
Abstract
Recent development of DNA sequencing technologies has enabled successful construction of thousands of mitochondrial genomes. Nevertheless, only 33 mitochondrial genomes have been reported for species in Bacillariophyta, which includes harmful algal bloom (HAB) species. In this study, we determined the complete mitogenome of the Bacillariophyta HAB species Odontella regia. For comparison, we also constructed the mitogenome of Lithodesmium undulatum, another Bacillariophyta species. Both strains were isolated in the Jiaozhou Bay, an epitome of China’s coastal ecosystem and an ideal site for HAB research. These two mitogenomes were characterized as 37,617 bp and 37,057 bp circular-mapping molecules with AT content of 73.4% and 75.3%, respectively. The phylogenetic tree based on 32 protein-coding genes of the mitogenome encoded in 35 Bacillariophyta species revealed that the class Mediophyceae consisted of multi-phyletic clades. While O. regia formed an independent clade, L. undulatum was closely related to Thalassiosira pseudonana and Skeletonema marinoi. Synteny comparison of O. regia and L. undulatum mitogenomes and mitogenomes of three closely related species displayed substantial differences among lineages in Mediophyceae by a series of gene translocations and/or inversion with many conserved gene blocks such as rpl2-rps19-rps3-rpl16-atp9. These analyses suggested a complex evolutionary relationship in Mediophyceae in which the mitogenome of O. regia was the least conservative compared with the mitogenomes of four other Bacillariophyta species. Further studies are needed to clarify detailed phylogenetic relationships in Bacillariophyta.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Harmful algal blooms (HABs) are a worldwide phenomenon caused by extreme successions of phytoplankton communities, which have attracted close attention due to serious consequences of HABs on human health, as well as economic loss and social disruption (Hoagland et al. 2002; Jin et al. 2008; Dyson and Huppert 2010; McPartlin et al. 2017; Crosman et al. 2019; Moore et al. 2020). HAB species produce potent toxins such as paralytic shellfish poisoning (PSP) from Alexandrium acatenella, diarrhetic shellfish poisoning (DSP) from Dinophysis acuta, and neurotoxic shellfish poisoning (NSP) from Gymnodinium breve, which are harmful to humans through the food chain (Hallegraeff 1993; Balmer-Hanchey et al. 2003). In 2012 alone, HABs in China resulted in a direct economic loss of more than 2 billion RMB (Chen et al. 2015). As a result of climate change and intensifying human activities, HABs have gained new features with a larger scale, a longer duration, more serious consequences, and more notable global expansion (Yu and Chen 2019). Therefore, studies on HAB species and HABs are becoming urgently important.
Diatoms are a diverse association of unicellular, autotrophic, eukaryotic algae that play an important role in aquatic food webs (Pogoda et al. 2019). Diatoms have the most effective RuBisCO enzyme within autotrophs (Giordano et al. 2005) and work as a vital part in the cycling of CO2 that accounts for ∼ 40% of marine carbon export (Armbrust et al. 2004; Shibl et al. 2020). Diatoms are the most diverse kind of algae with approximately 200,000 species (Mann and Droop 1996; Mann 1999), many of which cause HABs (Qi et al. 2004).
Odontella regia is a HAB species of the genus Odontella, family Odontellaceae, order Eupodiscales, and class Mediophyceae (Hegde et al. 2011). It is a planktonic and bipolar centric diatom that reproduces by sexual reproduction with 64 sperm cells per cell instead of sporulation. Odontella regia is distributed in coastal waters of warm temperate zones and tropics and appears in all seas of China (Yang and Dong 2006) and has been reported to form HABs in estuarine and coastal waters (Badylak 2004; Carstensen et al. 2015). In Jiaozhou Bay, China, O. regia HABs have brought adverse effects on marine fisheries and coastal aquaculture (Han et al. 2004; Wu et al. 2005). Multiple other species in the genus Odontella including Odontella aurita and Odontella sinensis can cause HABs as well (Guo 2004; Ibrahim and Imad 2017). In particular, O. aurita has been found to be rich in eicosapentaenoic acid and has attracted widespread attention (Haimeur et al. 2012; Mimouni et al. 2012; Xia et al. 2013). In contrast, research on O. regia was limited to morphological observation and only a few molecular markers including 18S ribosomal DNA (18S rDNA) gene and the ribulose-1,5-bisphosphate carboxylase (rbcL) gene.
Driven by the recent development of DNA sequencing technologies, mitochondrial genomes of numerous species have been sequenced and fully constructed, uncovering valuable knowledge about gene function and evolutionary trajectories (Smith 2016). Furthermore, comparative analysis of mitochondrial genomes has been applied as an effective method to develop high-resolution molecular markers (Chen et al. 2019). However, our knowledge about the mitogenomes of diatoms, especially those that cause HABs, is very limited. Until now there are only 33 published mitogenomes of species in Bacillariophyta, which consist of three main classes in Mediophyceae, Coscinodiscophyceae, and Bacillariophyceae. Comparative analysis of mitogenomes can help us understand the complex evolutionary relationships of algal species.
In this study, we determined the mitogenome of the HAB species in O. regia. We also constructed the mitogenome of another Bacillariophyta species Lithodesmium undulatum for comparative analysis. Both strains were isolated in the Jiaozhou Bay, which is connected to the Yellow Sea with a small opening. We analyzed morphological features of two species and determine their taxonomic positions using common molecular markers. For accurate comparison, we re-annotated 33 published mitogenomes downloaded from GenBank at NCBI. Those genes that were missing in the GenBank annotation have been re-annotated, and several annotation errors have also been corrected. Of the 35 diatom mitogenomes here, 31 are a complete circular-mapping molecule, whereas the four remaining are incomplete mitogenomes.
Materials and methods
Strain isolation, culturing, and phylogeny-based species characterization
Odontella regia strain CNS00380 and Lithodesmium undulatum strain CNS00316 were isolated from Jiaozhou Bay (120° 10′ 839″ E, 36° 06′ 105″ N; 120° 13′ 971″ E, 36° 04′ 023″ N), respectively. The strains were isolated with the method of single-cell capillary and cultured in L1 medium with 1‰ volume fraction Na2SiO3 with H2O added (Guillard and Hargreaves 1994). The culture temperature was 18–20 °C, and the irradiance was from 27 to 40 μmol photons m−2 s−1 with a photoperiod of 12 h light-12 h dark. Species identification was according to morphological features and phylogenetic analyses based on full-length 18S rDNA and ribulose-1,5-bisphosphate carboxylase (rbcL) genes.
The phylogenetic trees were constructed using MEGA7 (Kumar et al. 2016). Phylogenetic relationships were inferred using the neighbor-joining method (Saitou and Nei 1987). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches (Felsenstein 1985). The tree was drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the maximum composite likelihood method (Tamura et al. 2004) and are in the units of the numbers of base substitutions per site. The analyses involved 41 and 18 nucleotide sequences in the 18S rDNA and rbcL genes, respectively. The codon positions included were 1st + 2nd + 3rd + non-coding. All positions containing gaps and missing data were eliminated. There were a total of 1450 and 1289 positions in the final dataset of 18S rDNA and rbcL gene.
DNA library preparation and whole-genome sequencing
Total DNA was extracted with DNAsecure Plant Kit (Tiangen Biotech, China). The genomic DNA sample was fragmented by sonication (Covaris S220, Covaris, USA) to a size of 350 bp. DNA fragments were then end polished, A-tailed, and ligated with the full-length adapters for Illumina sequencing, followed by PCR (MiniAmp thermal cycler, Thermo Fisher, USA) enrichment using generic adapter P5 and P7 oligos. The PCR products were purified by the AMPure XP system (Beckman Coulter, USA); libraries were analyzed for size distribution by NGS3K/Caliper and quantified by real-time PCR (Qubit 3.0 fluorometer, Invitrogen, USA). Qualified libraries were sequenced on an Illumina platform according to the effective concentration and data volume at Novogene (Beijing, China). Sequencing was conducted using NovaSeq PE150 (Illumina, USA), generating paired-end reads in size of 150 bp. The sequencing results have been submitted to NCBI (BioProject number PRJNA685980).
Construction and annotation of mitochondrial genomes
Raw data was trimmed using Trimmomatic-0.39 with the parameters LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:75 (Bolger et al. 2014). Clean data were assembled using SPAdes v3.14 with default parameters spades/bin/spades.py -t 32 (Bankevich et al. 2012). The mitogenome of Skeletonema marinoi (NC_028615) was used as a query to search for scaffolds corresponding to the mitogenome sequences of strain CNS00380 and strain CNS00316 from the resulting assembly scaffolds using BLAST with default parameters (Johnson et al. 2008), respectively. The mitogenome sequences were validated using BWA, SAMtools, and IGV. Briefly, reads were aligned to the draft mitogenome using BWA (0.7.17) (Li and Durbin 2009). SAMtools (1.10) (Li et al. 2009) was used to extract the alignment results, and IGV (Thorvaldsdottir et al. 2013) was used to inspect the alignments for validation and error correction. Annotations (the accession numbers for O. regia and L. undulatum are MW018491 and MW023083, respectively) were made with MFannot (https://github.com/BFL-lab/Mfannot) and NCBI’s ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/), completed in NCBI’s Sequin 15.10 (https://www.ncbi.nlm.nih.gov/projects/Sequin/) using the genetic code of Mold, Protozoan, and Coelenterate Mitochondrial; Mycoplasma/Spiroplasma.
Phylogenetic analysis using concatenated mitochondrial protein-coding genes
Thirty-two mitochondrial protein-coding genes (atp6, 8, 9; cob; cox1, 2, 3; nad1-7, 4L, 9, 11; rpl2, 5, 6, 14, 16; rps3, 4, 7, 8, 10, 11, 13, 14, 19; and tatC) from each diatom species were extracted and concatenated for phylogenetic analysis. The amino acid sequences of each of the 32 genes from different diatom mitochondria were individually aligned using MAFFT with default parameters (Katoh and Standley 2013). The regions that were ambiguously aligned in each alignment were deleted using trimAl 1.2rev59 (Capella-Gutierrez et al. 2009) with the parameters gt = 1, and all amino acid sequences were concatenated using Phyutility (Smith and Dunn 2008). The concatenated dataset of amino acid that was 6424 characters was partitioned by gene position. The incongruence length difference test (ILD, also called the partition homogeneity test) was conducted via PAUP4.0 with the following parameters: number of replications = 100; optimality criterion = maximum parsimony (Swofford 2002). The p value (0.01) indicated that combining data would not affect the phylogenetic accuracy. When the ILD detected p values lower than 0.001, the combined data suffered relative to the individual partitions (Cunningham 1997). The evolutionary models and partitioning of the amino acid data were determined using ModelFinder (Kalyaanamoorthy et al. 2017). The phylogenetic tree was constructed with IQ-TREE using default parameters (Trifinopoulos et al. 2016). The ultrafast bootstrap analysis with 1000 replicates of the dataset and approximate Bayes test was performed to estimate statistical reliability (Anisimova et al. 2011; Minh et al. 2013). Phytophthora ramorum and Saprolegnia ferax in Oomycota were used as out-groups.
Synteny analysis of closely related mitochondrial genomes
Synteny analysis of five mitogenomes sequences from class Mediophyceae was carried out with the program Mauve v2.3.1 using progressiveMauve with default parameters (Darling et al. 2010). The comparative illustration of mitochondrial genomes was performed using circos-0.69 (Krzywinski et al. 2009).
Results
Morphological and molecular characterization of the harmful algal species Odontella regia
The diatom strain CNS00380 of Odontella regia was generally rectangular with the middle part of the valve surface being flat or slightly concaved with elongated rodlike protrusions extending from the four corners of the cell and obvious small ridges on the shell surface inside the protrusions with hollow spines on it (Fig. 1a). Such morphological features were in accordance with that of the species Odontella regia (Ashworth et al. 2013). In the full-length 18S rDNA-based phylogenetic tree, the strain CNS00380 clustered with KC309502.1 Odontella regia (Ashworth et al. 2013) and HQ912564.1 Odontella sinensis (Theriot et al. 2010) with PID 100% and 99.94%, respectively (Fig. 1c). Phylogenetic analysis of ribulose-1,5-bisphosphate carboxylase (rbcL) genes suggested that the rbcL gene of CNS00380 clustered well with KC309576.1 Odontella regia (Ashworth et al. 2013) with PID 100% in a monophyletic clade (Fig. 1d). The combined morphological and molecular features of CNS00380 confirmed that this strain was Odontella regia.

Representative micrographs of diatom species studied in this project. a Odontella regia (strain CNS00380). b Lithodesmium undulatum (strain CNS00316). c Phylogenetic analysis based on 18S ribosomal DNA (18S rDNA) gene. d Phylogenetic analysis based on ribulose-1,5-bisphosphate carboxylase (rbcL) gene. Numbers at the branches represent bootstrap values. Branch lengths are proportional to the genetic distances, which are indicated by the scale bar
The strain Lithodesmium undulatum CNS00316 was short columnar with a quadrangular valve surface, with adjacent cells connected into groups by the membranous valve membrane (Fig. 1b), and was consistent with that of L. undulatum (Karp-Boss et al. 2014). This taxonomical annotation was also confirmed by phylogenetic analysis using the full-length 18S rDNA and rbcL sequences as molecular markers (Fig. 1c, d).
General characteristics of mitochondrial genomes
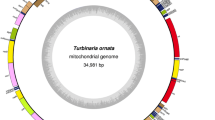
Diatom mitogenomes vary widely in size, ranging from 32,777 bp in Melosira undulata to 103,605 bp in Halamphora calidilacuna (Pogoda et al. 2019). AT contents vary from 65.0% in Phaeodactylum tricornutum to 78.4% in Melosira undulata (Oudot-Le Secq and Green 2011; Pogoda et al. 2019) (Table 1). The number of tRNA genes ranged from 22 to 28. The number of introns ranged from 0 to 20. Start codons of genes were usually ATG. The start codons of atp8 in different diatom mitogenomes could be different, including ATT, ATA, and TTG (Table 2). The complete mitochondrial genomes of O. regia and L. undulatum were 37,057 bp (Fig. 2a) and 37,617 bp (Fig. 2b) in size, respectively. The AT content of O. regia and L. undulatum was 73.4% and 75.3%, respectively, which were relatively higher than those of most of other diatom mitogenomes such as Skeletonema marinoi (70.3%) (An et al. 2017) and Thalassiosira pseudonana (69.9%) (Armbrust et al. 2004) (Table 1).

Complete mitogenomes of a Odontella regia (strain CNS00380) and b Lithodesmium undulatum (strain CNS00316). Genes shown on the inside of the map are transcribed in the clockwise direction, whereas those on the outside of the map are transcribed counterclockwise. The assignment of genes into different functional groups is indicated by colors. The ring of bar graphs on the inner circle shows the GC content in dark gray
Typical diatom mitogenomes contain a set of 35 core genes, including 33 protein-coding genes (PCGs) and two non-coding rRNA genes (rns and rnl) that are completely conserved throughout the taxa (Table 2). The genomes of O. regia and L. undulatum contained 24 and 25 tRNA genes, respectively. The start codons of atp8 in O. regia and L. undulatum were ATG and ATT, respectively. The rn5 gene, which is missing from many other diatoms (Guillory et al. 2018), was present in both O. regia and L. undulatum. The nad11 split coding region is present in many diatoms, which makes the nad11 gene undergo fission into two separate submits with its own start and stop codons respectively and corresponds to the iron-sulfur binding (nad11a) and molybdopterin-binding domains (nad11b) (Imanian et al. 2012; Guillory et al. 2018). However, this gene was absent from both O. regia and L. undulatum. The loss of rps2 is deep and ancient within angiosperms (Adams and Palme 2003). Even though the loss of rps2 is uncommon in diatoms, it was absent from both O. regia and L. undulatum.
Phylogenetic analysis of O. regia and other diatom species
To explore the evolutionary relationship between O. regia and other diatom species, we constructed a phylogenetic tree using 32 mitochondrial protein-coding genes shared by many species of Bacillariophyta and Oomycota using the maximum likelihood method with amino acid sequences (Fig. 3). The phylogenetic tree revealed three distinct classes, Bacillariophyceae, Mediophyceae, and Coscinodiscophyceae. Our results indicated that Bacillariophyceae was non-monophyletic (Fig. 3), which is inconsistent with a previous report reporting a monophyletic relationship based on 13 conserved mitochondrial protein-coding genes (Pogoda et al. 2019). The difference may be caused by the different numbers of genes used for constructing phylogenetic trees. Our results indicated that Mediophyceae was a sister group of Bacillariophyceae, which is consistent with the result based on the amino acid dataset of 34 mitochondrial PCGs (An et al. 2017). Odontella regia and Toxarium undulatum, which are both Mediophyceae, each formed an independent clade (Fig. 3). The close relationship between L. undulatum and Thalassiosira pseudonana was consistent with a previous report that Thalassiosirales and Lithodesmiales (which included Lithodesmium) were sisters (Williams 2007). Thalassiosira pseudonana was closely related to Skeletonema marinoi, as reported previously (An et al. 2017). Previous phylogenetic analyses using amino acid sequences of 30 mitochondrial PCGs suggested that Melosira undulata and T. pseudonana were sister species (Liu et al. 2019). Such discrepancy may be caused by different number of genes used in constructing phylogenetic trees and by the lack of a sufficiently large number of representative mitogenomes for phylogenetic analysis in Bacillariophyta.

Maximum likelihood (ML) phylogenetic tree based on concatenated amino acid sequences of 32 mitochondrial protein-coding genes (atp6, 8, 9; cob; cox1, 2, 3; nad1-7, 4L, 9, 11; rpl2, 5, 6, 14, 16; rps3, 4, 7, 8, 10, 11, 13, 14, 19; and tatC). Mitochondrial protein-coding genes of Phytophthora ramorum and Saprolegnia ferax were used as out-group taxa. Numbers on the left and right sides at the branches represent Bayesian posterior probabilities and bootstrap values. Branch lengths were proportional to the amount of sequence change, which are indicated by the scale bar below the trees
Synteny analysis revealed extensive rearrangement events
The mitogenomes of closely related species L. undulatum, T. pseudonana, S. marinoi, T. undulatum, and O. regia exhibited a high level of gene rearrangement (Fig. 4a), which was consistent with previous studies showing that mitogenomes have a variety of rearrangements in class Bacillariophyceae (Pogoda et al. 2019). Lithodesmium undulatum and O. regia displayed a new architecture that was different from other lineages in Mediophyceae by a series of gene translocation and inversion events.

a Synteny relationships among five mitogenomes based on Mauve analysis. Rectangular blocks of the same color indicate collinear regions. b Mitogenome gene arrangements of five diatom species. Blocks with the same color represent the same type of genes
The arrangements of five mitogenomes are shown in Fig. 4b. Notably, all genes of O. regia were located on a single strand, which is different from the other four mitogenomes that encode genes on both strands. This gene arrangement in O. regia was similar to that in Trachydiscus minutus, Nannochloropsis oceanica, and Nannochloropsis oculata, in which all mitochondrial genes are located on single strands except the gene tatC (Wei et al. 2013; Starkenburg et al. 2014; Sevcikova et al. 2016).
Although gene arrangements varied, many conserved multi-gene blocks can be identified. For example, the arrangement of tatC and nad1 genes was shared by the mitogenomes of Bacillariophyceae, Eustigmatophyceae, Raphidophyceae, Phaeophyceae, and Chrysophyceae (Synura synuroidea) (Oudot-Le Secq et al. 2001; Goer and Olsen 2006; Oudot-Le Secq and Green 2011; Burger and Nedelcu 2012; Liu et al. 2014), and tatC-nad1 was shared by four mitogenomes except O. regia because of the translocation of nad1 gene in this study. The conserved back-to-back arrangement of trnH-rnl-rns-trnM was present at all five mitogenomes. Among diatom mitochondrial genomes, rps7 was missing as in Synedra acus and Melosira undulata (Guillory et al. 2018; Pogoda et al. 2019), and rpl5-rpl14-trnR-rps7-rps12 was shared by the four mitogenomes except O. regia because of the translocation trnR gene.
In P. tricornutum, the two genes nad9 and rps14 have been found to be fused by an in-frame insertion and are cotranscribed (Oudot-Le Secq and Green 2011). However, these two genes were not adjacent in O. regia. In the mitogenomes of L. undulatum, T. pseudonana, S. marinoi, and T. undulatum, these two genes were adjacent but were not fused together.
The gene blocks rps8-rpl6-rps2-rps4 and rpl2-rps19-rps3-rpl16 were conserved in previously reported algae including Bacillariophyceae but were interrupted in Eustigmatophyceae (Wei et al. 2013; Sevcikova et al. 2016). In this study, the conserved block rps8-rpl6-rps2-rpl4 was absent from O. regia and L. undulatum because of the missing rps2 gene, which may have been transferred to their nuclear genomes (Ravin et al. 2010). The gene block trnP-trnY-rps11-rpl2-rps19-rps3-rpl16-atp9 was shared by all 5 mitogenomes. Odontella regia possessed the least conserved gene arrangement, suggesting that the evolutionary rate of its mitogenome may be in divergence with other four mitogenomes in Mediophyceae.
The comparative analysis of L. undulatum and T. pseudonana mitochondrial genomes showed that there were eight conserved back-to-back arrangements of genes including trnH-rnl-rns-trnM, rps10-trnF-rps8-rpl6, rps4-trnN, trnL-1-trnL-2-rps12-rps7-rpl14-rpl5-trnG, tatC-nad1, tatA-trnW, trnP-trnY-rps11-rpl2-rps19-rps3-rpl16-atp9-trnK-nad4L-trnD-nad11, and nad7-nad9-rps14 (Fig. 5a).

a The comparative analysis of Lithodesmium undulatum and Thalassiosira pseudonana mitochondrial genomes. b The comparative analysis of Skeletonema marinoi and Thalassiosira pseudonana mitochondrial genomes. The assignment of genes into different functional groups is indicated by colors. The conserved back-to-back arrangement of genes is all shown with the same color
The highest synteny conservation was identified between the mitochondrial genomes of S. marinoi and T. pseudonana, which belonged to two different families Skeletonemataceae and Thalassiosiraceaea, respectively, with rearrangements involving only the five genes cox2, cox3, trnW-2, trnM-3, and trnV (Fig. 5b). This high synteny conservation suggested that the evolutionary distance between S. marinoi and T. pseudonana may be smaller than current positions.
Discussion
In this study we analyzed mitochondrial genome characteristics of the HAB species O. regia and L. undulatum which would provide useful information for understanding the phylogenetic relationships among mitogenomes of diatoms, especially the HAB species. Phylogenetic analysis based on the protein-coding genes of 35 stains diatoms improved our understanding of their complex genetic relationships. Previous studies suggested that increasing the number of taxonomic samples was a suitable way to improve the result of phylogenetic analyses (Pollock et al. 2002). Therefore, further studies to obtain more mitochondrial genomes of diatoms and especially HAB species would be beneficial.
Rearrangements of DNA include events such as inversion, translocation, deletion, and duplication (Pogoda et al. 2019). Synteny comparison of five mitogenomes in Mediophyceae suggested that the gene arrangements were not conserved among different classes. All genes of the O. regia mitochondrial genome were on the same strand, which may represent a more economic transcription mode, as suggested previously in Ulva pertusa (Liu et al. 2017). Obviously, further studies are needed to explore the role of this transcription mode in the formation of O. regia blooms. It was not clear whether the conserved gene blocks, which were shared by all five mitogenomes in species in Mediophyceae, perform by working together. Further studies are needed to help us understand the function of these blocks.
Mitochondrial genomes usually show fast evolutionary rates (Ward et al. 1981; Palmer et al. 2000; Chen et al. 2014), which makes them an appropriate platform for developing molecular markers with high resolution. The availability of the O. regia mitochondrial genome will enable development of molecular markers for tracking the genetic diversity of O. regia, as done recently (Song et al. 2020).
Conclusion
Odontella regia is the first species in the family Odontellaceae to have its complete mitogenome sequenced. Among all sequenced Bacillariophyta mitogenomes, the feature that all mitochondrial genes are located on the same strand has only been found in O. regia so far. The genetic relationship of the O. regia mitogenome is far from other the four mitogenomes in Mediophyceae; meanwhile, the genes of the O. regia mitogenome have experienced the most rearrangements. These results provide insight into the evolution of mitochondrial genomes in Bacillariophyta, especially class Mediophyceae, and help us improve our understanding of the HAB species O. regia. The O. regia mitochondrial genome can be used to develop high-resolution molecular markers for tracking O. regia genetic diversity and O. regia HAB development.
References
Adams K, Palme D (2003) Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol 29:380–395
An SM, Noh JH, Choi DH, Lee JH, Yang EC (2016) Repeat region absent in mitochondrial genome of tube-dwelling diatom Berkeleya fennica (Naviculales, Bacillariophyceae). Mitochondrial DNA A 27:2137–2138
An SM, Kim SY, Noh JH, Yang EC (2017) Complete mitochondrial genome of Skeletonema marinoi (Mediophyceae, Bacillariophyta), a clonal chain forming diatom in the west coast of Korea. Mitochondrial DNA A 28:19–20
Anisimova M, Gil M, Dufayard J-F, Dessimoz C, Gascuel O (2011) Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst Biol 60:685–699
Armbrust EV, Berges JA, Bowler C, Green BR, Martinez D, Putnam NH, Zhou SG, Allen AE, Apt KE, Bechner M, Brzezinski MA, Chaal BK, Chiovitti A, Davis AK, Demarest MS, Detter JC, Glavina T, Goodstein D, Hadi MZ, Hellsten U, Hildebrand M, Jenkins BD, Jurka J, Kapitonov VV, Kroger N, Lau WWY, Lane TW, Larimer FW, Lippmeier JC, Lucas S, Medina M, Montsant A, Obornik M, Parker MS, Palenik B, Pazour GJ, Richardson PM, Rynearson TA, Saito MA, Schwartz DC, Thamatrakoln K, Valentin K, Vardi A, Wilkerson FP, Rokhsar DS (2004) The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306:79–86
Ashworth MP, Nakov T, Theriot EC (2013) Revisiting Ross and Sims (1971): toward a molecular phylogeny of the Biddulphiaceae and Eupodiscaceae (Bacillariophyceae). J Phycol 49:1207–1222
Aunins AW, Hamilton D, King TL (2018) The complete mitochondrial genome of the stalk-forming diatom Didymosphenia geminata. Mitochondrial DNA B 3:676–677
Badylak S (2004) Spatial and temporal patterns of phytoplankton composition in subtropical coastal lagoon, the Indian River Lagoon, Florida, USA. J Plankton Res 26:1229–1247
Balmer-Hanchey EL, Jaykus L-A, Jaykus L-A, McClellan-Green P (2003) Marine biotoxins of algal origin and seafood safety. J Aquat Food Prod Technol 12:29–53
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Son P, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Burger G, Nedelcu AM (2012) Mitochondrial genomes of algae. In: Bock R, Knoop V (eds) Genomics of chloroplasts and mitochondria. Springer, London, pp 127–157
Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973
Carstensen J, Klais R, Cloern JE (2015) Phytoplankton blooms in estuarine and coastal waters: seasonal patterns and key species. Estuar Coast Shelf Sci 162:98–109
Chen S-C, Wei D-D, Shao R, Shi J-X, Dou W, Wang J-J (2014) Evolution of multipartite mitochondrial genomes in the booklice of the genus Liposcelis (Psocoptera). BMC Genomics 15:861
Chen B, Xie E, Gao Y, Ji W, Zhou Q (2015) Toxic effects of red tide caused by Karenia mikimotoi on marine organisms. J Fujian Fish 37:241–250 (In chinese, abstract in english)
Chen L, Huang JR, Dai J, Guo YF, Sun JT, Hong XY (2019) Intraspecific mitochondrial genome comparison identified CYTB as a high-resolution population marker in a new pest Athetis lepigone. Genomics 111:744–752
Crosman KM, Petrou EL, Rudd MB, Tillotson MD (2019) Clam hunger and the changing ocean: characterizing social and ecological risks to the Quinault razor clam fishery using participatory modeling. Ecol Soc 24:16
Crowell RM, Nienow JA, Cahoon AB (2019) The complete chloroplast and mitochondrial genomes of the diatom Nitzschia palea (Bacillariophyceae) demonstrate high sequence similarity to the endosymbiont organelles of the dinotom Durinskia baltica. J Phycol 55:352–364
Cunningham CW (1997) Can three incongruence tests predict when data should be combined? Mol Biol Evol 14:733–740
Darling AE, Mau B, Perna NT (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147
Dyson K, Huppert DD (2010) Regional economic impacts of razor clam beach closures due to harmful algal blooms (HABs) on the Pacific coast of Washington. Harmful Algae 9:264–271
Felsenstein J (1985) Confidence-limits on phylogenies - an approach using the bootstrap. Evolution 39:783–791
Gastineau R, Kim S-Y, Lemieux C, Turmel M, Witkowski A, Park J-G, Kim B-S, Mann DG, Theriot EC (2019) Complete mitochondrial genome of a rare diatom (Bacillariophyta) Proschkinia and its phylogenetic and taxonomic implications. Mitochondrial DNA B 4:25–26
Giordano M, Beardall J, Raven JA (2005) CO2 concentrating mechanisms in algae: mechanisms, environmental modulation, and evolution. Annu Rev Plant Biol 56:99–131
Goer M-PO-LSSL-d, Olsen WTSJL (2006) Complete mitochondrial genome of the three brown algae (Heterokonta, Phaeophyceae) Dictyota dichotoma, Fucus vesiculosus and Desmarestia viridis. Curr Genet 49:47–58
Guillard RRL, Hargreaves PE (1994) Stichochrysis immobilis is a diatom, not a chrysophyte. Phycologia 32:234–236
Guillory WX, Onyshchenko A, Ruck EC, Parks M, Nakov T, Wickett NJ, Alverson AJ (2018) Recurrent loss, horizontal transfer, and the obscure origins of mitochondrial introns in diatoms (Bacillariophyta). Genome Biol Evol 10:1504–1515
Guo H (2004) Illustrations of planktons responsible for the blooms in Chinese coastal waters. Ocean Press, Beijing (In chinese)
Haimeur A, Ulmann L, Mimouni V, Gueno F, Pineau-Vincent F, Meskini N, Tremblin G (2012) The role of Odontella aurita, a marine diatom rich in EPA, as a dietary supplement in dyslipidemia, platelet function and oxidative stress in high-fat fed rats. Lipids Health Dis 11:147
Hallegraeff GM (1993) A review of harmful algae blooms and their apparent global increase. Phycologia 32:79–99
Han X, Zou J, Zhang Y (2004) Harmful algae bloom species in Jiaozhou Bay and the features of distribution. Mar Sci 28:49–54
Hegde S, Narale DD, Anil AC (2011) Sexual reproduction in Odontella regia (Schultze) Simonsen 1974 (Bacillariophyta). Curr Sci 101:222–225
Hoagland P, Anderson DM, Kaoru Y, White AW (2002) The economic effects of harmful algal blooms in the United States: estimates, assessment issues, and information needs. Estuaries 25:819–837
Ibrahim, Imad A-S (2017) Study of phytoplankton blooms incident in Shatt Al-Arab river and marine coast Iraqi line. Mesop Environ J 3:18–25
Imanian B, Pombert J-F, Dorrell RG, Burki F, Keeling PJ (2012) Tertiary endosymbiosis in two dinotoms has generated little change in the mitochondrial genomes of their dinoflagellate hosts and diatom endosymbionts. PLoS One 7:e43763
Jin D, Thunberg E, Hoagland P (2008) Economic impact of the 2005 red tide event on commercial shellfish fisheries in New England. Ocean Coast Manag 51:420–429
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL (2008) NCBIBLAST: a better Web interface. Nucleic Acids Res 36:W5–W9
Kalyaanamoorthy S, Bui Quang M, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Kamikawa R, Azuma T, Ishii K, Matsuno Y, Miyashita H (2018) Diversity of organellar genomes in non-photosynthetic diatoms. Protist 169:351–361
Karp-Boss L, Gueta R, Rousso I (2014) Judging diatoms by their cover: variability in local elasticity of Lithodesmium undulatum undergoing cell division. PLoS One 9:e109089
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data P (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079
Liu F, Pang S, Li X, Li J (2014) Complete mitochondrial genome of the brown alga Sargassum horneri (Sargassaceae, Phaeophyceae): genome organization and phylogenetic analyses. J Appl Phycol 27:469–478
Liu F, Melton JT III, Bi Y (2017) Mitochondrial genomes of the green macroalga Ulva pertusa (Ulvophyceae, Chlorophyta): novel insights into the evolution of mitogenomes in the Ulvophyceae. J Phycol 53:1010–1019
Liu F, Liu S, Huang T, Chen N (2019) Construction and comparative analysis of mitochondrial genome in the brown tide forming alga Aureococcus anophagefferens (Pelagophyceae, Ochrophyta). J Appl Phycol 32:441–450
Mann DG (1999) The species concept in diatoms. Phycologia 38:437–495
Mann DG, Droop SJM (1996) Biodiversity, biogeography and conservation of diatoms. Hydrobiologia 336:19–32
McPartlin DA, Loftus JH, Crawley AS, Silke J, Murphy CS, O'Kennedy RJ (2017) Biosensors for the monitoring of harmful algal blooms. Curr Opin Biotechnol 45:164–169
Mimouni V, Ulmann L, Pasquet V, Mathieu M, Picot L, Bougaran G, Cadoret JP, Morant-Manceau A, Schoefs B (2012) The potential of microalgae for the production of bioactive molecules of pharmaceutical interest. Curr Pharm Biotechnol 13:2733–2750
Minh BQ, Nguyen MAT, von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30:1188–1195
Moore SK, Dreyer SJ, Ekstrom JA, Moore K, Norman K, Klinger T, Allison EH, Jardine SL (2020) Harmful algal blooms and coastal communities: socioeconomic impacts and actions taken to cope with the 2015 U.S. West Coast domoic acid event. Harmful Algae 96:101799
Oudot-Le Secq MP, Green BR (2011) Complex repeat structures and novel features in the mitochondrial genomes of the diatoms Phaeodactylum tricornutum and Thalassiosira pseudonana. Gene 476:20–26
Oudot-Le Secq MP, Fontaine JM, Rousvoal S, Kloareg B, Loiseaux-De Goer S (2001) The complete sequence of a brown algal mitochondrial genome, the ectocarpale Pylaiella littoralis (L.) Kjellm. J Mol Evol 53:80–88
Palmer JD, Adams KL, Cho YR, Parkinson CL, Qiu YL, Song KM (2000) Dynamic evolution of plant mitochondrial genomes: mobile genes and introns and highly variable mutation rates. Proc Natl Acad Sci U S A 97:6960–6966
Pogoda CS, Keepers KG, Hamsher SE, Stepanek JG, Kane NC, Kociolek JP (2019) Comparative analysis of the mitochondrial genomes of six newly sequenced diatoms reveals group II introns in the barcoding region of cox1. Mitochondrial DNA A 30:43–51
Pollock DD, Zwickl DJ, McGuire JA, Hillis DM (2002) Increased taxon sampling is advantageous for phylogenetic inference. Syst Biol 51:664–671
Prasetiya FS, Gastineau R, Poulin M, Lemieux C, Turmel M, Syakti AD, Hardivillier Y, Widowati I, Risjani Y, Iskandar I, Subroto T, Falaise C, Arsad S, Safitri I, Mouget J-L, Leignel V (2019) Haslea nusantara (Bacillariophyceae), a new blue diatom from the Java Sea, Indonesia: morphology, biometry and molecular characterization. Plant Ecol Evol 152:188–202
Qi Y, Zou J, Liang S (2004) Red tide along the coast of China. Science Press, Beijing (In chinese)
Ravin NV, Galachyants YP, Mardanov AV, Beletsky AV, Petrova DP, Sherbakova TA, Zakharova YR, Likhoshway YV, Skryabin KG, Grachev MA (2010) Complete sequence of the mitochondrial genome of a diatom alga Synedra acus and comparative analysis of diatom mitochondrial genomes. Curr Genet 56:215–223
Saitou N, Nei M (1987) The neighbor-joining method-a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sevcikova T, Klimes V, Zbrankova V, Strnad H, Hroudova M, Vlcek C, Elias M (2016) A comparative analysis of mitochondrial genomes in eustigmatophyte algae. Genome Biol Evol 8:705–722
Shibl AA, Isaac A, Ochsenkuhn MA, Cardenas A, Fei C, Behringer G, Arnoux M, Drou N, Santos MP, Gunsalus KC, Voolstra CR, Amin SA (2020) Diatom modulation of select bacteria through use of two unique secondary metabolites. Proc Natl Acad Sci U S A 117:27445–27455
Smith DR (2016) The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief Funct Genomics 15:47–54
Smith SA, Dunn CW (2008) Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24:715–716
Song H, Liu F, Li Z, Xu Q, Chen Y, Yu Z, Chen N (2020) Development of a high-resolution molecular marker for tracking Phaeocystis globosa genetic diversity through comparative analysis of chloroplast genomes. Harmful Algae 99:101911
Starkenburg SR, Kwon KJ, Jha RK, McKay C, Jacobs M, Chertkov O, Twary S, Rocap G, Cattolico RA (2014) A pangenomic analysis of the Nannochloropsis organellar genomes reveals novel genetic variations in key metabolic genes. BMC Genomics 15:212
Swofford DL (2002) PAUP*: Phylogenetic Analysis Using Parsimony. Version 40b10 Sinauer Associates, Sunderland
Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A 101:11030–11035
Tang X, Bi G (2016) Complete mitochondrial genome of Fistulifera solaris (Bacillariophycidae). Mitochondrial DNA A 27:4405–4406
Theriot EC, Ashworth M, Ruck E, Nakov T, Jansen RK (2010) A preliminary multigene phylogeny of the diatoms (Bacillariophyta): challenges for future research. Plant Ecol Evol 143:278–296
Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192
Trifinopoulos J, Lam-Tung N, von Haeseler A, Minh BQ (2016) W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res 44:W232–W235
Villain A, Kojadinovic M, Puppo C, Prioretti L, Hubert P, Zhang Y, Gregori G, Roulet A, Roques C, Claverie J-M, Gontero B, Blanc G (2017) Complete mitochondrial genome sequence of the freshwater diatom Asterionella formosa. Mitochondrial DNA B 2:97–98
Ward BL, Anderson RS, Bendich AJ (1981) The mitochondrial genome is large and variable in a family of plants (Cucurbitaceae). Cell 25:793–803
Wei L, Xin Y, Wang D, Jing X, Zhou Q, Su X, Jia J, Ning K, Chen F, Hu Q, Xu J (2013) Nannochloropsis plastid and mitochondrial phylogenomes reveal organelle diversification mechanism and intragenus phylotyping strategy in microalgae. BMC Genomics 14:534
Williams DM (2007) Diatom phylogeny: fossils, molecules and the extinction of evidence. C R Palevol 6:505–514
Wu Y, Sun S, Zhang Y (2005) Long-term change of environment and it’s influence on phytoplankton community structure in Jiaozhou Bay. Oceanol Limnol Sin 36:487–498 (In chinese, abstract in english)
Xia S, Wang K, Wan LL, Li AF, Hu Q, Zhang CW (2013) Production, characterization, and antioxidant activity of fucoxanthin from the marine diatom Odontella aurita. Mar Drugs 11:2667–2681
Yang S, Dong S (2006) A map of common planktonic diatoms in the waters of China. Ocean University of China Press, Qingdao (In chinese)
Yu Z, Chen N (2019) Emerging trends in red tide and major research progresses. Oceanol Limnol Sin 50:474–486 (In chinese, abstract in english)
Yuan X-L, Cao M, Bi G-Q (2016) The complete mitochondrial genome of Pseudo-nitzschia multiseries (Bacillariophyta). Mitochondrial DNA A 27:2777–2778
Acknowledgments
We are thankful to all staffs of the marine ecological environment genomics research group in the Institute of Oceanology, Chinese Academy of Sciences. We also want to express our gratitude to the two anonymous reviewers for their critical comments and suggestions.
Funding
This research was supported by the Marine S & T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (No. 2018SDKJ0504); the Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB42000000) (to Nansheng Chen); the Chinese Academy of Sciences Pioneer Hundred Talents Program (to Nansheng Chen); the Taishan Scholar Project Special Fund (to Nansheng Chen); the Qingdao Innovation and Creation Plan (Talent Development Program-5th Annual Pioneer and Innovator Leadership Award to Nansheng Chen, 19-3-2-16-zhc); the Key Research Program of Frontier Sciences, Chinese Academy of Sciences (No. QYZDB-SSW-DQC023) (to Feng Liu); and the Major Scientific and Technological Innovation Project of Shandong Province (No. 2019JZZY020706) (to Feng Liu).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, Y., Chen, Y., Wang, J. et al. Mitochondrial genome of the harmful algal bloom species Odontella regia (Mediophyceae, Bacillariophyta). J Appl Phycol 33, 855–868 (2021). https://doi.org/10.1007/s10811-020-02364-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-020-02364-1