
Abstract
We report the complete mitochondrial sequences of three brown algae (Dictyota dichotoma, Fucus vesiculosus and Desmarestia viridis) belonging to three phaeophycean lineages. They have circular mapping organization and contain almost the same set of mitochondrial genes, despite their size differences (31,617, 36,392 and 39,049 bp, respectively). These include the genes for three rRNAs (23S, 16S and 5S), 25–26 tRNAs, 35 known mitochondrial proteins and 3–4 ORFs. This gene set complements two previously studied brown algal mtDNAs, Pylaiella littoralis and Laminaria digitata. Exceptions to the very similar overall organization include the displacement of orfs, tRNA genes and four protein-coding genes found at different locations in the D. dichotoma mitochondrial genome. We present a phylogenetic analysis based on ten concatenated genes (7,479 nucleotides) and 29 taxa. Stramenopiles were always monophyletic with heterotrophic species at the base. Results support both multiple primary and multiple secondary acquisitions of plastids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The stramenopiles (section Heterokonta) encompass both unicellular, e.g., the Bacillariophyceae (diatoms), and multicellular lineages, e.g., the Phaeophyceae (brown algae). They also comprise both heterotrophic species such as Bicosoecida and Oomycetes and autotrophic species including the Bacillariophyceae, Chrysophyceae and Phaeophyceae (Leipe et al. 1994). The plastids of the photosynthetic species arose from a secondary endosymbiosis event involving algae related to extant red algae (Delwiche and Palmer 1997; Medlin et al. 1997). The origin of the host cell that originally acquired the plastid remains unclear. Two main hypotheses have been proposed: first that stramenopiles acquired their photosynthetic lineage late (Saunders et al. 1995; Leipe et al. 1996; Blackwell and Powell 2000); and second that chloroplast acquisition occurred earlier, in a common ancestor of alveolates and stramenopiles, and prior to the stramenopile divergence (Cavalier-Smith 1998; Fast and Keeling 2001; Yoon et al. 2004). This hypothesis implies subsequent multiple losses of chloroplasts within the heterotrophic lineage.
Phylogenetic relationships within the stramenopiles and/or within heterokont algae have been studied using different molecular markers and methods, such as the nuclear small subunit rDNA (e.g., Leipe et al. 1996; Van de Peer et al. 1996; Potter et al.1997; Guillou et al. 1999; Van de Peer et al. 2000), the chloroplast gene rbcL (Daugbjerg and Andersen 1997; Daugbjerg and Guillou 2001), or a combination of both (Sorhannus 2001; Goertzen and Theriot 2003). In these analyses, the nuclear data support the heterokont algae as monophyletic with generally the Bacillariophyceae (including Bolidophyceae) as the most basal group. Exceptionally Oomycetes have been included within the heterokont algae (Van de Peer et al. 2000). Based on further analyses using chloroplast genes, the monophyletic origin of heterokont algal plastids was confirmed with the Bacillariophyceae occupying an intermediate position, associated with the Chrysophyceae.
As the relationship between the photosynthetic and the heterotrophic stramenopiles is still unclear, we decided to reevaluate their phylogeny using a molecular marker common to all of them, e.g., a suite of ten genes from the mitochondrial (not plastidial) genome. Mitochondrial genes provide a valuable alternative to nuclear genes, since the origin of mitochondria is probably concomitant with that of the eukaryotic cell (Martin and Müller 1998; Vellai et al. 1998) and thus are more likely to reflect the evolutionary history of the original host cell. Two recent studies also utilized mitochondrial genes (four genes, 3,708 nucleotides: Sanchez Puerta et al. 2004; five proteins sequences, 1,791 amino acids: Armbrust et al. 2004) and were able to confirm monophyly of the stramenopiles. However, the focus of these studies was the phylogenetic placement of Emiliania huxleyi and Thalassiosira pseudonana, respectively. Neither of these studies addressed the late–early hypotheses of secondary chloroplast acquisition.
In addition to refining the stability of the broader phylogeny, we were also interested in the evolution of the brown algae and their mitochondrial genomes. The brown algae are characterized by diverse morphologies, ranging from microscopical filaments to huge kelps with complex fronds, tens of meters in length (Patterson 1999). In an earlier study, two brown algal mitochondrial genomes were sequenced, Pylaiella littoralis [Ectocarpales] (Oudot-Le Secq et al. 2001), which had been viewed as a representative of the brown algal ancestral condition, and Laminaria digitata [Laminariales] (Oudot-Le Secq et al. 2002), which was hypothesized to have evolved more recently. Both were found to be very similar in gene content as well as in gene arrangement, but were quite different with respect to the presence (in P. littoralis only) of group II introns and a large insertion of DNA of an unknown origin. In the present study, we sequence and analyze the complete mitochondrial sequences of three additional brown algae Dictyota dichotoma [Dictyotales], viewed now as the most ancestral brown algal lineage (Draisma et al. 2001; Rousseau et al. 2001), Fucus vesiculosus [Fucales] and Desmarestia viridis [Desmarestiales].
We characterize these three new phaeophycean mtDNAs in detail and reassess phylogenetic relationships within the stramenopiles.
Material and methods
Algal sources
Dictyota dichotoma (Hudson) J.V. Lamouroux was cultivated from an isolated individual collected in 1996 at the Pointe de Mousterlin, Brittany, France, by H. Pakker. Fucus vesiculosus Linnaeus was collected on 22 October 2001 in front of the Marine Biological Station, in Roscoff, Brittany, France, by M.-P. Oudot-Le Secq. Desmarestia viridis (O.F. Müller) J.V. Lamouroux was collected in 1978 in Helgoland by K. Lüning. Frozen tissue from the original algal culture was used.
DNA preparation and PCR procedures
Algal tissue was ground in liquid nitrogen and total DNA extracted in a buffer containing 100 mM Tris–HCl pH 8.0, 1.4 M NaCl, 20 mM EDTA, 0.1% (w/v) PVPP, 0.2% (v/v) β-mercaptoethanol and 2% (w/v) CTAB. After two chloroform-isoamyl alcohol (24:1, v/v) extractions, the aqueous DNA solution was purified with the Sephaglas BandPrep Kit (Pharmacia), following the manufacturer’s instructions. These total DNAs were used as a template to amplify the mitochondrial DNAs. Primer design was based initially on Pylaiella littoralis and Laminaria digitata mitochondrial sequences and on multiple alignments of mitochondrial and bacterial sequences of homologous genes; and secondarily on the specific algal sequence under study. PCR experiments were performed either in a Cetus DNA thermo cycler (Perkin Elmer) or in a Mastercycler gradient cycler (Eppendorf) with Ready-to-Go beads (Taq, Amersham Pharmacia Biotech), 2 mM of each primer and 0.5–2 μL of total DNA (1–20 ng). The reaction profile was as follows: initial denaturation at 95°C for 1–3 min; ten cycles of denaturation at 95°C for 30 s, annealing at 46–62°C (depending on primer sequences) for 1 min, and elongation step at 72°C for 20 s–3 min (depending on the expected size of the amplified fragment); followed by 20–30 cycles of denaturation at 95°C for 30 s, annealing step at 46–62°C for 30 s, and elongation at 72°C for 20 sec–3 min; and a final elongation at 72°C for 10 min. When the expected size of the amplified fragment was above 4 kb the Expand Long Template PCR System (Roche) was used, according to the manufacturer protocols. PCR products were loaded on 0.8–1.5% agarose gels; fragments were cut from the gel. The DNA was extracted from gel slices and was purified using the Wizard® PCR Preps DNA Purification System (Promega) following the manufacturer’s directions.
DNA sequencing
Direct sequencing of PCR products was performed using the PCR primers and additional internal primers with the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems). Sequencing reactions were run on an ABI 377 automated sequencer (PE Applied Biosystems). Sequences, read on both strands, were assembled using the BioEdit Sequence Alignment Editor version 5.0.9 (Hall 1999).
Sequences and alignments
The complete sequences of the three mitochondrial genomes are available from GenBank under the following accession number: F. vesiculosus, AY494079; D. dichotoma, AY500368; and D. viridis AY500367. The three new mitochondrial sequences were manually aligned with those of P. littoralis and L. digitata using BioEdit Sequence Alignment Editor versions 5.0.9 and 6.0.7 (Hall 1999). The nucleotide sequences of the ten mitochondrial genes (atp6, atp9, cob, cox1–3, nad1, nad3–4 and nad4L) from 22 selected organisms and the bacterial counterparts of these genes from two α-proteobacteria (Table 1) were aligned with those of the five brown algal mitochondrial sequences. Regions too ambiguous to align were excluded. The dataset is 7,479 nucleotide positions long with atp6 (543), atp9 (213), cob (1,083), cox1 (1,437), cox2 (726), cox3 (780), nad1 (906), nad3 (339), nad4 (1,200) and nad4L (252).
Phylogenetic analysis
Bayesian maximum likelihood analysis was performed using MrBayes version 3 (Ronquist and Huelsenbeck 2003). Models of DNA evolution were determined with Modeltest 3.06 (Posada and Crandall 1998) and PAUP (Swofford 2003), based on Hierarchical Likelihood Ratio Tests, run on each partition (e.g., individual gene alignment). Table 2 summarizes the models chosen. The following settings were used: nst (number of substitution types) =6 (models TVM and GTR), with gamma-distributed rates across sites and proportion of invariable sites when required. We allowed the set of parameters to be different for each partition. Introduced gaps were treated as missing data in subsequent analyses. The different parameters from the phylogenetic models were estimated during the phylogenetic analysis. Four chains were run; trees were sampled every 100 of the 2,000,000 generations and the 1,000 first trees were discarded as burn-in.
Results and discussion
Characterization of mt genomes in Dictyota dichotoma, Fucus vesiculosus and Desmarestia viridis, with references to those of Pylaiella littoralis and Laminaria digitata
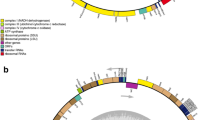
The D. dichotoma, F. vesiculosus and D. viridis mtDNAs have a circular mapping organization. The three mtDNA maps are depicted in Fig. 1. Their sizes are: 31,617 bp (D. dichotoma), 36,392 bp (F. vesiculosus) and 39,049 bp (D. viridis). Different characteristics of the genomes, such as their A+T content for different categories of sequences and figures about spacer sequences and overlaps are summarized in Table 3. All these values are in the same range as those of P. littoralis and L. digitata (Oudot-Le Secq et al. 2001, 2002).

Physical map and gene organization of the Dictyota dichotoma, Fucus vesiculosus and Desmarestia viridis mtDNAs (a color version is provided as supplementary data). Genes and orfs are depicted as blocks, with gene abbreviations listed in Table 4. The gene blocks shown outside and inside are transcribed clockwise and counter-clockwise, respectively. Transfer RNA genes (trn) are indicated as white boxes surrounded by black lines, their names are indicated by the amino acid (one-letter code) they specify (see Fig. 3). Thin black bars between gene blocks indicate gene overlaps. The three inner rings show the restriction fragments generated by BamH1, EcoR1 and Sal1, respectively. On the black circle, between genes and restriction maps, the scale size is shown (in kilobases)
Two of the overlapping regions are exactly conserved among the three mtDNAs described here, as well as in P. littoralis and L. digitata; these are found between the genes encoding the ribosomal proteins rps8, rpl6 and rps2. Both involve the start codon ATG and a stop codon (TGA or TAA); the rps8–rpl6 overlap is 4 bp long, ATGA, and the rpl6–rps2 overlap is only 1 bp long, A (in the context: taAtg).
More than 60 putative coding regions were identified in each of the three mtDNAs (Table 4), basically the same as in P. littoralis and L. digitata mtDNAs. These are the genes for three rRNAs (23S, 16S and 5S), for 25 tRNAs in the case of D. dichotoma or 26 for F. vesiculosus and D. viridis, for 35 proteins identified by sequence homology, as well as for one common ORF (also encoded in P. littoralis and L. digitata mtDNAs). Fucus vesiculosus and D. viridis share another related ORF, despite its size difference (379 and 622 amino acids, respectively). Finally, D. dichotoma and D. viridis each encode one more unique unknown orf, orf54 and orf211, respectively. None of the three new mtDNAs contain introns, nor any insertion with a phage-like RNA polymerase gene as has been described in the P. littoralis genome (Costa et al. 1997; Rousvoal et al. 1998; Oudot-Le Secq et al. 2001). At present P. littoralis is the only known brown alga that shows these unusual features in its mitochondrial genome.
The gene order is very well-conserved between the three new brown algae mtDNAs and those of P. littoralis and L. digitata, although exceptions exist (Fig. 2). These are mainly due to orfs not present in all the mtDNAs (see gray triangles in Fig. 2), to tRNA genes (see dashed lines) and to a few genes coding for proteins (atp8, atp9, rpl31 and rps10) in the D. dichotoma mtDNA compared to the other mtDNAs (see black lines).

Gene order comparison between the mtDNAs of Dictyota dichotoma (Dd), Fucus vesiculosus (Fv), Desmarestia viridis (Dv), Laminaria digitata (Ld) and Pylaiella littoralis (Pl) mtDNAs (colored version in supplementary material). On these linear representations of mtDNA gene contents, only the gene order is taken into account; spaces between two genes are there only to allow horizontal alignment and to help visual comparison. Genes on a same horizontal line are homologous. White blocks depict ribosomal protein encoding genes; light gray blocks (black writing) depict genes coding for proteins involved in respiratory chains; dark gray blocks (white writing) depict other protein encoding genes and orfs; black blocks depict RNA genes (tRNA and rRNA genes). Arrows indicate transcription direction. Gray triangles highlight presence/absence of a gene between two mtDNAs. Thick black lines highlight the different locations of protein encoding genes between D. dichotoma and other mtDNAs. Dashed black lines highlight trn different locations. The two asterisks in P. littoralis mtDNA indicate that although tRNA Leu and orf40 are not coded anymore, the corresponding regions still share high sequence homology with the L. digitata sequence
In order to calculate pairwise identity scores, a nucleotide alignment of the five complete mitochondrial genomes was restricted to the genes, conserved orfs and corresponding spacers, found at the same location in all the brown algal mtDNAs (i.e., those that are found at the same horizontal level in the five maps, Fig. 2). The resulting alignment contains 34,501 positions. Four out of the five mtDNAs share from 69 to 75% identity (Table 5), whereas the D. dichotoma sequence is only 57–58% identical to the others. When the cox2 insertion region, absent in D. dichotoma (see below), is removed from the alignment it reaches 64% identity with the others (not shown).
Genetic code
The universal genetic code seems to be used in all of these mtDNAs, based on protein sequence alignments. RNA editing does not appear to be necessary.
A gene, rps14 in D. dichotoma and a putative orf, orf379 in F. vesiculosus, use GTG as initiation codon, and two other orfs, orf211 in D. viridis and orf37 in D. dichotoma use TTG. GTG and TTG are commonly used as alternative start codons in the mitochondrial genetic code of different animal groups (e.g., invertebrates, molds, protozoans and coelenterates), in the plant plastidial code and in the bacterial code (Elzanowski and Ostell 2000). All the codons are used to encode proteins (Supplementary material). Those ending in A or T out-numbered the synonymous codons ending in G or C, as expected for A+T rich genomes. All of the three stop codons are used (see Table 5), with a marked preference for TAA of 64% (D. dichotoma) and 83% (F. vesiculosus and D. viridis).
rRNA genes
The 5S rRNA gene is encoded in the three new mtDNAs. These three genes share with the L. digitata 5S gene, a putative insertion between stems V and I that may form a hairpin that is absent in P. littoralis (a representation of the potential folding of the five rrn5 is provided as Supplementary Material). Stems I are shorter and weaker than usual, as was the case for the P. littoralis and L. digitata genes. The rns genes are well conserved and their potential folding is in general accordance with the eubacterial model (Wuyts et al. 2002). The same can be said about the rnl genes although some of their variable areas differ from one species to the other.
trn genes
The D. dichotoma, F. vesiculosus and D. viridis mtDNAs share 24 potential trn genes with L. digitata mtDNA (Fig. 3) that can fold, following the standard cloverleaf secondary structure (Sprinzl et al. 1998) and fit the L-shape, except in the tRNAArg [ucu] from D. dichotoma, where the position 48 is not a pyrimidine but an adenine (Zagryadskaya, Kotlova and Steinberg 2004). The tRNALeu [caa] gene cannot be depicted in the P. littoralis mtDNA (see the star in Fig. 2), though the region still shares high sequence homology with the trn sequence, it lacks the main conserved features (Oudot-Le Secq et al. 2002). Another region, common to the five mtDNAs, but labeled differentially in the different algal mtDNAs (Figs. 2 and 4a), “K?” in D. dichotoma, D. viridis and L. digitata, “X?” in F. vesiculosus and L. digitata, or “F?” in P. littoralis, can fold according to the cloverleaf model. As shown in Fig. 4a, this region has a high similarity level with the trn Glu sequence, but some of the so-called invariant positions do not follow the consensus (gray shading in Fig. 4a). In addition, the anticodon positions are not the same in all the five mtDNAs. They are TTT for a trn Lys in D. dichotoma and D. viridis. The anticodon in F. vesiculosus mtDNA is TTA, which should “decode” the stop codon TAA (the most frequently used stop codon in the genome). It is even more complex in the P. littoralis and L. digitata cases; in the first, the anticodon loop is one nucleotide longer than usual, leading to “AAAA”, the trn Phe (F), while one nucleotide is missing in the second, leading to two possibilities for the anticodon position: TTT (trn Lys) as in D. dichotoma and D. viridis or TTA as in F. vesiculosus. The P. littoralis and L. digitata sequences also have an insertion of 16 and 15 nucleotides, respectively, in the D loop (see Fig. 4). Finally, since the five mtDNAs encode well-conserved, canonical genes for both tRNA Lys and Phe, this noncanonical gene should not be necessary to encode a tRNA. On the other hand, the fact that the cloverleaf folding is conserved, when the other tRNA’s mandatory positions are not, may suggest that this pseudogene has now gained a new function. It could serve as punctuation marks, for the process of a large mRNA encompassing the surrounding genes. It is known that trns play such a role in fungi mt genome (Burger et al. 1985) and animal mt genomes (Clayton 1984). The case of a pseudogene that could have gained such a function has been described recently in the mt genome of a fish (Mabuchi et al. 2004). Furthermore, a long mRNA encompassing at least cox2, nad4 and nad5 genes has been evidenced by a northern experiment (M.-P. Oudot-Le Secq and Loiseaux-de Goër, unpublished data) in P. littoralis.

Nucleotide sequences of the 24 common tRNA genes found in the mitochondrial genomes of D. dictyota (Dd), D. viridis (Dv) and F. vesiculosus (Fv). Anticodons are shown in bold. Black shading highlights identities between the three sequences for each trn. The upper line (Consensus) shows the invariant and semi-invariant nucleotides found in tRNA genes, gray shading indicates positions that do not fit the consensus

Nucleotide sequences of two putative tRNA genes. a Comparison between the trn Glu (E) sequences of the five brown algal mtDNAs with a putative tRNA gene, found within the five genomes at a same location. b Sequence of an additional trn Tyr (Y) found in D. viridis and F. vesiculosus mtDNAs
Fucus vesiculosus and D. viridis mtDNA share one additional putative tRNA gene (Fig. 4b), that of tRNATyr [aua]. Both sequences display deviations from the consensus (gray shading). Another tRNATyr gene, but with [gua] as anticodon, is shared by the five mtDNAs. We do not know if these noncanonical trns are transcribed and used or if they are remains or intermediates of the evolutionary process. In the first case, the sequences seem to be related to those of trn Glu and could have gained a new function as a processing signal, but in the latter (trn Tyr), there is no such obvious origin (not shown). Since all the codons are necessary to code the protein genes, the trn set encoded in the brown algal mtDNAs is not sufficient to decode them all; the lacking tRNAs have to be imported from the cytosol to mitochondria, as it is the case in many organisms (Schneider and Maréchal-Drouard 2000).
Protein-encoding genes and orfs
The identified mitochondrial protein-encoding gene set is the same in the five mtDNAs studied so far (Table 4). These are: genes for the three first subunits of the cytochrome oxidase (cox1–3); cob, which encodes apocytochrome B; ten genes encoding ten subunits of the NADH dehydrogenase nad1–7, 4L, 9 and a short nad11 gene; three genes encoding three subunits of the ATPase, atp6, 8 and 9; and seventeen ribosomal protein-coding genes. The cox2 gene contains a large in-frame insertion of 2,994 bp in D. viridis, comparable in size and related to those found in the cox2 genes of P. littoralis (2,973 bp) and L. digitata (2,979 nucleotides). The insert also exists in the F. vesiculosus cox2 gene, with a slightly smaller size (2,283 bp) but is absent in the D. dichotoma gene. The insert is also absent in Sphacellaria (unpublished data). These insertions show a conserved leucine-zipper-like region and may prove useful as phylogenetic markers within brown algae. Pearson et al. (2001) sequenced a 648 bp long fragment identical to part of the F. vesiculosus cox2-insertion, starting 61 bp after the beginning of the insertion (only the first 3 bp are not the same) and finishing just upstream from the leucine-zipper-like region. This fragment was found among the clones obtained by the screening of a cDNA library generated by suppression subtractive hybridization for F. vesiculosus undergoing mild desiccation stress. Northern analysis did not show a clear over-expression after desiccation (Pearson et al. 2001). This suggests a possible excision of the insertion in the cox2 mRNA, but would be in contradiction with the results obtained previously (Oudot-Le Secq et al. 2001). An AT-rich sequence downstream of the cloned fragment on the F. vesiculosus genome, i.e., AAAAAAAATTAGAAAAA could explain the presence of this clone after selection of full-length poly-A+ mRNAs.
The gene encoding a protein transporter component of the secY-independent pathway, tatC (previously called ymf16), is present, always in the reverse transcriptional orientation, in the five brown algal mtDNAs. These genomes all encode one related orf, orf111 (D. dichotoma), orf131 (F. vesiculosus), orf143 (D. viridis), orf129 (L. digitata) and orf127 (P. littoralis), which seems specific to brown algal mtDNAs. No similarity with other orfs was found in other algae or other organisms that have been found up till now. Between the trn Trp and trn Ile, another small orf is present in four out of the five genomes. These orf43 (F. vesiculosus), orf39 (D. viridis) and orf40 (L. digitata), even if small, are very well conserved, while orf37 (D. dichotoma) is different. No similarity with other orfs has been found by Blast. In P. littoralis three longer orfs precede a region that could be a pseudogene of this ORF. F. vesiculosus and D. viridis share another common orf with L. digitata, despite size differences (379, 622 and 384 amino acids encoded, respectively). Finally, D. dichotoma and D. viridis each encode one more unique unknown orf, orf54 and orf211, respectively.
Repeats
Despite the great similarity between the brown algal mtDNAs, they do not share any common inverted repeats. The longest repeats found in each mtDNA are mutually different, both in size and location (not shown). Two of these harbor a different stem–loop of 23 nucleotides, one between trn Trp and trn Ile (in orf37) in D. dichotoma, with a eight nucleotide long loop, and the other between the trn Glu and nad9 genes in the case of D. viridis, with a six nucleotide long loop. At the same location, L. digitata also has an imperfect inverted repeat (one bulge) of 15 nucleotides. But the largest inverted repeat is found at the end of the D. dichotoma rps7 gene. This gene, which is about 100 nucleotide longer that its counterparts, harbors a 30 nucleotide long stem-loop, covering the end of the gene in such way that the stop codon is located in the middle of the second repeat.
Phylogenetic relationships of stramenopiles based on mt genes
Phylogenetic reconstructions were carried out using a concatenation of ten mitochondrial protein-encoding genes (atp6, atp9, cob, cox1–3, nad1, nad3–4 and nad4L) from a set of 29 species (Table 1). The 12 stramenopiles comprise: four heterotrophs (one Bicosoecida, one Labyrinthulida and two Oomycetes), two Chrysophyceae, the Bacillariophyceae Thalassiosira pseudonana (Armbrust et al. 2004) and the five Phaeophyceae, D. dichotoma, F. vesiculosus, D. viridis, L. digitata and P. littoralis. These represent all of the complete stramenopile mtDNAs currently available. Other representatives from eukaryotic lineages included: the Haptophyceae Emiliania huxleyi (Sanchez Puerta et al. 2004), the Cryptophyceae Rhodomonas salina, three Rhodophyceae, two Chlorophyta, two Streptophyta, one Jakobidae, one Malawimonanididae, the Choanoflagellida Monosiga brevicollis (Burger et al. 2003), two fungi and the amoeboid protozoon Acanthamoeba castellanii (Burger et al. 1995); finally, two α-proteobacteria were used as outgroup.
The overall topology of the tree (Fig. 5) is very well supported by posterior probabilities of 1.00 for a majority of nodes. The stramenopiles form a monophyletic group in which the photosynthetic species cluster together. The Chrysophyceae emerge first, followed by T. pseudonana and the Phaeophyceae (posterior probability=0.92). The relationships between phaeophycean species are in accordance with previous studies based on nuclear rDNA data (Rousseau and de Reviers 1999; Rousseau et al. 2001) and on nuclear and plastidial combined data (Draisma et al. 2001).

Phylogenetic tree constructed from mtDNA nucleotide sequences of ten genes (atp6, atp9, cob, cox1-3, nad1, nad3-4, nad4L, 7,479 positions) from 29 species, using Bayesian phylogenetic inference. Posterior probabilities are indicated at the nodes. The dataset was partitioned into genes, and each partition analyzed with the appropriate DNA evolutionary model, as determined by Modeltest (Table 2). The light gray area highlights the stramenopiles and the dark gray area the heterokont algae. Single and double underlinings highlight organisms with primary and secondary plastids, respectively
Within the heterotrophic stramenopile species, Cafeteria roenbergensis (Bicosoecida) is the most basal followed by Thraustochytrium aureum (Labyrinthulida) and Oomycetes (posterior probability = 0.96) as a sister group of the algal species.
A complex sister lineage to that of the heterokonts comprises the “green” lineage (Viridiplantae) plus R. americana and M. jakobiformis on one hand, and the red algae plus the cryptophyte Rhodomonas salina on the other. The haptophyte Emiliania huxleyi diverges before these, with a posterior probability of 0.96, separated from the well-conserved fungi-choanoflagellate clade and A. castellanii (Acanthamoebidae). In this analysis, the association of R. americana and M. jakobiformis with the green lineage separated from the parallel association between the cryptophyte and the red lineage is an argument in favor of two primary endosymbiotic events at the origin of primary chloroplasts, since Viridiplantae and Rhodophyceae do not form a monophyletic group. The presence of this clade (Viridiplantae, R. Americana–M. jakobiformis, Rhodophyceae, Cryptophyceae) between stramenopiles and haptophytes does not support the single acquisition of secondary plastids by a common ancestor of these lineages. The monophyly of the heterokont algae also suggests a secondary endosymbiotic event, which would have occurred in a heterotrophic stramenopile.
Until additional mitochondrial genomic data become available for more stramenopiles—such as representatives from the labyrinthulid/thraustochytrid group, the opalinids or the free living Developayella elegans, which are supposed to be close to the Oomycetes (Massana et al. 2002), for the heterotrophic heterokonts, and the Synurophyceae, Eustigmatophyceae, Xanthophyceae, Raphidophyceae and Pelagophyceae, for the algae—this conclusion remains provisional.
Conclusion
Although brown algae display great morphological and physiological differences (e.g., diversity of life cycles), the brown algal mitochondrial genomes studied so far are similar and alignable. The main common features of P. littoralis and L. digitata mtDNAs are shared by the other brown algal mtDNAs. These are the presence of rare mitochondrial encoded genes, such as rrn5, the presence of many ribosomal protein subunit encoded genes, and the unusually short nad11 gene, where only the first third, corresponding to the well-known Fe–S binding domain of the protein is encoded. A large in-frame insertion is found in the cox2 gene, although of variable size, in all but the D. dichotoma mtDNA. It will be interesting to investigate more species and determine if this insertion was acquired after the D. dichotoma divergence or earlier and lost secondarily.
Phylogenetic relationships based on a set of ten mitochondrial genes support previous analyses based on single and double-gene studies (Rousseau and de Reviers 1999; Draisma et al. 2001) that utilized nuclear and chloroplast loci. This result is also corroborated by Fig. 2, showing the rearrangements necessary to switch from one genome organization to the others and indicating that gene order may be a phylogenetically informative character (Boore and Brown 1998; Rokas and Holland 2000; Sankoff et al. 2000). Indeed the phylogenetic order of modifications implied by Fig. 2 is the same as that given by the trees, i.e., D. dichotoma, F. vesiculosus, D. viridis and L. digitata–P. littoralis.
As genome sequencing becomes faster and cheaper, the switch to longer alignments as well as the utilization of gene order and indels should provide greater resolution in deeper branches. Finally, the finding that phaeophycean mitogenomes are remarkably similar provides encouragement for the development of useful genetic markers for species and population-level studies.
Electronic supplementary material
An electronic appendix to this article provides color versions of Figs. 1 and 2, codon usage of mitochondrial encoded protein genes and the potential folding of 5S rRNAs, on the Current Genetics’ web site.
Abbreviations
- Mt:
-
Mitochondrial
References
Armbrust EV, Berges JA, Bowler C, Green BR, Martinez D, PutnamNH, Zhou, Allen AE, Apt KE, Bechner M, Brzezinski MA, Chaal BK, Chiovitti A, Davis AK, Demarest MS, Detter JC, Glavina T, Goodstein D, Hadi MZ, U. Hellsten M. Hildebrand, Jenkins BD, Jurka J, Kapitonov VV, Kroger N, Lau WW, Lane TW, Larimer FW, Lippmeier JC, Lucas S, Medina M, Montsant A, Obornik M, Parker MS, Palenik B, Pazour GJ, Richardson PM, Rynearson TA, Saito MA, Schwartz DC, Thamatrakoln K, Valentin K, Vardi A, Wilkerson FP, Rokhsar DS (2004) The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306:79–86
Blackwell WH, Powell MJ (2000) A review of group filiation of Stramenopiles, additional approaches to the question. Evol Theor 12:49–88
Boore JL, Brown WM (1998) Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev 8:668–674
Burger G, Citterich MH, Nelson MA, Werner S, Macino G (1985) RNA processing in Neurospora crassa mitochondria: transfer RNAs punctuate a large precursor transcript. EMBO J 4:197–204
Burger G, Forget L, Zhu Y, Gray MW, Lang BF (2003) Unique mitochondrial genome architecture in unicellular relatives of animals. Proc Natl Acad Sci USA 100:892–897
Burger G, Plante I, Lonergan KM, Gray MW (1995) The mitochondrial DNA of the amoeboid protozoon, Acanthamoeba castellanii: complete sequence, gene content and genome organization. J Mol Biol 245:522–537
Cavalier-Smith T (1998) A revised six-kingdom system of life. Biol Rev 73:203–266
Clayton DA (1984) Transcription of the mammalian mitochondrial genome. Ann Rev Biochem 53:573–594
Costa M, Fontaine J-M, Loiseaux-de Goër S, Michel F (1997) A group II self-splicing intron from the brown alga Pylaiella littoralis is active at unusually low magnesium concentrations and forms populations of molecules with a uniform conformation. J Mol Biol 274:353–364
Daugbjerg N, Andersen RA (1997) A molecular phylogeny of the heterokont algae based on analyses of chloroplast-encoded rbcL sequence data. J Phycol 33:1031–1041
Daugbjerg N, Guillou L (2001) Phylogenetic analyses of Bolidophyceae (Heterokontophyta) using rbcL gene sequences support their sister group relationship to diatoms. Phycologia 40:153–161
Delwiche CF, Palmer JD (1997) The origin of plastids and their spread via secondary symbiosis. In: Bhattacharya D (ed) Origins of algae and their plastids. Springer Verlag, pp 53–86
Draisma SGA, Prud’ homme van Reine WF, Stam WT, Olsen JL (2001) A reassessment of phylogenetic relationships within the Phaeophyceae based on rubisco large subunit and ribosomal DNA sequences. J Phycol 37:586–603
Elzanowski AA, Ostell J (2000) The genetic codes. NCBI web site. http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi
Fast NM, Keeling PJ (2001) Alpha and beta subunits of pyruvate dehydrogenase E1 from the microsporidian Nosema locustae: mitochondrion-derived carbon metabolism in microsporidia. Mol Biochem Parasit 117:201–209
Goertzen LR, Theriot EC (2003) Effect of taxon sampling, character weighting, and combined data on the interpretation of relationships among the heterokont algae. J Phycol 39:423–439
Guillou L, Chrétiennot-Dinet M-J, Medlin LK, Claustre H, Loiseaux-de Goër S, Vaulot D (1999) Bolidomonas: a new genus with two species belonging to a new algal class, the Bolidophyceae (Heterokonta). J Phycol 35:368–381
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acid Symp Ser 41:95–98
Leipe DD, Tong SM, Goggin CL, Slemenda SB, Pieniazek NJ, sogin ML (1996) 16S-like rDNA sequences from Developayella elegans, Labyrinthuloides haliotidis, and Proteromonas lacertae confirm that the stramenopiles are a primarily heterotrophic group. Eur J Protistol 32:449–458
Leipe DD, Wainright PO, Gunderson JH, Porter D, Patterson DJ, Vlois F, Himmerich S, Sogin ML (1994) The stramenopiles from a molecular perspective: 16S-like rRNA sequences from Labyrinthuloides minuta and Cafeteria roenbergensis. Phycologia 33:369–377
Mabuchi K, Miya M, Satoh TP, Westneat MW, Nishida M (2004) Gene rearrangements and evolution of tRNA pseudogenes in the mitochondrial genome of the parrotfish (Teleostei: Perciformes: Scaridae). J Mol Evol 59:287–297
Martin WF, Müller M (1998) The hydrogen hypothesis for the first eukaryote. Nature 392:37–41
Massana R, Guillou L, Diez B, Pedros-Alio C (2002) Unveiling the organisms behind novel eukaryotic ribosomal DNA sequences from the ocean. Appl Environ Microbiol 68:4554–4558
Medlin LK, Kooistra WHCF, Potter D, Saunders GW, Andersen RA (1997) Phylogenetic relationships of the “golden algae” (haptophytes, heterokont chromophytes) and their plastids. Pl Syst Evol Suppl 11:187–219
Oudot-Le Secq M-P, Fontaine J-M, Rousvoal S, Kloareg B, Loiseaux-de Goër S (2001) The complete sequence of a brown algal mitochondrial genome, the Ectocarpale Pylaiella littoralis (L.) Kjellm. J Mol Evol 53:80–88
Oudot-Le Secq M-P, Kloareg B, Loiseaux-de Goër S (2002) The mitochondrial genome of the brown alga Laminaria digitata: a comparative analysis. Eur J Phycol 37:163–172
Patterson DJ (1999) The Diversity of Eukaryotes. Am Nat 154:S96–S124
Pearson G, Serrão EA, Cancela ML (2001) Suppression subtractive hybridization for studying gene expression during aerial exposure and desiccation in fucoid algae. Eur J Phycol 36:359–366
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Potter D, Saunders GW, Andersen RA (1997) Phylogenetic relationships of the Raphidophyceae and Xanthophyceae as inferred from nucleotide sequences of the 18S ribosomal RNA gene. Am J Bot 84:966–972
Rokas A, Holland PW (2000) Rare genomic changes as a tool for phylogenetics. Trends Ecol Evol 15:454–459
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rousseau F, Burrowes R, Peters AF, Kuhlenkamp R, de Reviers B (2001) A comprehensive phylogeny of the Phaeophyceae based on nrDNA sequences resolves the earliest divergences. CR Acad Sci III-Vie 324:305–319
Rousseau F, de Reviers B (1999) Phylogenetic relationships within the Fucales (Phaeophyceae) based on combined partial SSU + LSU rDNA sequence data. Eur J Phycol 34:53–64
Rousvoal S, Oudot M-P, Fontaine J-M, Kloareg B, Loiseaux-de Goër S (1998) Witnessing the evolution of transcription in mitochondria: the mitochondrial genome of the primitive brown alga Pylaiella littoralis (L.) Kjellm. encodes a T7-like RNA polymerase. J Mol Biol 277:1047–1057
Sanchez Puerta MV, Bachvaroff TR, Delwiche CF (2004) The complete mitochondrial genome sequence of the haptophyte Emiliania huxleyi and its relation to heterokonts. DNA Res 11:1–10
Sankoff D, Bryant D, Deneault M, Lang BF, Burger G (2000) Early eukaryote evolution based on mitochondrial gene order breakpoints. J Comput Biol 7:521–535
Saunders GW, Potter D, Paskind MP, Andersen RA (1995) Cladistic analyses of combined traditional and molecular data sets reveal an algal lineage. Proc Natl Acad Sci USA 92:244–248
Schneider A, Maréchal-Drouard L (2000) Mitochondrial tRNA import: are there distinct mechanisms? Trends Cell Biol 10:509–513
Swofford DL (2003) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland, Massachusetts
Sorhannus U (2001) A “total evidence” analysis of the phylogenetic relationships among the photosynthetic stramenopiles. Cladistics 17:227–241
Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S (1998) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res 26:148–153
Van de Peer Y, Baldauf SL, Doolittle WF, Meyer A (2000) An updated and comprehensive rRNA phylogeny of (crown) eukaryotes based on rate-calibrated evolutionary distances. J Mol Evol 51:565–576
Van de Peer Y, Van der Auwera G, De Wachter R (1996) The evolution of stramenopiles and alveolates as derived by “substitution rate calibration” of small ribosomal subunit RNA. J Mol Evol 42:201–210
Vellai T, Takacs K, Vida G (1998) A new aspect to the origin and evolution of eukaryotes. J Mol Evol 46:499–507
Wuyts J, Van de Peer Y, Winkelmans T, De Wachter R (2002) The European database on small subunit ribosomal RNA. Nucleic Acids Res 30:183–185
Yoon HS, Hackett JD, Ciniglia C, Pinto G, Bhattacharya D (2004) A molecular timeline for the origin of photosynthetic eukaryotes. Mol Biol Evol 21:809–818
Zagryadskaya EI, Kotlova N, Steinberg S (2004) Key elements in maintenance of the tRNA L-shape. J Mol Biol 340:435–444
Acknowledgements
We thank S.A. Boele-Bos and J. Veldsink, for technical assistance, as well as G. Hoarau and M. Chevolot for helpful discussions. We also would like to thank two anonymous reviewers for useful comments to improve the manuscript. M.-P. O.-L.S. was supported by a Marie Curie Individual Fellowship (programme “Quality of Life and Management of Living Resources” QLK3-CT-2000-52053).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Ralph Bock
Nucleotide sequence data reported are available in the GenBank databases under the accession numbers AY494079, AY500368 and AY500367.
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Secq, MP.OL., Goër, S.Ld., Stam, W.T. et al. Complete mitochondrial genomes of the three brown algae (Heterokonta: Phaeophyceae) Dictyota dichotoma, Fucus vesiculosus and Desmarestia viridis . Curr Genet 49, 47–58 (2006). https://doi.org/10.1007/s00294-005-0031-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-005-0031-4