
Abstract
A novel docking challenge has been set by the Drug Design Data Resource (D3R) in order to predict the pose and affinity ranking of a set of Farnesoid X receptor (FXR) agonists, prior to the public release of their bound X-ray structures and potencies. In a first phase, 36 agonists were docked to 26 Protein Data Bank (PDB) structures of the FXR receptor, and next rescored using the in-house developed GRIM method. GRIM aligns protein–ligand interaction patterns of docked poses to those of available PDB templates for the target protein, and rescore poses by a graph matching method. In agreement with results obtained during the previous 2015 docking challenge, we clearly show that GRIM rescoring improves the overall quality of top-ranked poses by prioritizing interaction patterns already visited in the PDB. Importantly, this challenge enables us to refine the applicability domain of the method by better defining the conditions of its success. We notably show that rescoring apolar ligands in hydrophobic pockets leads to frequent GRIM failures. In the second phase, 102 FXR agonists were ranked by decreasing affinity according to the Gibbs free energy of the corresponding GRIM-selected poses, computed by the HYDE scoring function. Interestingly, this fast and simple rescoring scheme provided the third most accurate ranking method among 57 contributions. Although the obtained ranking is still unsuitable for hit to lead optimization, the GRIM–HYDE scoring scheme is accurate and fast enough to post-process virtual screening data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Molecular docking is still the most straightforward computational technique to predict three-dimensional (3-D) atomic coordinates of a protein–ligand complex [1, 2]. Any docking attempt is aimed at simultaneously solving two questions: (i) What is the conformation of the ligand when bound to its target?, (ii) What is the relative orientation of the ligand with respect to its host protein? Almost 35 years of practice and the development of over 60 different docking methods [3] have considerably helped the community to enhance the quality of the answers to the above two questions, and to better depict the precise applicability domain of the method. Very soon, computational chemists have realized that posing a drug-like low molecular weight ligand to a structurally druggable and reasonably rigid cavity is reachable in the large majority of cases [4]. However, ranking these docking solutions to prioritize the most relevant one in first place is still a major issue [5]. Pushed by massive interest of the pharmaceutical industry in structure-based drug design approaches, the docking community has then organized various resources to challenge computational chemists to refine both their practices and methods. Among the most important resources are the Community Structure Activity Resource (CSAR) [6] and the Drug Design Data Resource (D3R) [7] which periodically organize challenges aimed at predicting protein–ligand coordinates prior to the release of their crystal structures, discriminating X-ray poses from decoys and scoring/ranking a set of ligands for well-defined targets [5, 8,9,10,11,12,13,14].
The first D3R Grand Challenge [13], launched in 2015, was a good opportunity to test in real life conditions, our recently-developed graph matching algorithm (GRIM) [15] to rank docking poses. GRIM converts protein–ligand atomic coordinates into a graph where each node is matched to an interaction pseudoatom (IPA) describing a single protein–ligand interaction (apolar contact, aromatic interaction, hydrogen bond, ionic bond, metal chelation). For each interaction, three IPAs are derived, one on the ligand-interacting atom, one on the protein-interacting atom and one at the geometric barycenter of the later two IPAs. Since the graph precisely depicts the corresponding protein–ligand interaction pattern, two different protein–ligand complexes can thus be easily compared by using standard graph matching techniques to determine a clique, in other words, the largest maximal common subgraph which describes the protein–ligand interaction pattern shared by the two investigated complexes. This simple graph alignment-based method produces a quantitative estimate of the similarity of two protein–ligand interaction patterns. It can therefore be used to rank docking poses by similarity to any reference interaction pattern (e.g. stored in the Protein Data Bank) or to post-process virtual screening docking data. In the latter two scenarios and starting from the same set of docking poses, GRIM significantly outperformed standard scoring functions in either posing a set of protein ligands or enriching virtual screening hits in true ligands [15].
In the first D3R challenge, GRIM rescoring of Surflex-poses was ranked 3rd out of 42 submissions for the pose-prediction of HSP90α and MAP4k4 inhibitors, respectively [16]. To check the target and ligand set dependency of our method, we applied the same strategy to the D3R second challenge consisting in (i) pose prediction of 36 agonists of the Farnesoid X nuclear receptor (FXR), (ii) rank/score a set of 102 FXR agonists by decreasing binding affinity. This novel competition represents a real challenge for several reasons: (i) the FXR receptor structure exhibits a flexible binding site whose conformation depends on the bound-ligand chemotype [17], (ii) some but not all ligands require a bound water [18], (iii) about one-third of the new ligands whose pose need to be predicted exhibit a chemotype never co-crystallized with the target, (iv) FXR ligands may adopt almost non overlapping poses due to the large size and hydrophobic nature of the ligand-binding cavity.
Computational methods
FXR dataset
The structures of the FXR apo-receptor (PDB format) and 102 FXR ligands (SD files and SMILES strings, Supplementary Table 1) were downloaded from the D3R Grand Challenge 2 website [7] as a zipped archive file (417_data_517096.zip). In addition, 26 agonist-bound FXR X-ray structures were defined as templates (Supplementary Table 2) by searching the RCSB Protein Data Bank [19] for the text keyword ‘FXR’ and a known bound ligand, confirmed as a true FXR agonist. Existing hydrogen atoms were removed and added again while optimizing both the protonation and tautomeric states of all atoms using Protoss [20]. Water molecules were explicitly conserved at the condition that the water oxygen atom was closer than 6.5 Å from any ligand heavy atom and that at least two hydrogen bonds with either the protein and/or the ligand could be identified assuming a donor–acceptor distance lower than 3.5 Å and a donor–hydrogen–acceptor angle higher than 120°. Protein (including water atoms) and ligand atoms were then separately saved in MOL2 file format in SYBYL-X 2.1.1 (Certera LP, Princeton, NJ, USA). Last, atomic coordinates (main chain atoms only) of the 26 protein templates were structurally aligned to that of the apostructure provided by the organizers of the D3R Grand Challenge, using the ‘Align_Structures’ module of SYBYL-X 2.1.1.
The SD files of the 102 ligands to dock were converted in three-dimensional (3-D) atomic coordinates with Corina v3.40 (Molecular Networks GmbH. Erlangen, Germany). The protonation state of every ligand was assigned at pH 7.4 with Filter v.2.5.1.4 (OpenEye Scientific Software, Santa Fe, NM, USA) and verified manually. 3-D coordinates were saved in MOL2 file format.
Docking poses generation
Ligands were docked to the above mentioned 26 protein structures using Surflex v.3066 [21]. For each protein input structure, a protomol was first generated using a list of binding site residues (including bound waters) for which at least one heavy atom was closer than 6.5 Å from one co-crystallized ligand heavy atom. The protomol is a pseudo-ligand used as a target to generate putative alignments of fragments of an input ligand. It utilizes three probes (C=O, NH, CH4) whose positions correspond to energetically favored locations for hydrogen bond acceptors, hydrogen bond donors and apolar atoms, respectively.
In order to better define the docking zone, an additional 3 Å voxels of space around the volume specified by the protomol (proto_bloat parameter set to 3) was chosen. The degree of buriedness (proto_thresh parameter) for the primary volume used to generate the protomol was set to a value of 0.3. The protein residues side-chains were kept rigid. Other parameters were assigned as default. The docking accuracy parameter set –pgeom was used. The ‘pgeom’ option starts each docking from four initial and different poses to ensure good search coverage, turns on ligand minimization prior to docking and after docking (in-pocket minimization), ensures that the returned poses are different from one another by at least 0.5 Å rmsd, and saves a total of 20 poses (ranked by Surflex energy score, from 000 to 019). Each of the 102 FXR ligands was docked to each of the 26 protein structure templates, thereby generating a set of 53,040 (102 × 20 × 26) docking poses. Only poses with a predicted Surflex score (in pKd unit) higher than 2.0 were kept for further analysis.
GRIM rescoring
Protein–ligand interaction patterns were generated with IChem [22] for both the 26 X-ray structures and the docking poses, and further compared to that of 26 FXR-agonist X-ray templates with GRIM [15]. Each alignment was quantified using the empirical GRIMscore [15] (Eq. 1).
where NLig is the number of matched ligand-based IPAs, Ncenter is the number of matched centered IPAs, NProt is the number of matched protein-based IPAs, SumCl: \(\frac{{\mathop \sum \nolimits^ {\text{pair~weights~in~clique}}}}{{\mathop \sum \nolimits^ {\text{all~possible~pair~weights~}}}}\), RMSD is the root mean square deviation (in Å) of the matched clique, DiffI is the absolute value of the difference in the number of IPAs between reference and query.
For every ligand, the five poses with the highest GRIMscore (GRIM-1 to GRIM-5) were retained.
In a previous study [15], the GRIMscore has been determined by fitting the above described six parameters to the IShape similarity score [15] parametrized on a training set of 1800 protein–ligand X-ray structures. In contrast to IShape, the GRIM score shows an excellent and sharp discrimination between known similar and known dissimilar complexes [15]. Please note that GRIM assigns a weight to each IPA (used in the SumCl parameter), whose value is inversely proportional to the frequency of the corresponding interaction among 9877 sc-PDB protein–ligand complexes [13].
Root-mean square deviations to atomic coordinates
Root mean square deviations (rmsd) of predicted poses to the X-ray structures, released at the end of the first stage of the challenge, were computed using the rms routine of the Surflex package. Rmsd were computed only for heavy atoms and included symmetry operations to explicitly account for equivalent atoms (e.g. carboxylate oxygen atoms). One compound (FXR_33) was excluded from the pose prediction phase of the challenge due to an oxidation during the co-crystallization process, generating a structure in the crystal that did not correspond to that initially provided.
Ligand ranking
For each of the 102 FXR ligands (FXR_1–FXR_102) to rank, the corresponding protein–ligand complex with the absolute best GRIMscore was selected and its binding free energy was estimated using the HYDE scoring function [23] (see Eq. 2) implemented in seeSAR (BioSolveIT GmbH, Sankt Augustin, Germany).
Briefly, the HYDE scoring function relies on atom type-specific hydration and desolvation terms that have been carefully calibrated using octanol/water partition coefficients of small molecules. For more details on the HYDE scoring energy terms, the reader is referred to the original article [23].
Results and discussion
Suitability of known PDB interaction patterns for FXR agonist docking
We first examined the currently available protein–ligand interaction patterns in the PDB by selecting 25 entries (one having two chains considered) corresponding to FXR structures co-crystallized with 26 agonists of three different chemotypes (Supplementary Table 2). At this step, we only considered FXR structures bound to a known agonist for two main reasons: (i) the FXR receptor, like most nuclear hormone receptors, is known to change its overall conformation with respect to the functional effect of the ligand [24], (ii) the purpose of the current D3R challenge was focused on FXR agonists only. The FXR structure in complex with ivermectin (PDBID 4wvd) was discarded from the list because of the very unique binding mode of the latter compound [25]. Ivermectin exhibits both a peculiar structure and protein–ligand interaction pattern with respect to the 26 X-ray templates as well as the 102 FXR agonist to dock. We therefore decided to remove this structure, as we believe it would add more noise than true signal to the training set. The remaining 26 X-ray templates, despite evident structural similarities, were all kept since the computing effort for docking and GRIM rescoring remains quite negligible.
For each of the 26 PDB templates, a protein–ligand interaction pattern graph was obtained with IChem [22] and a full pairwise similarity matrix was generated, considering GRIMscore as a similarity descriptor. The corresponding heat map (Fig. 1) clearly shows three groups according to the chemotypes of the bound ligands. Benzimidazoles appear to bind to FXR with a very homogeneous interaction pattern evidenced by very high pairwise similarities (GRIMscore > 1.0). For isoxazoles, the observed interaction patterns are clearly more diverse with a few highly similar complexes (e.g. 3rut, 3ruu, and 3p89; pairwise GRIMscore > 1.0) and much more complexes with still statistically significant interaction patterns but lower GRIMscores (between 0.7 and 1.0). The binding mode observed in the 3fxv entry seems to be unique to this group (Fig. 1). Last, the miscellaneous class is characterized by two different interaction patterns corresponding to FXR structures crystallized with either cholic acids (1osv, 1ot7_A, 1ot7_B, 4qe6) or tetrahydroazepinoindoles (3fli, 1l1b; Fig. 1).

Heat map of protein–ligand interaction pattern similarity (GRIMscore) among 26 reference FXR-agonist X-ray structures. Entries are grouped according to the three chemotypes (benzimidazoles, isoxazoles, miscellaneous) of the bound FXR-agonists. Pairwise similarity GRIMscores are calculated with IChem v5.2.6, and colored as follows: blue, no similarity (GRIMscore < 0.7); yellow, medium similarity (0.7 < GRIMscore < 1.0); red, high similarity (GRIMscore > 1.0)
To ascertain that the 26 X-ray structures are indeed suitable templates to select high-quality poses for the 102 FXR agonists to dock within this challenge, we next cross-docked each of the 26 template ligands into the remaining 25 available protein template structures (self-docking was not permitted here). For each docking pose, the corresponding interaction pattern graph was compared and matched, with the GRIM alignment method [15], to the 25 interaction pattern graphs used as references. The benefit of rescoring Surflex poses by GRIM is nicely visible by comparing the root mean square deviations (rmsd) to the X-ray structure of the best predicted poses with or without GRIM rescoring (Fig. 2). Very high quality poses are picked by the GRIM scoring function for all 26 ligands (Fig. 2). With a single exception (PDB entry 1ot7, chain B), all corresponding ligands could be docked within 1.0 Å rmsd (median rmsd = 0.607 Å), thereby validating both the docking engine and the GRIM rescoring method as suitable for this particular dataset. Conversely, the best poses selected by the native Surflex score exhibit a higher and wider distribution of rmsd across the 26 reference ligands (Fig. 2). The GRIMscores associated to the GRIM-selected poses are also very high (median GRIMscore = 1.040) and demonstrate that the set of interaction patterns available in the PDB is sufficient to precisely pick high-quality docking solutions for previously uncharted ligands.Footnote 1

Distribution of root mean square deviations (in Å) from the true X-ray pose, for 26 reference FXR agonists docked to 25 FXR X-ray structures. Poses were scored according to GRIMscore or the native Surflex scoring function. The boxes delimit the 25th and 75th percentiles, the whiskers delimit the 5th and 95th percentiles. The median and mean values are indicated by a horizontal line and a square, respectively. Crosses delimit the 1% and 99th percentiles, respectively. Minimum and maximum values are indicated by a dash
GRIM rescues Surflex for selecting accurate poses for 36 novel FXR agonists
The first stage of the D3R Grand Challenge 2 aimed at predicting the binding mode of 36 novel FXR agonists (Supplementary Table 1) prior to the release of their FXR-bound X-ray coordinates. Out of the 36 agonists, 25 ligands exhibit chemotypes already crystallized in complex with FXR and thus in the list of 26 PDB templates (21 benzimidazoles, two isoxazoles, one tetrahydroazepinoindole, one cholic acid) whereas 9 ligands (3 spiro[indoline-3,4′-piperidine]-2-ones, 3 sulfonamides, 3 miscellaneous) exhibit chemotypes not yet crystallized with FXR. One compound (FXR_33) was excluded from the dataset following organizers’ request due to an oxidation of the compound during the co-crystallization process.
Following our strategy successfully applied in the previous challenge [16], all ligands were first docked with Surflex [21] to all PDB templates. We eliminated the poses with a Surflex score inferior to 2.0. The remaining poses were then converted into interaction pattern graphs and further compared to the 26 reference interaction patterns. For each ligand, the five poses with the highest GRIMscore values, whatever their initial Surflex rank, are last retained. The overall quality of the docking/rescoring protocol is judged by the rmsd of the ligand heavy atoms coordinates to the later released X-ray coordinates. GRIM rescoring clearly aids the Surflex docking engine to find high quality poses as evidenced by the mean rmsd which is lower for highest ranked GRIM poses, GRIM-1 (rmsd = 3.25 Å), with respect to highest ranked Surflex poses, Surflex-1 (rmsd = 4.12 Å; Fig. 3). As already observed in the previous challenge [16], the GRIM-1 pose is usually the most accurate. If one considers the absolute lowest rmsd pose out of the five proposed by GRIM, there is no significant decrease of the mean rmsd (3.08 vs. 3.25 Å; Table 1; Fig. 3), thereby confirming that GRIM rescoring does not necessitate to output more than a single solution. However, we have to admit that the obtained results are not entirely satisfactory since the mean rmsd is clearly above the threshold value (rmsd = 2.0 Å) generally considered as acceptable by the docking community. Considering the absolute lowest rmsd pose out of the 520 (20 × 26) generated for each ligand (Table 1; Fig. 3), we clearly evidence that the present challenge was not difficult from a pure docking viewpoint since the mean rmsd of the best possible pose is really low (rmsd = 1.34 Å; Fig. 3). The overall quality of the poses is certainly due to the use of multiple protein structures (26 in the present case) accounting for the ligand-dependent flexibility of the binding site. Scoring these excellent poses appears to be more challenging since neither GRIM nor the Surflex scoring function were able to rank them high enough to be selected. GRIM rescoring is however definitely better than relying on the original docking score provided by Surflex.

Mean root mean square deviations (in Å) from the true X-ray pose, for 35 new FXR agonists docked to 26 FXR X-ray structures. Poses were scored according to GRIMscore or the native Surflex scoring function. Top ranked poses by GRIM and Surflex are labelled GRIM-1 and Surflex-1, respectively. The lowest rmsd pose identified by GRIM and Surflex are labelled GRIM-best and Surflex-best, respectively. The median values are indicated by a horizontal line, the standard deviation to the mean by a whisker
Analyzing the obtained rmsd values by chemotype, we clearly see that only agonists from the benzimidazole series were accurately docked with 18 out of 23 compounds posed with a rmsd below 2.0 Å (Fig. 4). None of the two isoxazoles (FXR_4, FXR_23) was properly docked although this series had already been co-crystallized with the target. Agonists bearing a new chemotype (sulfonamides, spiro compounds, miscellaneous) were systematically misdocked, considering either GRIM or the Surflex scoring function. Only the novel tetrahydroazepinoindole (FXR_5) was accurately posed (Fig. 4).

Root mean square deviations (in Å) of the GRIM-1 pose from the true X-ray pose for 35 novel FXR agonists
Since GRIM rescoring is a knowledge-based approach, we next looked at which preexisting protein–ligand interaction patterns have been used to select the final poses. As to be expected, GRIM has chosen available interaction patterns from the benzimidazole series to rescore docking poses of compounds sharing this chemotype (Table 2). Since this series adopt a very homogenous binding mode (Fig. 1), it is therefore no surprise that most of the new compounds from this series were docked with low rmsd values. Similarly, the two isoxazoles were also rescored according to interaction patterns from the same chemical series. Nevertheless, our knowledge-based scoring approach failed in selecting accurate poses (Table 2). We previously demonstrated that known interaction patterns for this chemotype were more diverse (Fig. 1), thereby explaining the difficulty to dock such compounds (Table 2). Interestingly, the low two-dimensional (2-D) similarity of the new isoxazoles to the existing templates (Table 2) may explain the hard attempt of GRIM to find out the existing good poses generated by Surflex (Table 1) but not scored high enough. For agonists exhibiting a novel chemotype (sulfonamides and spiro compounds), none of the existing interaction patterns was adequate for rescoring (Table 2). In most cases, these compounds were rescored considering only shape similarity to known templates, as evidenced by the Npol value, which accounts for the number of polar nodes in the selected alignment subgraph (Table 2). In GRIM, nodes have seven possible pharmacophoric properties corresponding to the encoded interaction type (hydrophobic, aromatic, hydrogen bond donor, hydrogen bond acceptor, negative ionisable, positive ionisable, and metal complexation). Whereas apolar nodes (hydrophobic, aromatic) describe mostly the shape of the bound ligand, polar nodes (remaining five properties) are placed at the location of polar protein–ligand interactions (hydrogen and ionic bonds), therefore increasing the confidence for the selected pose.
In the present case, the Npol descriptor exhibited quite often a null value, demonstrating that the GRIM alignment method has not found a single common protein–ligand polar interaction with the template. Among the miscellaneous series, only the novel tetrahydroazepinoindole FXR_5 was accurately docked, by similarity to the interaction pattern observed for the 3fli template of the same series (Table 2). Last, the cholic acid analog FXR_34 was misdocked, even if four interaction pattern templates from the same series were available (1osv, 1ot7_A, 1ot7_B, 4qe6) but not chosen by GRIM (Table 2).
Defining good practices for GRIM rescoring: are docking failures predictable?
To better delineate the applicability domain of GRIM rescoring, we plotted the observed rmsd of the the top-1 GRIM pose with respect to three simple descriptors: GRIMscore, 2-D similarity to the X-ray ligand chosen for interaction template matching, and number of conserved polar interactions between the selected pose and the template pose (Npol parameter). The corresponding scatter plots (Fig. 5) clearly show some conditions that favor GRIM selection. They are here recapitulated by the following three rules:

Variation of the root mean square deviations (in Å) to the X-ray poses for 35 FXR agonists as a function of three properties: a GRIMscore of the pose, b 2-D similarity of the FXR agonist to the FXR-bound template ligand used by GRIM, c number of polar nodes (polar interactions, Npol) in the clique common to the protein-FXR ligand docked pose and the protein–ligand PDB template chosen by GRIM for pose selection. Docking poses complying to a precise rule are enclosed by a red circle
Rule 1: a very high GRIMscore (>1.0) generally leads to a low rmsd (<2.0). There are only four exceptions to this rule, one for which the GRIMscore is below the 1.0 threshold (FXR_9) although the rmsd is <2.0, and three (FXR_13, FXR_18 and FXR_35) for which the rmsd is >2.0 although the GRIMscore is >1.0. (Fig. 5a).
Rule 2: a high 2-D similarity to the template ligand (Tc > 0.6) generally leads to a low rmsd (<2.0 Å). This rule is verified for 31 out of 35 ligands analyzed. There are no cases for which GRIM could prioritize a good pose from a template in which the X-rayed ligand had a low 2-D similarity to the compound to dock (Fig. 5b). In four cases (FXR_8, FXR_13, FXR_18, FXR-35), a sufficiently high 2-D similarity to the template ligand could not guide GRIM to find out a good pose. When this 2-D similarity is low (<0.60), GRIM fails with no exception. Interestingly, such a strong 2-D chemical similarity to PDB templates was not required to properly dock HSP90α and MAP4K4 inhibitors in the previous D3R challenge [16]. This rule might thus be dataset-dependent.
Rule 3: Good poses correspond to interaction patterns with at least two conserved polar interactions (Npol ≥ 2). In this challenge, we prioritized pose selection by the overall GRIMscore. This strategy clearly fails in case the common interaction pattern subgraph chosen for pose selection is lacking enough polar nodes (NPol parameter). Assuming only shape conservation (Npol = 0), there is only one ligand (FXR_7) out of nine that is well docked by GRIM (Fig. 5c). In case Npol is equal to 1 (one common FXR-agonist polar interaction), no docking success (rmsd < 2.0) could be reported as well. It is only when Npol is greater or equal to 2 that good rmsd values begin to be observed (Fig. 5c). There are only three ligands (FXR_13, FXR_34, FXR_35) for which the rule is not verified despite a Npol value of 3.0. These three rules clearly define a strict applicability domain for GRIM rescoring, at least for the present dataset of 36 agonists. New challenges will enable to precise whether these rules are transferable to new datasets or not.
Reasons for failure
After the submission of the first set of predictions (stage1: prediction of up to five poses for each ligand), X-ray structure of the 36 new agonists in complex with FXR were released. The comparison of the corresponding protein–ligand interaction patterns with that of our 26 PDB templates illustrates the difficulty of the present challenge (Fig. 6a). With the exception of all benzimidazoles and one tetrahydroazepinoindole (FXR_5), all novel FXR agonists adopt an unprecedented binding mode (Fig. 6a). A molecular explanation to this observation lies in the large (750–1025 Å3) and hydrophobic cavity in FXR that is able to accommodate very different and almost non-overlapping agonists. Despite these significant hurdles, observed failures to predict binding modes of these ligands are not due to real docking issues. Among the set of proposed poses by Surflex, true docking failures (Surflex-best rmsd <3.0 Å) were only reported for three ligands (FXR_1, FXR_18 and FXR_23). The case of the isoxazole FXR_23 is perfectly illustrative. The top-1 GRIM pose (Fig. 6b) was generated by analogy of the corresponding interaction pattern found on the 3hc5 template of the same chemical series, nicely overlapping the aryl-isoxazole moieties of both compounds. Surprisingly, the disclosed X-ray pose of FXR_23 departs considerably from that already seen for FXR isoxazole agonists (Fig. 6b). None of the solutions generated by Surflex are similar to this unexpected pose. It is interesting to notice that two out of these three ligands are among the less potent agonists of the dataset, with in vitro binding affinities in the micromolar range.

Predicted versus X-ray protein–ligand interaction patterns for 35 FXR agonists. a A posteriori observed binding mode similarity of 35 novel FXR agonists to 26 known PDB ligands, as a function of the root mean square deviations (in Å) of the GRIM-1 pose to the true X-ray structure. For each novel FXR-agonist X-ray structure, the highest GRIMscore considering all 26 PDB templates is used to quantify binding mode similarity. b Predicted (cyan sticks) versus X-ray pose (tan sticks) of the agonist FXR_23 bound to the FXR receptor (white ribbons). GRIM-1 pose was obtained by docking FXR_23 to the 3ruu atomic coordinates and selected according to the similarity of its interaction pattern to that of the 3hc5 ligand (light green sticks). Heteroatoms are colored in blue (nitrogen), red (oxygen), yellow (sulfur) and dark green (chlorine). c Predicted (cyan sticks) versus X-ray pose (tan sticks) of the agonist FXR_8 bound to the FXR receptor (white ribbons). GRIM-1 pose was obtained by docking FXR_8 to the 3oki atomic coordinates and selected according the similarity of its interaction pattern to that of the 3oki ligand (light green sticks). Heteroatoms are colored in blue (nitrogen) and red (oxygen)
GRIM failed to propose reliable poses for three benzimidazoles (FXR_8, FXR_13, FXR_35) although they exhibit the canonical benzimidazole-binding mode (Fig. 6a) and despite the existence of suitable poses generated by Surflex (Surflex-best rmsd <2.0 Å, Table 1). The overall cross-like shape of the ligands as well as the exact binding amino acids are found, but the benzimidazole ring switched to the area normally occupied by a cyclohexyl ring (Fig. 6c). In these examples, the GRIMscore is dominated by the conservation of apolar contacts (in other words, the overall ligand shape) at the cost of an important hydrogen bond between the benzimidazole and Tyr373 side chain. The same observation applies to most of the scoring failures for which the GRIM-selected pose has been dominated by shape conservation (Fig. 5c). The herein described pose selection protocol, biased towards the highest GRIMscore is therefore not suited for hydrophobic ligands binding to predominantly apolar cavities. Removing poses with no common polar interactions to the chosen template (Npol = 0) before final GRIM ranking should avoid some of the above reported failures in the future.
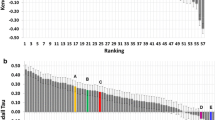
Altogether, combining Surflex for pose generation and GRIM for ranking remains a robust docking strategy. When compared to the 51 other contributions to the first phase of the D3R Grand Challenge 2, the Surflex/GRIM protocol lies among the top contributions sharing a posing accuracy in the 2–3 Å rmsd range (Fig. 7).

Accuracy of 52 contributions to the stage 1 (pose prediction) of the D3R Grand Challenge 2. The accuracy of each contribution is described by the mean rmsd of the highest-ranked pose to the X-ray coordinates of 35 test FXR agonists, released after closing the challenge. Incomplete submissions are indicated by white bars
Ranking GRIM poses by the HYDE scoring function: estimating the binding affinity of 102 FXR agonists
In a second step, the binding affinity of 102 FXR agonists (Supplementary Table 2), including the 36 ligands (FXR_1-FXR_36) considered for the pose prediction phase and 66 additional ligands (FXR_37-FXR_102), was predicted and compared to the experimental affinity data released once the challenge was closed. The GRIM-1 pose of each ligand was thus rescored with the HYDE scoring function [23]. This scoring function was chosen because of the very careful estimation of the desolvation contributions to the overall binding free energy of a wide array of diverse protein–ligand complexes. Importantly, HYDE is not parametrized against any particular training set and provides a balanced assessment of the energetics of desolvation considering three atomic physicochemical properties: local hydrophobicity, solvent accessible surface, and contact surface area. When applied to the FXR dataset of 102 ligands, HYDE scoring of the GRIM-1 pose yielded the third most accurate ranking with a Kendall’s τ ranking coefficient of 0.442 (Fig. 8a) and a Pearson’s ρ correlation coefficient of 0.593 (Fig. 8b). Predicted HYDE binding free energies were systematically higher than that derived from in vitro experimental binding affinity data but an overall trend could be confirmed. The current performance in affinity ranking is slightly better than that obtained for the previous D3R challenge for two different datasets (180 HSP90α inhibitors, 18 MAP4K4 inhibitors) [13], but far from being usable for hit to lead optimization, thereby confirming most previous attempts to accurately predict binding free energies for a set of heterogeneous compounds [11, 26]. Since the observed ranking and correlation coefficients were not improved for the subset of benzimidazoles for which docking poses are of good quality, consistent failures in predicting binding free energies cannot be attributed to error in atomic coordinates but merely to inconsistent treatment of energetics.

Assessment of the GRIM–HYDE rescoring protocol for ranking and affinity prediction of 102 FXR agonists. a Comparison of the GRIM–HYDE protocol with respect to 57 competing contributions in this challenge, estimated by the Kendall’s τ ranking coefficient. Error bars were obtained by re-computing all statistics in 10,000 rounds of resampling with replacement, where, in each sample, the experimental IC50 data were randomly modified based on the experimental uncertainties (data provided by the organizers). b Predicted (ΔGpred) versus experimental (ΔGexp) absolute binding free energies in kJ/mol. The red line indicates a linear regression fit of predicted to experimental values
Conclusions
We have herein applied a knowledge-based graph matching method (GRIM) to rank docking poses of 36 novel FXR agonists. Although the current poses have been generated by Surflex, the rescoring protocol is fully independent on any docking tool, at the condition that correct MOL2 file formats can be generated. This second D3R docking challenge was significantly more demanding than the previous one for the major reason that the large and apolar ligand-binding site of the chosen target (FXR receptor) is able to accommodate chemically heterogeneous ligands in many different ways. Docking predictions were reasonably accurate (mean rmsd = 1.97 Å) for the class of 21 benzimidazoles already co-crystallized with FXR, which exhibited a conserved binding mode. For other chemical classes, the existence (isoxazoles) or not (spiroindolines and sulfonamides) of known similar PDB templates, did not impact GRIM posing, which was very unsatisfactory (rmsd above 4 Å). Several reasons could be invoked to explain these systematic rescoring failures: (i) the inability of the docking engine to provide a single accurate pose for further rescoring, (ii) the impossibility to predict completely novel binding modes, (iii) the bias in GRIM rescoring towards poses with the highest protein–ligand interaction pattern similarity to that of existing PDB templates, thereby favoring quite often shape matching for ligands with a predominant hydrophobic character.
However, the current challenge has taught us interesting rules to improve future predictions. First, the obtained data confirm the well-known bias of fast energy-based scoring functions to rank improper poses among the top-ranked solutions. Any knowledge-based rescoring scheme [27,28,29,30] similar in spirit to GRIM is therefore preferable to relying only on docking scores. In our current rescoring protocol, poses with very low predicted binding energies (predicted pkd <2) were filtered out and not subjected to GRIM rescoring. Later, the comparison of the experimental solutions with our predictions surprisingly identified several cases where reasonably good poses (rmsd to X-ray pose <2 Å) were rejected for the simple reason that the predicted pkd was below 2. We will therefore remove this filter in our next rescoring protocol.
Second, GRIM rescoring relies on the existence of a similar protein–ligand interaction pattern in the PDB. For unbalanced interaction patterns dominated by apolar contacts, we identified a bias in prioritizing shape overlap over the conservation of a few but very important hydrogen bonds or salt bridges. As a workaround, we thus propose to eliminate from the set of GRIM selections, any pose for which the number of aligned graph polar nodes to the chosen template is below a value of 2.
Third, rescoring the GRIM highest ranked pose using the HYDE scoring function provided one of the most accurate affinity ranking strategy submitted to this challenge. Due to the overall speed of the procedure, we therefore propose to apply this rescoring protocol to virtual screening where GRIM is used to prioritize a few thousand ligands to be further rescored with HYDE within a reasonable amount of cpu time (ca. 10 s/compound).
GRIM presents several advantages over alternative knowledge-based rescoring strategies: (i) it can be coupled to any docking algorithm, (ii) it does not constrain ligand docking but rewards interaction patterns already present among PDB templates, (iii) it takes advantage of ligands with similar binding modes and not necessarily similar chemical structures [16], (iv) it can be applied in a target family-biased pose selection process in which PDB templates from the same protein but also from similar targets can be used to store reference interaction patterns, (v) it permits to directly quantify binding mode similarity between a predicted protein–ligand complex and any PDB template at a very high throughput.
GRIM is part of the IChem toolkit and available for non-profit research at http://bioinfo-pharma.u-strasbf.fr/labwebsite/download.html.
Notes
Previous experiences suggest that GRIMscore values above 0.70 correspond to a statistically significant similarity of the two interaction patterns under comparison.
References
Brooijmans N, Kuntz ID (2003) Molecular recognition and docking algorithms. Annu Rev Biophys Biomol Struct 32:335–373
Rognan D (2017) The impact of in silico screening in the discovery of novel and safer drug candidates. Pharmacol Ther 175:47–66
Chen YC (2015) Beware of docking! Trends Pharmacol Sci 36:78–95
Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS (2006) A critical assessment of docking programs and scoring functions. J Med Chem 49:5912–5931
Smith RD, Dunbar JB Jr, Ung PM, Esposito EX, Yang CY, Wang S, Carlson HA (2011) CSAR benchmark exercise of 2010: combined evaluation across all submitted scoring functions. J Chem Inf Model 51:2115–2131
Community Structure Activity resource. http://www.csardock.org. Accessed 30 July 2010
Drug Design Data Resource. https://drugdesigndata.org/about/grand-challenge-2. Accessed 30 July 2010
Dunbar JB Jr, Smith RD, Yang CY, Ung PM, Lexa KW, Khazanov NA, Stuckey JA, Wang S, Carlson HA (2011) CSAR benchmark exercise of 2010: selection of the protein–ligand complexes. J Chem Inf Model 51:2036–2046
Damm-Ganamet KL, Smith RD, Dunbar JB Jr, Stuckey JA, Carlson HA (2013) CSAR benchmark exercise 2011–2012: evaluation of results from docking and relative ranking of blinded congeneric series. J Chem Inf Model 53:1853–1870
Dunbar JB Jr, Smith RD, Damm-Ganamet KL, Ahmed A, Esposito EX, Delproposto J, Chinnaswamy K, Kang YN, Kubish G, Gestwicki JE, Stuckey JA, Carlson HA (2013) CSAR data set release 2012: ligands, affinities, complexes, and docking decoys. J Chem Inf Model 53:1842–1852
Carlson HA (2016) Lessons learned over four benchmark exercises from the Community Structure-Activity Resource. J Chem Inf Model 56:951–954
Carlson HA, Smith RD, Damm-Ganamet KL, Stuckey JA, Ahmed A, Convery MA, Somers DO, Kranz M, Elkins PA, Cui G, Peishoff CE, Lambert MH, Dunbar JB Jr (2016) CSAR 2014: a benchmark exercise using unpublished data from pharma. J Chem Inf Model 56:1063–1077
Gathiaka S, Liu S, Chiu M, Yang H, Stuckey JA, Kang YN, Delproposto J, Kubish G, Dunbar JB Jr, Carlson HA, Burley SK, Walters WP, Amaro RE, Feher VA, Gilson MK (2016) D3R Grand Challenge 2015: evaluation of protein-ligand pose and affinity predictions. J Comput Aided Mol Des 30:651–668
Smith RD, Damm-Ganamet KL, Dunbar JB Jr, Ahmed A, Chinnaswamy K, Delproposto JE, Kubish GM, Tinberg CE, Khare SD, Dou J, Doyle L, Stuckey JA, Baker D, Carlson HA (2016) CSAR benchmark exercise 2013: evaluation of results from a combined computational protein design, docking, and scoring/ranking challenge. J Chem Inf Model 56:1022–1031
Desaphy J, Raimbaud E, Ducrot P, Rognan D (2013) Encoding protein–ligand interaction patterns in fingerprints and graphs. J Chem Inf Model 53:623–637
Slynko I, Da Silva F, Bret G, Rognan D (2016) Docking pose selection by interaction pattern graph similarity: application to the D3R Grand Challenge 2015. J Comput Aided Mol Des 30:669–683
Richter HG, Benson GM, Bleicher KH, Blum D, Chaput E, Clemann N, Feng S, Gardes C, Grether U, Hartman P, Kuhn B, Martin RE, Plancher JM, Rudolph MG, Schuler F, Taylor S (2011) Optimization of a novel class of benzimidazole-based farnesoid X receptor (FXR) agonists to improve physicochemical and ADME properties. Bioorg Med Chem Lett 21:1134–1140
Bass JY, Caravella JA, Chen L, Creech KL, Deaton DN, Madauss KP, Marr HB, McFadyen RB, Miller AB, Mills WY, Navas F 3rd, Parks DJ, Smalley TL Jr, Spearing PK, Todd D, Williams SP, Wisely GB (2011) Conformationally constrained farnesoid X receptor (FXR) agonists: heteroaryl replacements of the naphthalene. Bioorg Med Chem Lett 21:1206–1213
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242
Bietz S, Urbaczek S, Schulz B, Rarey M (2014) Protoss: a holistic approach to predict tautomers and protonation states in protein-ligand complexes. J Cheminform 6:12
Jain AN (2007) Surflex-Dock 2.1: robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J Comput Aided Mol Des 21:281–306
Desaphy J, Da Silva F, Rognan D (2014) IChem v.5.2.6. http://bioinfo-pharma.u-strasbg.fr/labwebsite/downloads.
Schneider N, Lange G, Hindle S, Klein R, Rarey M (2013) A consistent description of HYdrogen bond and DEhydration energies in protein–ligand complexes: methods behind the HYDE scoring function. J Comput Aided Mol Des 27:15–29
Xu X, Xu X, Liu P, Zhu ZY, Chen J, Fu HA, Chen LL, Hu LH, Shen X (2015) Structural basis for small molecule NDB (N-Benzyl-N-(3-(tert-butyl)-4-hydroxyphenyl)-2,6-dichloro-4-(dimethylamino) Benzamide) as a selective antagonist of farnesoid X receptor alpha (FXRalpha) in stabilizing the homodimerization of the receptor. J Biol Chem 290:19888–19899
Jin L, Feng X, Rong H, Pan Z, Inaba Yu, Qiu L, Zheng W, Lin S, Wang R, Wang Z, Wang S, Liu H, Li S, Xie W, Li Y (2013) The antiparasitic drug ivermectin is a novel FXR ligand that regulates metabolism. Nat Commun 4:1937
Kim R, Skolnick J (2008) Assessment of programs for ligand binding affinity prediction. J Comput Chem 29:1316–1331
Gao C, Thorsteinson N, Watson I, Wang J, Vieth M (2015) Knowledge-based strategy to improve ligand pose prediction accuracy for lead optimization. J Chem Inf Model 55:1460–1468
Kelley BP, Brown SP, Warren GL, Muchmore SW (2015) POSIT: flexible shape-guided docking for pose prediction. J Chem Inf Model 55:1771–1780
Anighoro A, Bajorath J (2016) Three-dimensional similarity in molecular docking: prioritizing ligand poses on the basis of experimental binding modes. J Chem Inf Model 56:580–587
Kumar A, Zhang KY (2016) Application of shape similarity in pose selection and virtual screening in CSARdock2014 exercise. J Chem Inf Model 56:965–973
Acknowledgements
We thank the CAPES foundation and the Ministry of Education of Brazil for a postdoctoral fellowship to P.S.F.C.G (Computational Biology program Grant, Issue No. 51/2013). The Computing Center of the IN2P3 (CNRS, Villeurbanne, France) is acknowledged for allocation of computing time and excellent support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
da Silva Figueiredo Celestino Gomes, P., Da Silva, F., Bret, G. et al. Ranking docking poses by graph matching of protein–ligand interactions: lessons learned from the D3R Grand Challenge 2. J Comput Aided Mol Des 32, 75–87 (2018). https://doi.org/10.1007/s10822-017-0046-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-017-0046-1