
Abstract
A straightforward approach for the production of highly deuterated proteins labeled with 13C and 1H at Ile-γ2 methyl positions is described. The utility of the methodology is illustrated with an application involving the half proteasome (360 kDa). High quality 2D Ile 13Cγ2,1Hγ2 HMQC data sets, exploiting the methyl-TROSY principle, are recorded with excellent sensitivity and resolution, that compare favorably with Ile 13Cδ1,1Hδ1 spectra. This labeling scheme adds to a growing list of different approaches that are significantly impacting the utility of solution NMR spectroscopy in studies of supra-molecular systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The development of new and powerful ways of labeling biomolecules (Castellani et al. 2002; Kainosho et al. 2006; Sprangers and Kay 2007) has been critical for stimulating many of the advances in NMR methodology that in turn have led to applications involving increasingly complex molecular targets (Amero et al. 2009; Gelis et al. 2007; Hamel and Dahlquist 2005; Religa et al. 2010; Sprangers and Kay 2007; Velyvis et al. 2007). One labeling strategy focuses on methyl groups as key probes of structure and dynamics in proteins (Tugarinov and Kay 2005), in particular for studies of very high molecular weight systems, where many of the more traditional labeling approaches often fail. Ile, Leu and Val methyl groups can be selectively 13C-methyl labeled using α-keto acid precursors (Gardner and Kay 1997; Goto et al. 1999; Tugarinov and Kay 2005) that are available commercially. All three of the relevant methyl isotopomers can be obtained—13CH3, 13CH2D and 13CHD2—facilitating a wide range of experiments. More recently, Boisbouvier and coworkers have developed a synthetic protocol for the production of a form of acetolactate (2-[13C]methyl-3-oxo-4-[2H3]butanoate) that serves as the precursor for labeling with 13CH3 at exclusively the proS Leu, Val methyl positions in proteins (Gans et al. 2010); this precursor and the corresponding compound for the production of proR 13C,1H methyl label are now commercially available. Additional approaches for the production of either Ala-[13CH3] or Met-[13CH3] labeled proteins have also been published (Ayala et al. 2009; Fischer et al. 2007; Gelis et al. 2007; Isaacson et al. 2007). Herein we add to the list of available precursors for methyl labeling in proteins by introducing a reagent that generates Ile-[13CH3,γ2] molecules. The utility of the labeling protocol is demonstrated with an application to the half proteasome, α7α7, that consists of a pair of identical heptameric rings (α7) with an aggregate molecular weight of 360 kDa.
Ile methyl groups are useful probes of structure and dynamics in proteins (Gardner and Kay 1997), with the Ile 13Cδ1 and 13Cγ2 regions of 13C,1H correlation maps well dispersed. Initial labeling protocols targeted the Ile Cδ1 position using [13CH3] α-ketobutyrate as a precursor (Gardner and Kay 1997) but inspection of the Ile biosynthetic pathway, a portion of which is shown in Fig. 1, indicates that it should be straightforward to label the 13Cγ2 position as well by a judicious choice of metabolite. Here we have used the compound indicated by the dashed box in Fig. 1, α-aceto-α-hydroxybutyrate, with 13CH3 in the acetyl moiety (blue) and deuteration at all other positions, that was synthesized commercially (Sigma–Aldrich) as the ethyl ester. The ester derivatives of this precursor and of acetolactate are significantly more stable than the corresponding acid forms of these molecules that undergo decarboxylation over time (Crout and Hedgecock 1979).

Key steps in the biosynthetic pathway of Ile, highlighting the source of the γ2 position in blue (Stryer 1995). The precursor, α-aceto-α-hydroxybutyrate (2-hydroxy-2-ethyl-D5-3-oxobutanoate-4-13C), used in the production of U-[2H],Ile-[13CH3,γ2]-α7α7 is indicated in the red box. This precursor is purchased (Sigma–Aldrich, Isotech; $2000/g) in the ethyl-ester form (ethyl-2-hydroxy-2-ethyl-D5-3-oxobutanoate-4-13C) that is significantly more stable than the acid (Crout and Hedgecock 1979). The acid can be generated from the ester by base hydrolysis that is followed by 1D NMR (the preferred method; see text) or by the addition of Bacillus stearothermophilus esterase (a recombinant enzyme produced in E. coli, Sigma catalogue #69509, 44 units/mg). For proteasome growth in 100 mL D2O-M9, 12.7 μL (14.0 mg) of the precursor ester and 5.5 mg of esterase were dissolved in 0.5 mL of 0.5 M sodium phosphate, pH 7.4, and incubated at 37°C. De-esterification was 80–90% complete in 35 min as observed by 1D NMR. The reaction mixture was stored at 4°C until addition to D2O-M9 media after brief centrifugation with no further processing. Attempts to produce γ2-labeled protein by addition of the ester to the growth medium were unsuccessful indicating that E. coli are unable to process the ester form of the precursor
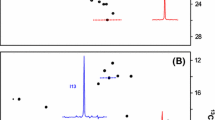
Figure 2a, b compare 13C,1H HMQC spectra recorded of 1.6 mM (monomer concentration) U-[2H],Ile-[13CH3,δ1]- and U-[2H],Ile-[13CH3,γ2]-α7α7, 50°C, 800 MHz. In addition to the Ile labeling we have included [13CH3]-Met in the growth medium as an ‘internal’ standard so that any (slight) difference in protein concentration in the pair of samples can be taken into account in a comparison of signal–noise (s/n), see below. The concentrations of protein within each sample differ by 10% and both data sets are plotted with the same noise floor in the figure. By inspection, the s/n of γ2 and δ1 correlations appear qualitatively to be similar (see below), with the resolution of cross-peaks in the γ2 spectrum arguably superior to that in the δ1 data set. It is worth noting that Ile γ2-methyl or δ1-methyl isotopomers of the form 13CH2D or 13CHD2 are not observed in any spectra that have been recorded.

a Portion of the 13C,1H HMQC spectrum of U-[2H],Ile-[13CH3,δ1]-α7α7, 50°C, 800 MHz. The data set was recorded using a standard HMQC pulse scheme (90° excitation pulse) with a relaxation delay of 1.5 s for a total measurement time of 44 min. The sample concentration was 114 μM in α7α7 (1.6 mM in monomer) in 100% D2O, 25 mM potassium phosphate pH 6.8, 50 mM NaCl, 1 mM EDTA, 0.03% NaN3 and 2 mM DTT. The sample was prepared as described in detail previously (Sprangers and Kay 2007), using 60 mg/L α-ketobutyric acid ([methyl-13CH3]) 1 h prior to induction of protein expression. Protein was harvested after 12 h of production. b Expanded region of the 13C,1H HMQC dataset of U-[2H],Ile-[13CH3,γ2]-α7α7, 50°C, 800 MHz recorded as in (a). The sample was generated using 140 mg/L ethyl-2-hydroxy-2-ethyl-D5-3-oxobutanoate-4-13C (α-aceto-α-hydroxybutyrate) that was prepared as described in the text and in the legend to Fig. 1. Note that only 70 mg, ½, of the added precursor has the correct stereochemistry at position 2 to be processed by E. coli enzymes. A concentration of 70 mg/L of the ‘correct stereochemistry’ γ2 precursor is equivalent to 50 mg/L of α-ketobutyric acid (δ1 precursor) on a molar basis that was found to be optimal for incorporation of δ1 label (90–95%) in a previous study (Gardner and Kay 1997). Since α-ketobutyric acid immediately precedes α-aceto-α-hydroxybutyrate (Fig. 1) a similar level of 90–95% incorporation of 13CH3 γ2 label can be expected. c Full spectrum of U-[2H],Ile-[13CH3,γ2]-α7α7, 50°C, 800 MHz illustrating the small amount of ‘scrambling’ to Leu/Val proR positions that reflects a labeling level of between 2 and 7% (mean of 4%). Shown also are a number of traces through peaks indicated by arrows; traces for Leu/Val correlations are scaled by a factor of five prior to plotting
One drawback of γ2 labeling using the precursor introduced here, at least for production of U-[2H],Ile-[13CH3,γ2]-α7α7, is that small cross-peaks at the proR (δ1/γ1) methyl positions of Leu/Val were observed for approximately 70% of these residues, Fig. 2c. These proR Leu/Val correlations varied in intensity by a factor of four, with the strongest 17%(2.6%) as intense as the weakest(strongest) γ2 signal. By comparing these peaks with the corresponding correlations measured in a spectrum of U-[2H],Leu,Val-[proR 13CH3,proS 12CD3]-α7α7 the level of ‘accidental’ labeling for the Leu/Val methyls in the γ2 sample ranges between 2 and 7% (mean 4%) for those correlations that could be quantified.
Non-specific labeling has not been observed previously in the production of Ile-[13CH3,δ1]- or Leu,Val-[13CH3,12CD3]-samples. A comparison of NMR spectra recorded of the precursor for Ile-[13CH3,γ2] with that for proR stereospecific methyl labeling of Leu/Val (purchased from Sigma–Aldrich, Isotech) establishes that there is no cross contamination. Although the process of removing the ester involves hydrolysis with NaOH (pH 12.5, 25°C, <5 min) which does lead to a number of small impurities, the proR Leu/Val precursor is not among them, as established by NMR. In attempt to minimize side reactions we have also carried out the de-esterification under very mild conditions by an enzymatic process (see legend to Fig. 1). The near quantitative formation of the product carboxylic acid produced by the enzyme-catalyzed reaction (80–90%) was monitored by NMR. The precursor generated in this way was added approximately 1 h prior to the induction of protein over-expression, as is done in a standard manner for all methyl precursors used (Goto et al. 1999), and protein production allowed to proceed over a period of 12 h prior to harvesting. Similar levels of incorporation into Leu/Val were observed as for the precursor prepared from base hydrolysis. As a further attempt to minimize ‘scrambling’ we have also limited the protein production period to 4 h, however, there was little change in the relative intensities of Ile γ2 vs proR Leu/Val correlations in comparison to protein prepared with longer inductions times. The presence of these minor peaks is an annoyance but not a significant deterrent to the use of the γ2 labeling strategy. Certainly, in cases where Leu,Val-[13CH3,13CH3]-, non-stereospecific Leu,Val-[13CH3,12CD3]- or Leu,Val-[proR 13CH3,proS 12CD3]-labeled protein is produced in addition to the Ile-γ2 label, these extra minor peaks would be ‘obscured’ by correlations resulting from the intended Leu/Val label. It is also likely that in the production of Ile-[13CH3,γ2],Leu,Val-[proS 13CH3,proR 12CD3]-labeled protein the added Leu/Val precursor would ‘outcompete’ the small amount of impurity associated with Ile γ2 precursor so that label would not be generated at proR Leu/Val positions. If necessary, the Leu/Val correlations in spectra of Ile-[13CH3,γ2] labeled protein can be removed through the addition of d7,α-ketoisovalerate, a precursor for both Leu and Val methyl groups (Ayala et al. 2009), that is available for purchase. When used in the production of perdeuterated Ala-[13CH3]-labeled proteins following the protocol of Ayala et al. (2009), d7,α-ketoisovalerate eliminates Leu/Val correlations that otherwise would be at a level close to 25% if only 13Cβ-Ala is added.
Figure 3 plots histograms of Ile 1Hδ1 T1 (a) and T2 (b) relaxation times for U-[2H],Ile-[13CH3,δ1]-α7α7, 50°C, 800 MHz, along with the corresponding values for Ile Hγ2 from the U-[2H],Ile-[13CH3,γ2]-labeled protein (c,d) that have been measured using pulse schemes described previously (Ollerenshaw et al. 2005). Most striking are the significant differences in 1H T1 times, close to two-fold, that arise largely from two effects. First, on average, methyl axis order parameters are significantly smaller for Ile Cδ1-Cγ1 bonds than for Cγ2-Cβ bonds (Mittermaier et al. 1999), lengthening the 1Hδ1 T1, and second, the increased rate of rotation about the methyl three-fold axis for Hδ1 (Kay et al. 1996) also decreases the relative efficiency of 1H relaxation at this position.

Histograms of Ile 1Hδ1 (1Hγ2) T1 and T2 relaxation times for U-[2H],Ile-[13CH3,δ1]-α7α7 (U-[2H],Ile-[13CH3,γ2]-α7α7), 50°C, 800 MHz. Median values are indicated by vertical arrows
The substantial differences in Ile 1Hδ1/1Hγ2 T1 values have been taken into account in recording ‘optimal’ 13C,1H HMQC experiments. Based on average 1H T1 values of 1.8 and 0.74 s for 1Hδ1 and 1Hγ2 in α7α7 (50°C, 800 MHz) respectively, SOFAST-HMQC data sets (Amero et al. 2009) have been obtained with θ, the angle of the excitation pulse, set to 60° and recycle times of 1.0 s and 0.5 s for samples of U-[2H],Ile-[13CH3,δ1]-α7α7 and U-[2H],Ile-[13CH3,γ2]-α7α7. Acquisition times were (t1,t2) = (40 ms, 64 ms) and data sets were measured for identical durations. The slight difference in protein concentrations in each of the two samples has been taken into account to give the distributions of s/n values shown in Fig. 4a, b. Only correlations that were well resolved were included in the quantification, so that slightly different sets of peaks were used for the Ileδ1, Ileγ2 distributions in the figure. Median s/n values of 400(360) were obtained for γ2(δ1) correlations; such values are rather sensitive to the shape of each distribution. An alternative way of assessing relative sensitivities of the δ1,γ2 13C,1H HMQC data sets would be to compare s/n values for δ1/γ2 cross-peaks on a per-residue basis, as is done in Fig. 4c. For the set of 11 residues that are well resolved and can be quantified in both data sets a median s/n ratio of 1.3 is obtained (in favor of γ2) with a standard deviation of 0.6; 8 of the peaks show higher s/n values in the γ2 data set.

Histogram of s/n of correlations measured in spectra of U-[2H],Ile-[13CH3,δ1]-α7α7 (a) and U-[2H],Ile-[13CH3,γ2]-α7α7 (b), 50°C, 800 MHz recorded using SOFAST HMQC (Amero et al. 2009), as described in the text. The relative protein concentrations in the two samples have been established by recording fully relaxed Met spectra since each of the proteins is also Met-13Cε labeled. Median values are indicated with vertical arrows. c Ratio of s/n values for Ileγ2/Ileδ1 correlations on a per-residue basis. A horizontal line is placed at a ratio of 1
Prior to exploiting the high sensitivity and resolution of Ile γ2 methyl correlations in HMQC spectra, the cross-peaks must first, of course be assigned. Assignments can be carried out as for Ile δ1, Leu δ, Val γ methyl groups by a divide and conquer strategy (Gelis et al. 2007; Sprangers and Kay 2007), via mutations (Gelis et al. 2007; Sprangers and Kay 2007; Velyvis et al. 2009), NOE and/or pseudo contact shift measurements in concert with a high resolution X-ray structure (John et al. 2007; Velyvis et al. 2009), or by using a combination of experimental and computational approaches (Xu et al. 2009). In the case of α7α7 we have exploited the fact that the Ile δ1 methyl groups are already assigned, facilitating the assignment of Ile γ2 methyls by recording NOE spectra correlating intra-residue Ile δ1-γ2 methyl groups. To this end we have prepared a U-[2H],Ile-[13C,1H] labeled α7α7 sample by adding U-13C,1H Ile to the growth medium. A short mixing time (75 ms) HSQC-NOESY-HSQC data set (see Supporting Information for details of the pulse sequence) was obtained (Sprangers and Kay 2007), providing firm assignments for over half of the γ2 methyls and tentative assignments for the remainder. Figure 5a illustrates the utility of the experiment for Ile 70 and 109, highlighting intra-residue NOE connectivities between Hδ1 and Hγ2 protons. In addition, a methyl-TROSY based 3D NOESY data set with a mixing time of 500 ms (Sprangers and Kay 2007) has been recorded on the U-[2H],Ile-[13CH3,γ2]-α7α7 sample to measure inter-residue Hγ2-Hγ2 NOEs to confirm the assignments, Fig. 5b. A large mixing time was chosen so as to correlate methyl groups well outside of the typical range of 5–6 Å, by exploiting the high level of deuteration of the sample (Sounier et al. 2007). Figure 5c shows a small region of the X-ray structure of the α-subunit (Lowe et al. 1995) that was used in the analysis of the NOESY data set, focusing on the region of structure that is of interest in the analysis of the strips of Fig. 5b. Ile side-chains are highlighted in red with the Cγ2 positions denoted by red balls. Solid(dashed) lines between methyl groups indicate correlations that could(could not) be observed in the data set, with the distances between 13Cγ2 carbons indicated. In total, assignments for all of the Ile γ2 correlations observed in spectra (corresponding to 16 of 17 Ile residues) have been obtained from analysis of this pair of NOESY experiments.

a Strip-plots from a 75 ms mixing time HSQC-NOESY-HSQC data set (see SI for details) recorded on a sample of U-[2H],Ile-[13C,1H]-α7α7, 50°C, 800 MHz, illustrating intra-residue NOEs between Hδ1-Hγ2 of Ile 70 and Ile 109. Diagonal peaks are denoted with ‘d’, while ‘*’ indicates that peak intensity is greater in an adjacent plane. b Strip-plots from a 500 ms mixing time HSQC-NOESY-HSQC data set recorded on a sample of U-[2H],Ile-[13CH3,γ2]-α7α7, 50°C, 800 MHz correlating proximal inter-residue Ile Hγ2 protons, as illustrated in the structure of a portion of the α-subunit of the proteasome in (c). See text for details
As a final note it is worth commenting on the expense of labeling a protein at the Ile γ2 position relative to Ile δ1. The cost of expression of a labeled, U-[2H]-protein derives from a number of factors, including the expense of D2O, U-[2H]-glucose and the appropriate precursors. For the Ile(δ1), Leu, Val methyl labeling scheme described previously (Tugarinov and Kay 2004) the relative cost of the precursors is small; α-ketobutyrate(Ile) and α-ketoisovalerate(Leu/Val) combined contribute only slightly more than 10% of the total cost of protein production. However, the Ile γ2 methyl precursor as purchased, ethyl-2-hydroxy-2-ethyl-D5-3-oxobutanoate-4-13C (Sigma–Aldrich, Isotech) is much more expensive than α-ketobutyrate (Ile δ1), by approximately a factor of 7 that includes the fact that the compound is available as a racemic mixture at position 2 (see Fig. 1) so that only 50% of the precursor can be used by the E. coli biosynthetic machinery for protein synthesis. The net effect is that the cost of an Ile γ2 labeled sample is approximately 40% higher than for Ile δ1, with the precursor accounting for 30% of the expense. In contrast, the Ile δ1 label contributes only 6% to the total sample cost.
In summary, a precursor has been introduced for the production of Ile-[13CH3,γ2] labeled proteins. Ile γ2 methyl groups are powerful probes for studies of very high molecular weight proteins because they often give rise to spectra with excellent sensitivity and dispersion, as demonstrated here. Drawbacks include the fact that a small amount of labeling (~4%) is produced at the proR Leu/Val methyl positions and the increased expense in sample production relative to Ile δ1-labeled samples. Nevertheless, Ile γ2 labeling serves as a useful addition to the increasing number of selective labeling methods that continue to impact favorably on the utility of solution NMR for studies of supra-molecular systems.
References
Amero C, Schanda P, Dura MA, Ayala I, Marion D, Franzetti B, Brutscher B, Boisbouvier J (2009) Fast two-dimensional NMR spectroscopy of high molecular weight protein assemblies. J Am Chem Soc 131:3448–3449
Ayala I, Sounier R, Use N, Gans P, Boisbouvier J (2009) An efficient protocol for the complete incorporation of methyl-protonated alanine in perdeuterated protein. J Biomol NMR 43:111–119
Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H (2002) Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature 420:98–102
Crout D, Hedgecock C (1979) Base-catalyzed rearrangement of alpha-acetolactate-novel carboxylate ion migration in a tertiary ketol rearrangement. J Chem Soc Perkin Trans 1(8):1982–1989
Fischer M, Kloiber K, Hausler J, Ledolter K, Konrat R, Schmid W (2007) Synthesis of a 13C-methyl-group-labeled methionine precursor as a useful tool for simplifying protein structural analysis by NMR spectroscopy. Chembiochem 8:610–612
Gans P, Hamelin O, Sounier R, Ayala I, Dura MA, Amero C, Noirclerc-Savoye M, Franzetti B, Plevin MJ, Boisbouvier J (2010) Stereospecific isotopic labeling of methyl groups for NMR spectroscopic studies of high molecular weight proteins. Angew Chem Int Ed 49:1958–1962
Gardner KH, Kay LE (1997) Production and incorporation of 15N, 13C, 2H (1H-δ1 methyl) isoleucine into proteins for multidimensional NMR studies. J Am Chem Soc 119:7599–7600
Gelis I, Bonvin AM, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, Economou A, Kalodimos CG (2007) Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell 131:756–769
Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE (1999) A robust and cost-effective method for the production of Val, Leu, Ile (δ1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J Biomol NMR 13:369–374
Hamel DJ, Dahlquist FW (2005) The contact interface of a 120 kD CheA-CheW complex by methyl TROSY interaction spectroscopy. J Am Chem Soc 127:9676–9677
Isaacson RL, Simpson PJ, Liu M, Cota E, Zhang X, Freemont P, Matthews S (2007) A new labeling method for methyl transverse relaxation-optimized spectroscopy NMR spectra of alanine residues. J Am Chem Soc 129:15428–15429
John M, Schmitz C, Park AY, Dixon NE, Huber T, Otting G (2007) Sequence-specific and stereospecific assignment of methyl groups using paramagnetic lanthanides. J Am Chem Soc 129:13749–13757
Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Mei Ono A, Guntert P (2006) Optimal isotope labelling for NMR protein structure determinations. Nature 440:52–57
Kay LE, Muhandiram DR, Farrow NA, Aubin Y, Forman-Kay JD (1996) Correlation between dynamics and high affinity binding in an SH2 domain interaction. Biochemistry 35:361–368
Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R (1995) Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science 268:533–539
Mittermaier A, Kay LE, Forman-Kay JD (1999) Analysis of deuterium relaxation-derived methyl axis order parameters and correlation with local structure. J Biomol NMR 13:181–185
Ollerenshaw JE, Tugarinov V, Skrynnikov NR, Kay LE (2005) Comparison of 13CH3, 13CH2D, and 13CHD2 methyl labeling strategies in proteins. J Biomol NMR 33:25–41
Religa TL, Sprangers R, Kay LE (2010) Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science 328:98–102
Sounier R, Blanchard L, Wu Z, Boisbouvier J (2007) High-accuracy distance measurement between remote methyls in specifically protonated proteins. J Am Chem Soc 129:472–473
Sprangers R, Kay LE (2007) Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature 445:618–622
Stryer L (1995) Biochemistry, 4th edn. W. H. Freeman and Company, New York
Tugarinov V, Kay LE (2004) An isotope labeling strategy for methyl TROSY spectroscopy. J Biomol NMR 28:165–172
Tugarinov V, Kay LE (2005) Methyl groups as probes of structure and dynamics in NMR studies of high-molecular-weight proteins. Chembiochem 6:1567–1577
Velyvis A, Yang YR, Schachman HK, Kay LE (2007) A solution NMR study showing that active site ligands and nucleotides directly purturb the allosteric equilibrium in aspartate transcarbamolyase. Proc Natl Acad Sci USA 104:8815–8820
Velyvis A, Schachman HK, Kay LE (2009) Assignment of Ile, Leu, and Val methyl correlations in supra-molecular systems: an application to aspartate transcarbamoylase. J Am Chem Soc 131:16534–16543
Xu Y, Liu M, Simpson PJ, Isaacson R, Cota E, Marchant J, Yang D, Zhang X, Freemont P, Matthews S (2009) Automated assignment in selectively methyl-labeled proteins. J Am Chem Soc 131:9480–9481
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR). L.E.K. holds a Canada Research Chair in Biochemistry.
Author information
Authors and Affiliations
Corresponding author
Additional information
Amy M. Ruschak and Algirdas Velyvis contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10858_2010_9449_MOESM1_ESM.pdf
Details of pulse scheme for recording the HSQC-NOESY-HSQC experiment on a U-[2H],Ile-[13C,1H] labeled α7α7 sample. (PDF 506 kb)
Rights and permissions
About this article
Cite this article
Ruschak, A.M., Velyvis, A. & Kay, L.E. A simple strategy for 13C,1H labeling at the Ile-γ2 methyl position in highly deuterated proteins. J Biomol NMR 48, 129–135 (2010). https://doi.org/10.1007/s10858-010-9449-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-010-9449-1