
Abstract
Genetic relationship among 25 rice accessions have been evaluated using 21 SSR markers. A total of 87 alleles were produced by 21 polymorphic SSRs with an average of 4 alleles per SSRs. The SSR RM514 produced maximum number of alleles (9) followed by RM125 (7) and RM271 and RM447 (6). The PIC value varied from 0.07 (RM507) to 0.83 (RM514) with an average of 0.46. Jaccard’s dissimilarity coefficient ranged 0.10–0.89 with an average of 0.65. The highest genetic dissimilarity was observed between Dub Raj & IPB—1 (0.89), and Pant—12 & IPB—1 (0.89) and minimum dissimilarity observed between Kalanamak and CSR—10 (0.10). By evaluating data from clustering, dissimilarity and admixture nature the most diverse accessions were IPB—1, Pant—12, Dub Raj, Ram Raj, NDR 2064 and Sobhini. The accessions IPB—1, NDR—8002, BPT—5204, NDR—2065, PB—1, PUSA Basmati, and NDR—359 was found highly diverse as these accessions showed highest dissimilarity with respect to rest of the accessions. The NJ clustering pattern revealed 3 clusters and supported by 3 subpopulations determined from STRUCTURE analysis. The analysis of molecular variance (AMOVA) showed that 85% of allelic diversity was found within the population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice is one of the most important staple crop for a large-scale human population over worldwide and India both. As a primary food source, it can meet the nutritional demands of many people. With the ever rising population and unfavorably changing climate there must be a keen step to follow to increase rice productivity with high quality. India has already been victim of such climate change problems so far. Some the abiotic stresses reducing rice crop productivity includes, drought, salinity, temperature, pollution etc. (Singha et al. 2021). The high yield and productivity have always been a concern to breeders for genetic improvement with desirable nutritional quality traits in rice (Sreenivasulu et al. 2015; Sun et al. 2018). In any genetic improvement program, the availability and knowledge about genetic diversity play important role for safeguarding competent use and also for choosing efficient breeding strategies (Chakravarthi and Naravaneni 2006). The overall success of breeding program depends on the consequence of genetic variability and how the beneficial make-up is heritable. Diversity in genes of rice accessions creates an option for the breeder to select desirable traits and use them in making new combinations (Garris et al. 2005). Many techniques support to evaluate genetic variation at both phenotypic and genotypic levels. For the genotypic level, one of the best methods for analyzing diversity in rice is by molecular markers which can tell huge differences among accessions at the DNA level, providing a more reliable, and well-planned aid in accession characterization, and genetic make-up. Many such methods are there, like RAPD, SSR, AFLP, ISSR, etc. The SSR is one of the widely and most commonly used, powerful, and low-cost methods for genetic characterization of germplasm. SSR markers can efficiently facilitate the establishment of genetic relationships due to their co-dominant and specific nature, high level of allelic diversity, relative polymorphic abundance, and extensive distribution across the genome (Powell et al. 1996; Mondini et al. 2009; Hoshino et al. 2012). SSR markers even in less number can give a better genetic diversity spectrum due to their multi-allelic and highly polymorphic nature (Singh et al. 2016). SSR markers have pivotal role in detecting high level of allelic diversity and genetic polymorphisms, and it is broadly used to know genetic variation among rice accessions at a finer level of variation among closely related accessions. The objective of this study was to understand and analyze the extent of genetic diversity and differences among 25 accessions of rice using SSR markers because they give favourable genetic information, help in grouping of accessions according to similarity and dissimilarity to their genetic relationship and identification of diverse accessions for future breeding programs which has the main aim of enhancing rice production balanced with adequate increase in rice quality.
Materials and methods
Plant material and DNA extraction
The plant materials for the present study consisted of 25 rice accessions of indica sub-species collected from Chandra Shekhar Azad University of Agriculture and Technology (CSAUA&T), Kanpur, Uttar Pradesh, and Acharya Narendra Dev University, Faizabad, Uttar Pradesh. The details of accessions are provided in Table 1. The plants were grown in experimental field at CSIR, NBRI, Lucknow during cropping season 2018–2019. The fresh and young leaves from 1 month old plant were collected and total genomic DNA extracted using CTAB method (Murray and Thompson, 1980). In brief, the leaf sample was ground with help of liquid nitrogen and powder was added in pre heated 2% extraction buffer (20 mM EDTA, 1.5 M NaCl, 100 mM Tris HCL, 2% CTAB & 1% BME). The mixture was then incubated in water bath at 65 °C for 45 min and shaken softly in between followed by treatment with Chloroform: Isoamyl alcohol (25:1), 100 mg/ml RNase, and 70% ethanol, then dissolved the pellet in 1X TE buffer. The isolated genomic DNA was quantified by Spectrophotometer Nanodrop (Thermo Scientific, Wilmington, DE, USA), and its quality was checked on 0.8% agarose gel. Finally, the DNA was diluted to 20 ng/µl in TE buffer for PCR amplification.
SSR genotyping and data analysis
A set of 30 SSR primers were selected from the panel of 50 standard SSR markers defined for rice diversity analysis (https://archive.gramene.org/markers/microsat/50_ssr.html). Initially, 4 genomic DNA were amplified with 30 SSR primers to check the amplification and appropriateness of each primer for further genotyping of remaining accessions. Out of 30 SSR primers, 21 primers were selected based on polymorphic amplified products. The PCR amplification was carried out in 10 µl reaction volume containing 20 ng DNA, 1X PCR master mix (Genei Labs, India), 5 pmol each forward and reverse primer. The amplification was carried in C1000 thermal cycler (Bio-Rad Laboratories Inc. USA) with following conditions: pre denaturation at 95 ºC for 5 min followed by 39 cycles of denaturation at 95 ºC for 30 s, annealing at 53–58 ºC (primer specific) for 45 s, extension at 72 ºC for 1 min and final extension at 72 ºC for 10 min. The amplified PCR product was separated on 6% non-denaturing PAGE and visualized by silver staining. The fragment size was calculated by fragment size calculator (http://biotools.nubic.northwestern.edu/SizeCalc.html). The SSR genotyping data were scored both as allele size and as binary matrix (0–1). The allelic data were subjected to calculation of the polymorphic information content (PIC) value, major allele frequency, number of alleles per locus, and gene heterozygosity using Power Marker Software (Liu and Muse 2005). The binary data matrix were subjected to calculate the distance matrix following Jaccard similarity coefficient using DARwin Software (version 6.0.021) (Perrier et al., 2003). The resultant distance matrix was used to construct neighbour joining dendrogram with 1000 bootstrap. To determine the number of subpopulation among the accessions, Bayesian clustering was carried out using software package STRUCTURE v 2.3.3 (Pritchard et al., 2000). The STRUCTURE program was run using admixture model with independent allele frequencies. The presumed population (K) was set from 2 to 10 with 3 independent runs per K and each run was carried out using 30,000 burn-in period and 100,000 iterations. The optimal value of K was determined by examining Delta K statistic and L (K) according to Evanno et al., 2005 using structure harvester (Earl and Holdt 2012). The number of observed alleles (Na), number of effective alleles (Ne), Shannon’s information index (I) and molecular variance (AMOVA) (Excoffier et al. 2009) was calculated with GenAlex 6.5 software.
Results
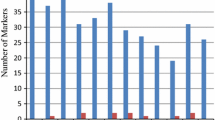
The 21 SSR primer produced a total of 87 alleles across the 25 rice accessions with an average of 4 alleles per SSR that ranged from 2 (RM507, RM237, RM283, and RM133) to 9 alleles (RM514) per SSR (Table 1). The maximum number of alleles were shown by RM514 (9 alleles) followed by RM125 (7 alleles) and RM271, RM447 (6 alleles). The gene diversity was in between 0.08 (RM507) to 0.84 (RM514) with an average of 0.51 ± 0.22. The 19 SSRs showed observed heterozygosity below 0.20. The observed heterozygosity ranged from 0.00 to 0.36 (RM125) with an average of 0.03 ± 0.09. The PIC value ranged from 0.07 (RM507) to 0.83 (RM514) with an average of 0.46 ± 0.21. Out of 21 SSRs, 23.8% viz. 5 SSRs showed moderate PIC value ranged between 0.25 to 0.50 while 4 SSRs (19.04%) showed lower PIC value (0.07–0.25) and 12 SSRs (57.1%) were highly informative with PIC value ranging from 0.50 to 0.83 Table 1. To understand the genetic dissimilarity among 25 accessions the allelic data were used to calculate a pairwise genetic distance based on the Jaccard coefficient. The pairwise genetic dissimilarity determines the level of relatedness among the accessions, which varied from 0.10 to 0.89. The maximum genetic dissimilarity (0.89) was noticed between accessions Dub Raj & IPB—1 (0.89), and Pant—12 & IPB—1 (0.89) while minimum (0.10) was noticed between Kalanamak and CSR—10. The genetic dissimilarity of one accession with respect to the rest 24 accession revealed that IPB—1 (0.77 ± 0.12) was found to be highly diverse followed by, NDR—8002 (0.74 ± 0.11), BPT—5204 (0.74 ± 0.08), NDR—2065 (0.73 ± 0.09), PB—1 (0.71 ± 0.07), PUSA Basmati (0.71 ± 0.09), & NDR—359 (0.71 ± 0.09) (Table 2). The NJ based dendrogram divided all the 25 accessions into 3 major clusters. The cluster I accommodated 9 (36%) accessions namely Pant–12, Azad Basmati, Kalmuhi, Tarvari Basmati, CSR—10, Kalanamak, CSR—27, Dub Raj and Shakkarchini, cluster II with 7 accessions (28%), NDR—2065, NDR—359, Pusa Basmati, PB—1, BPT—5204, DRR—42 and Sobhini, and cluster III with 9 accessions (36%), NDR—2064, NDR—3112, Swarna Sub–1, MTU – 7029, Sarjoo–52, NDR–8002, IPB—1, Ram Raj and CSR—43 (Fig. 1). Further, the clustering pattern was supported by Bayesian based population structure which also revealed 3 subpopulation based on ∆K value as it showed maximum for K = 3 (Fig. 2). The clustering pattern based on Bayesian statistics depicts consistency with NJ clustering. The bar plot cluster shows 10 accessions in subpopulation I, 6 accessions in subpopulation II and 9 accessions in subpopulation III and of which 7 accessions tend to be pure and 3 showed admixture nature in subpopulation I. All accessions of subpopulation II were pure while 2 accessions of subpopulation III were admixture type (Fig. 3). The cluster composition of NJ tree and bar plot was almost of similar types except the accession Sobhini which was clustered in II cluster in NJ tree and subpopulation I in bar plot. The structure analysis revealed that out of 25, five accessions (Pant 12, Dub Raj, Sobhini, NDR 2064, Ram Raj) were found to be of admixture types and rest have pure genetic makeup based on the present investigation using 21 SSRs. The AMOVA helps in knowing the significance of genetic structure by separating accessions based on genetic variation within and among population using 3 population based on NJ dendrogram. It showed that the 85% of the total genetic variation was found to be within the population and 15% among the population (Table 3) with an average pairwise ϕpt (similar to Fst) being 0.154. The maximum gene diversity (He) was noticed for population 1 (0.19 ± 0.01), followed by Population 3 (0.15 ± 0.02), and population 2 (0.13 ± 0.02) with an average of 0.15 ± 0.01 (Table 4). The other parameters, such as number of different alleles (Na), effective number of alleles (Ne) and Shannon’s information index (I) was noticed higher for population 1 having a mean value of 1.16 ± 0.09, 1.31 ± 0.03 and 0.31 ± 0.02 respectively (Table 4).

Dendrogram of 25 rice accessions constructed by NJ cluster analysis of genetic dissimilarity based on 21 SSR data

Estimation of subpopulation by using ∆K (k = 2 to 10)

Bar plot clustering performed in Structure for K = 3 populations of rice accessions
Discussion
The understanding of genetic diversity is feasible in adopting and incorporating suitable breeding strategies for developing high yielding varieties of rice to fulfill its increasing requirement. Genetic improvement is constantly progressing procedure so there must be a need of the knowledge and understanding of genetic variablity to perform genetic improvement strategy. For identification of diverse accessions there must be evaluation of different set of genetic material with appropriate tool which helps in utilization of these accession in breeding programs. The efficiency of any DNA marker for diversity analysis is mainly based on its polymorphism rate among the accessions to be evaluated. The knowledge and existence of genetic diversity in any other crop plant pave the foundation for breeders to devise suitable breeding strategies towards genetic improvement for developing high-yielding varieties. Many types of molecular markers such as RAPD, ISSR, SNP etc. apart from SSR marker can be used to access genetic variability in wide range of crop plants. The SSR markers are highly informative, co-dominant, multi–allelic, reproducible and transferable in nature (Mason 2015) and thus very useful for population genetics studies. The SSR provides the best tool in identification and characterization of genetically distinct accessions, it serves adequate power of resolution to discriminate among accessions. In rice, several set of genetic materials have been assessed for genetic variability and identification of potential accessions (Embate et al. 2021; Rohmawati et al. 2021). However, the informations obtained from one set of genetic materials could not be implemented on the other set, therefore, it is important to evaluate the genetic materials for variability afresh. A set of well characterized 50 SSRs have been reported for their utilization to evlauate the new sets of accessions (https://archive.gramene.org/markers/microsat/50_ssr.html). We have utilized 21 SSRs from the same set of SSRs to access the genetic diversity among 25 accessions of rice mainly cultivated in Indo-gangetic plains of Uttar Pradesh. A fair number of alleles (87 alleles) have been observed among the studied accessions. Similar range of allelic variability has also been reported earlier in rice with SSRs (Hossain et al. 2007; and Sajib et al. 2012; Ahmed et al. 2019). The average number of alleles detected were 4.14 with a range of 2 (RM507, RM237, RM283, RM133) to 9 (RM514). These findings are similar to those of Ahmed et al. (2019). According to Zhu et al. (2011) an allele observed in less than 5 percent are considered as rare allele. Rare alleles are very instructive in rice accessions fingerprinting (Jain et al. 2004). RM514 and RM125 have higher number of alleles which might be due to wider diversity in the accessions evaluated. The SSRs RM_514, RM_125, RM_447, RM_271 and RM_495 have been identified as potential ones as these showed considerable allelic variability and higher PIC values. These SSRs could be further utilized for diversity analysis in new set of accessions of rice. Gene diversity is the most suitable method of evaluating genetic variability within genotype (Toro and Caballero 2005). According to Rana et al. (2018), marker showing higher number of alleles has high genetic variability and so on. The Polymorphism information content (PIC) values shown by some of the SSRs in the present investigation was found to be higher than the previously reported (Borba et al. 2009; Upadhyay et al. 2011; Singha et al. 2021). However, similar range of PIC value was reported by Islam et al. (2018). The results of our study give out that RM514 is among screening in 25 rice accessions followed by RM125 and RM447. In general the SSRs which showed wide allelic range also showed higher PIC value indicated that the PIC value is an outcome of allele diversity and frequency among the varieties (Rashid et al. 2018). Similar observation have also been reported earlier by Yu et al. (2003) and Onaga et al. (2013). Considering the lowest range of pair-wise genetic similarity coefficient, the accessions PB—1, BPT—5204, PUSA Basmati were found to most diverse. These accessions were also distributed across the 3 cluster of NJ tree and bar plot of population structure. The neighbour-joining (NJ) based clustering and model based population structure analysis was done to understand the extent of genetic divergence which revealed similar pattern of accessions distribution except the accession Sobhini in both. This indicated the robustness of two clustering methods and suggests that any one could be utilized for diversity analysis. Apart from clustering pattern structure analysis also revealed admixture nature of accessions and here a total of 5 accessions showed different level of admixture indicating genetic relatedness and sharing of alleles among themselves. The genetic structure of populations previously reported in rice by Zhao et al. (2009), Jin et al. (2010), Huang et al. (2010). The present study exhibited 3 subpopulation having 5 admixture accessions (Pant 12, Dub Raj, Sobhini, NDR 2064, Ram Raj). The similar level of admixture in several rice accessions have also been reported earlier by Upadhyay et al. (2012). Considering each cluster as one population allowed the estimation of partitioning of genetic diversity as variation within population and variation between populations. The variance within population was much higher than the variance among population. Similar pattern of population variance was also reported by Jasim et al. (2018), Yogi et al. (2020). The main focus of the study is to screen out the diverse accessions of rice for maintaining the important yield attributing trait by selecting most diverse accessions as parents to perform breeding trials and is essential for commercial rice production.
Conclusion
The present investigation propose that the genetics of most of the popular rice accessions in India in the region of Indo Gangetic Plains is narrow, so there must be characterization of large set of commercial rice varieties which helps in its improvement. By evaluating data from clustering, dissimilarity and admixture nature the most diverse accessions were IPB—1, Pant—12, Dub Raj, Ram Raj, NDR 2064 & Sobhini. The accessions IPB—1, NDR—8002, BPT—5204, NDR—2065, PB—1, PUSA Basmati, & NDR—359 were found to be highly diverse as these accessions showed highest dissimilarity with respect to rest of the accessions. The identified potential SSR such as RM514, RM125, RM447, RM271 and RM495 could be utilized to investigate genetic diversity in new set of rice accessions and accessions identified here could be utilized in various breeding programs to create new lines. Microsatellite marker (SSR) offers good opportunity for assessing genetic diversity.
References
Ahmed S, Anik TR, Islam A, Uddin I and Haque MS (2019) Screening of Some Rice (Oryza sativa L.) Genotypes for Salinity Tolerance using Morphological and Molecular Markers. Biosciences Biotechnology Research Asia 16(2):377–390
Borba TCO, Brondani RPV, Rangel PHN, Brondani C (2009) Microsatellite marker-mediated analysis of the EMBRAPA rice core collection genetic diversity. Genetica 137(3):293–304
Chakravarthi, BK, Naravaneni R (2006) SSR marker based DNA fingerprinting and diversity study in rice (Oryza sativa. L). African Journal of Biotechnology, 5(9)
Earl DA, vonHoldt BM (2012) Structure Harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361
Embate MVG, Calayugan MIC, Gentallan RP, Cruz PCS, Hernandez JE and Borromeo TH (2021) Genetic diversity of selected pigmented traditional rice (Oryza sativa L.) varieties from Mindanao, Philippines using agromorphological traits and simple sequence repeats markers. J Crop Sci Biotechnol 24(3):259–277
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol 14(8):2611–2620
Excoffier L, Hofer T, Foll M (2009) Detecting loci under selection in a hierarchically structured population. Heredity 103:285–298
Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch S (2005) Genetic structure and diversity in Oryza sativa L. Genetics 169(3):1631–1638
Hoshino A A, Bravo J P, Nobile P M, Morelli K A (2012). Microsatellites as tools for genetic diversity analysis, genetic diversity in microorganisms. InTech Open: 382
Hossain MZ, Rasul MG, Ali MS, Iftekharuddaula KM, Mian MAK (2007) Molecular characterization and genetic diversity in fine grain and aromatic landraces of rice using microsatellite markers. Bangladesh J Genet Plant Breeding 20(2):1–10
Huang M, Xie F, Chen L, Zhao X, Jojee L, Madonna M (2010) Comparative analysis of genetic diversity and structure in rice using ILP and SSR markers. Rice Sci 17(4):257–268
Islam MZ, Khalequzzaman M, Prince MFRK, Siddique MA, Rashid ESMH, Ahmed MSU, Pittendrigh, BR and Ali MP (2018). Diversity and population structure of red rice germplasm in Bangladesh. PLoS One 13(5):e0196096
Jain S, Jain RK, McCOUCH SR (2004) Genetic analysis of Indian aromatic and quality rice (Oryza sativa L.) germplasm using panels of fluorescently-labeled microsatellite markers. Theoretical Appl Genet 109(5): 965–977
Jasim Aljumaili S, Rafii MY, Latif MA, Sakimin SZ, Arolu IW, Miah G (2018) Genetic diversity of aromatic rice germplasm revealed by SSR markers. Biomed Res Int 2018:7658032
Jin L, Lu Y, Xiao P, Sun M, Corke H, Bao J (2010) Genetic diversity and population structure of a diverse set of rice germplasm for association mapping. Theor Appl Genet 121(3):475–487
Liu K, Muse SV (2005) Power Marker: an integrated analysis environment for genetic marker analysis. Bioinform Appl Note 21(9):2128–2129
Mason AS (2015) SSR genotyping. In: Plant genotyping (pp 77–89). Humana Press, New York, NY
Mondini L, Noorani A, Pagnotta MA (2009) Assessing plant genetic diversity by molecular tools. Diversity 1(1):19–35
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucl Acids Res 8(19):4321–4325
Onaga G, Egdane J, Edema R, Ismail AM (2013) Morphological and genetic diversity analysis of rice accessions (Oryza sativa L.) differing in Iron toxicity tolerance. Journal of Crop Science and Biotechnology 16(1):85–95
Perrier X, Flori A, Bonnot F (2003) Data analysis methods. In: Hamon P, Seguin M, Perrier X, Glaszmann JC (eds) Genetic diversity of cultivated tropical plants. Enfield Science Publishers, Montpellier, pp 43–76
Powell W, Morgante M, Andre C, Hanafey M, Vogel J, Tingey S, Rafalski A (1996) The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol Breeding 2(3):225–238
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Rana MM, Islam MA, Imran S, Ruban S, Hassan L (2018) Genetic diversity analysis of NERICA lines and parents using SSR markers. Int J Plant Soil Sci 23(6):1–10
Rashid MM, Imran S, Islam MA and Hassan L (2018) Genetic diversity analysis of rice landraces (Oryza sativa L.) for salt tolerance using SSR markers in Bangladesh. Fundamental and Applied Agriculture, 3(2):460–466
Rohmawati I, Nursusilawati P and Abdullah S (2021) Genetic diversity of Some Indonesian Local Rice Varieties based on Simple Sequence Repeat (SSR) marker related to Aromatic Genes. In IOP Conference Series: Earth and Environmental Science (Vol. 715, No. 1, p. 012046)
Sajib AM, Hossain MdM, Mosnaz ATMJ, Hossain H, Islam MdM, Ali MdS, Prodhan SH (2012) SSR marker-based molecular characterization and genetic diversity analysis of aromatic landraces of rice (Oryza sativa L.). Journal of Biological Science and Biotechnology 1(2):107–116
Singh N, Choudhury DR, Tiwari G, Singh AK, Kumar S, Srinivasan K, Singh R (2016) Genetic diversity trend in Indian rice varieties: an analysis using SSR markers. BMC Genet 17:127
Singha T, Mahamud MA, Imran S, Paul NC, Hoque MN, Chakrobarty T, Al Galib MA, Hassan L (2021) Genetic diversity analysis of advanced rice lines for salt tolerance using SSR markers. Asian J Med Biol Res 7(2):214–221
Sreenivasulu N, Butardo VM Jr, Misra G, Cuevas RP, Anacleto R, Kavi Kishor PB (2015) Designing climate-resilient rice with ideal grain quality suited for high-temperature stress. J Exp Bot 66(7):1737–1748
Sun J, Yang L, Wang J, Liu H, Zheng H, Xie D, Zhang M, Feng M, Jia Y, Zhao H and Zou D (2018) Identification of a cold-tolerant locus in rice (Oryza sativa L.) using bulked segregant analysis with a next-generation sequencing strategy. Rice, 11(1): 1–12
Toro MA, Caballero A (2005) Characterization and conservation of genetic diversity in subdivided populations. Philosophical Trans R Soc b 360(1459):1367–1378
Upadhyay P, Neeraja CN, Kole C, Singh VK (2012) Population structure and genetic diversity in popular rice varieties of India as evidenced from SSR analysis. Biochem Genet 50(9):770–783
Upadhyay P, Singh VK, Neeraja CN (2011). Identification of genotype-specific alleles and molecular diversity assessment of popular rice (Oryza sativa L.) varieties of India. International Journal of Plant Breeding and Genetics 5(2):130–140
Yogi R, Kumar N, Kumar R and Jain RK (2020) Genetic diversity analysis among important rice (Oryza sativa L.) genotypes using SSR markers. Advances in Bioresearch 11(2):68–74
Yu SB, Xu WJ, Vijayakumar CHM, Ali J, Fu BY, Xu JL, Jiang YZ, Marghirang R, Domingo J, Aquino C and Virmani SS (2003) Molecular diversity and multilocus organization of the parental lines used in the International Rice Molecular Breeding Program. Theoretical and Applied Genetics, 108(1):131–140
Zhao W, Chung JW, Ma KH, Kim TS, Kim SM, Shin DI, Kim CH, Koo HM, Park YJ (2009) Analysis of genetic diversity and population structure of rice cultivars from Korea, China and Japan using SSR markers. Genes & Genomics 31(4):283–292
Zhu, C., Li, X. and Yu, J., 2011. Integrating rare-variant testing, function prediction, and gene network in composite resequencing-based genome-wide association studies (CR-GWAS). G3: Genes| Genomes| Genetics, 1(3), pp.233–243
Acknowledgements
The authors would like to thank the Director, CSIR-National Botanical Research Institute, Lucknow for the support and facilities to conduct the experiment. The funding from Department of Science and Technology (DST), New Delhi, India is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Naaz, S., Pandey, V. & Yadav, H.K. Evaluation of genetic diversity in rice (Oryza sativa L. ssp. Indica) accessions using SSR marker. Vegetos 35, 961–968 (2022). https://doi.org/10.1007/s42535-022-00378-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42535-022-00378-3