix analysis is required to extract precise distance restraints from diagonal signals and NOEs cross-peaks (Sounier et al. 2007). These long-range distances will be particularly useful for structure determination of large proteins and complexes.

2D [1H,13C] spectra of [U-2H], A-[13CH3]β, I-[13CH3]δ1, (LV)-[13CH3]proS MSG (82 kDa). MSG sample was produced with scrambling-free and incorporation optimized combinatorial labeling protocol described in “Materials and methods” section. Spectra were acquired at 37 °C on a spectrometer operating at a proton frequency of 800 MHz. The sample concentrations was 1 mM in 2H2O buffer at pH 7.1 containing 20 mM MES and 25 mM MgCl2. In blue circle Iδ1 signals, in red circle Aβ signals, in yellow circle VproS signals and green circle LproS signals

Detection of long-range nOes in the [U-D], A-[13CH3]β, I-[13CH3]δ1, (LV)-[13CH3]proS-MSG sample (82 kDa). 2D extracts of the 3D 13C-HMQC–NOESY spectrum are presented along with the extracted distances between each pair of methyl groups from the 3D structure of MSG (1D8C; Howard et al. 2000). The experiment was acquired on a 800 MHz spectrometer equipped with a cryoprobe at 37 °C for 3.5 days with recycling delay d1 = 1.3 s and NOE mixing time τm = 500 ms. The [U-2H, AβIδ1(LV)proS]-MSG sample was at 1 mM in a 2H2O buffer containing 20 mM MES, pH 7.1, and 25 mM MgCl2. Diagonal peaks are labeled by residue numbers in red and nOes cross-peaks in black
Conclusion
We have outlined an optimized protocol for scrambling-free AβIδ1(LV)proS labeling that will be widely applicable. Simultaneous labeling of the methyl groups of four different amino acids serves to increase the number of useful probes for measuring long-range NOEs in large or challenging proteins. Thanks to this labeling scheme, which optimises both resolution and sensitivity, we could detect long-range 1H–1H NOEs between methyl probes separated by up to 10 Å in high molecular weight proteins. This labeling scheme will be useful for structural characterization of large proteins and protein complexes.
References
Amero C, Asuncion Dura M, Noirclerc-Savoye M, Perollier A, Gallet B, Plevin MJ, Vernet T, Franzetti B, Boisbouvier J (2011) A systematic mutagenesis-driven strategy for site-resolved NMR studies of supramolecular assemblies. J Biomol NMR 50:229–236
Audin MJ, Dorn G, Fromm SA, Reiss K, Schütz S, Vorländer MK, Sprangers R (2013) The archaeal exosome: identification and quantification of site-specific motions that correlate with cap and RNA binding. Angew Chem Int Ed Engl 52:8312–8316
Ayala I, Sounier R, Use N, Gans P, Boisbouvier J (2009) An efficient protocol for the complete incorporation of methyl-protonated alanine in perdeuterated protein. J Biomol NMR 43:111–119
Ayala I, Hamelin O, Amero C, Pessey O, Plevin MJ, Gans P, Boisbouvier J (2012) An optimized isotopic labelling strategy of isoleucine-γ2 methyl groups for solution NMR studies of high molecular weight proteins. Chem Commun (Camb) 48:1434–1436
Bar-Ilan A, Balan V, Tittmann K, Golbik R, Vyazmensky M, Hubner G, Barak Z, Chipman DM (2001) Binding and activation of thiamin diphosphate in acetohydroxyacid synthase. Biochemistry 40:11946–11954
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Dumas R, Biou V, Halgand F, Douce R, Duggleby RG (2001) Enzymology, structure, and dynamics of acetohydroxy acid isomeroreductase. Acc Chem Res 34:399–408
Engel S, Vyazmensky M, Berkovich D, Barak Z, Chipman DM (2004) Substrate range of acetohydroxy acid synthase I from Escherichia coli in the stereoselective synthesis of alpha-hydroxy ketones. Biotechnol Bioeng 88:825–831
Fischer M, Kloiber K, Hausler J, Ledolter K, Konrat R, Schmid W (2007) Synthesis of a 13C-methyl-group-labeled methionine precursor as a useful tool for simplifying protein structural analysis by NMR spectroscopy. ChemBioChem 8:610–612
Gans P, Hamelin O, Sounier R, Ayala I, Dura MA, Amero CD, Noirclerc-Savoye M, Franzetti B, Plevin MJ, Boisbouvier J (2010) Stereospecific isotopic labeling of methyl groups for NMR spectroscopic studies of high-molecular-weight proteins. Angew Chem Int Ed Engl 49:1958–1962
Gardner KH, Kay LE (1997) Production and incorporation of 15N, 13C, 2H (1H-d1 methyl) isoleucine into proteins for multidimensional NMR studies. J Am Chem Soc 119:7599–7600
Gelis I, Bonvin AM, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, Economou A, Kalodimos CG (2007) Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell 131:756–769
Godoy-Ruiz R, Guo C, Tugarinov V (2010) Alanine methyl groups as NMR probes of molecular structure and dynamics in high-molecular-weight proteins. J Am Chem Soc 132:18340–18350
Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE (1999) A robust and cost-effective method for the production of Val, Leu, Ile (delta 1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J Biomol NMR 13:369–374
Gross JD, Gelev VM, Wagner G (2003) A sensitive and robust method for obtaining intermolecular NOEs between side chains in large protein complexes. J Biomol NMR 25:235–242
Howard BR, Endrizzi JA, Remington SJ (2000) Crystal structure of Escherichia coli malate synthase G complexed with magnesium and glyoxylate at 2.0 A resolution: mechanistic implications. Biochemistry 39:3156–3168
Isaacson RL, Simpson PJ, Liu M, Cota E, Zhang X, Freemont P, Matthews S (2007) A new labeling method for methyl transverse relaxation-optimized spectroscopy NMR spectra of alanine residues. J Am Chem Soc 129:15428–15429
Lichtenecker R, Ludwiczek ML, Schmid W, Konrat R (2004) Simplification of protein NOESY spectra using bioorganic precursor synthesis and NMR spectral editing. J Am Chem Soc 126:5348–5349
Lichtenecker RJ, Weinhaupl K, Reuther L, Schorghuber J, Schmid W, Konrat R (2013) Independent valine and leucine isotope labeling in Escherichia coli protein overexpression systems. J Biomol NMR 57:205–209
Mas G, Crublet E, Hamelin O, Gans P, Boisbouvier J (2013) Specific labeling and assignment strategies of valine methyl groups for NMR studies of high molecular weight proteins. J Biomol NMR 57:251–262
Miyanoiri Y, Takeda M, Okuma K, Ono AM, Terauchi T, Kainosho M (2013) Differential isotope-labeling for Leu and Val residues in a protein by E. coli cellular expression using stereo-specifically methyl labeled amino acids. J Biomol NMR 57:237–249
Plevin M, Boisbouvier J (2012) Isotope-labelling of methyl groups for NMR studies of large proteins. In: Clore M, Potts J (eds) Recent developments in biomolecular NMR. Royal Society of Chemistry, London
Religa TL, Sprangers R, Kay LE (2010) Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science 328:98–102
Rosenzweig R, Kay LE (2014) Bringing dynamic molecular machines into focus by methyl-TROSY NMR. Annu Rev Biochem 83:291–315
Ruschak AM, Velyvis A, Kay LE (2010a) A simple strategy for 13C, 1H labeling at the Ile-γ2 methyl position in highly deuterated proteins. J Biomol NMR 48:129–135
Ruschak AM, Religa TL, Breuer S, Witt S, Kay LE (2010b) The proteasome antechamber maintains substrates in an unfolded state. Nature 467:868–871
Shi L, Kay LE (2014) Tracing an allosteric pathway regulating the activity of the HslV protease. Proc Natl Acad Sci USA 111:2140–2145
Sounier R, Blanchard L, Wu Z, Boisbouvier J (2007) High-accuracy distance measurement between remote methyls in specifically protonated proteins. J Am Chem Soc 129:472–473
Sprangers R, Kay LE (2007) Probing supramolecular structure from measurement of methyl 1H–13C residual dipolar couplings. J Am Chem Soc 129:12668–12669
Sprangers R, Velyvis A, Kay LE (2007) Solution NMR of supramolecular complexes: providing new insights into function. Nat Methods 4:697–703
Tugarinov V, Kay LE (2004) An isotope labeling strategy for methyl TROSY spectroscopy. J Biomol NMR 28:165–172
Tugarinov V, Choy WY, Orekhov VY, Kay LE (2005) Solution NMR-derived global fold of a monomeric 82-kDa enzyme. Proc Natl Acad Sci USA 102:622–627
Umbarger HE (1996) Biosynthesis of the branched-chain amino acids. In: Neidhardt FC, Curtiss III R, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (eds) Escherichia coli and Salmonella typhimurium: cellular and molecular biology. American Society for Microbiology Press, Washington, DC, pp 442–457
Velyvis A, Ruschak AM, Kay LE (2012) An economical method for production of 2H, 13CH3-threonine for solution NMR studies of large protein complexes: application to the 670 kDa proteasome. PLoS One 7:e43725
Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 59:687–696
Vyazmensky M, Sella C, Barak Z, Chipman DM (1996) Isolation and characterization of subunits of acetohydroxy acid synthase isozyme III and reconstitution of the holoenzyme. Biochemistry 35:10339–10346
Acknowledgments
We would like to thank Dr. S. J. Remington for providing MSG plasmid, Dr. D. Chipman for providing AHAS II plasmid and Mrs I. Ayala as well as Dr. R. Sounier for stimulating discussions. This work used the high-field NMR and the isotopic labeling facilities at the Grenoble Instruct Centre (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). The research leading to these results has received funding from the European Research Council under the European Community’s Seventh Framework Program FP7/2007-2013 Grant Agreement no. 260887.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kerfah, R., Plevin, M.J., Pessey, O. et al. Scrambling free combinatorial labeling of alanine-β, isoleucine-δ1, leucine-proS and valine-proS methyl groups for the detection of long range NOEs. J Biomol NMR 61, 73–82 (2015). https://doi.org/10.1007/s10858-014-9887-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-014-9887-2