
Abstract
In this study, Trema orientalis was used as an example to explore chloroplast genome evolution and determine Cannabaceae’s phylogenetic relationship using phylogenetic analysis. Comparing six Trema species chloroplast genomes shows that gene order, gene content, and length are highly conserved yet dynamically evolve among species. The whole T. orientalis chloroplast genome is 157,134 bp long with accession number OQ871457 and includes a pair of inverted repeats (IRs) of 25,493 bp separated by a small single-copy region of 19,320 bp and a large single-copy region of 86,822 bp. The total content of GC is 36.3%. The chloroplast genome was annotated to include 129 genes, 84 of which code for proteins, 37 for tRNA, and 8 for rRNA. Regarding, there are 127 SSRs were found, with the highest concentration in p1 (60), whose length varied from 10 to 16 bp; these areas could serve as foundational molecular markers for the Trema genus. The IRS repeats were found: 17 were forward repeats (F), 25 were palindromic repeats (P), and five were reverse repetitions (R). T. orientalis and T. orientalis (NC_039734.1), with 99% similarity, were found in the same group in a phylogenetic analysis of Trema species. IR scope expansion and contraction were also determined and compared with 17 related species in this family. It is the first report of the chloroplast genome of T. orientalis collected from Western Desert, Saudi Arabia, providing an important data reference for future investigations into genetic diversity and plant evolution. Such information based on the complete chloroplast genomes facilitates the evolution of species-specific molecular tools to discriminate T. orientalis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Cannabaceae family has 10 genera and about 117 species (Sytsma et al. 2002; Bell et al. 2010; Byng et al. 2016). The Cannabaceae family consists primarily of woody plants. However, it does include at least one herb (Cannabis L.) and a few vines (Humulus L.). The family is found worldwide in both tropical and temperate climates. Species such as Aphananthe (Thunb.) Planch., Celtis L., and Trema Lour (Yang et al. 2013). Trema orientalis L. is a tree in the Cannabaceae family with leaves that stay green all year. The height of this tree changes depending on the weather and where it grows. There is a tendency for the leaf base to be different lengths and widths. The length can range from 2 to 20 cm, and the width from 1.2 to 7.2 cm. Even though the flowers are small, green, and not very noticeable, they are carried in dense bunches that are short and close together. The small fruits are round, dark green or purple, and turn black when ready; they are carried on very short stalks (Farzana et al. 2022). This plant has strong roots that help it stay alive during long periods of drought (Adinortey et al. 2013). Some common names for the plant are pigeon wood, hop out, charcoal tree, Indian charcoal tree, Indian nettle tree, and gunpowder tree. This tree species can be found worldwide (Orwa et al. 2009), and it can grow in different climate zones and soil types, from heavy clay to light sand (Smith 1966). T. orientalis is a potentially versatile animal feed. Nonetheless, adequate seed remains a significant obstacle for most fodder promotion attempts (Franzel et al. 2014). The seeds of T. orientalis are gathered from the wild, where populations have diminished in part due to the destruction of natural habitats and may also have a significant role in determining the distribution of pioneer species like T. orientalis (Goodale et al. 2014). Hence, stochastic alterations in the genetic integrity of the seeds of this promising fodder species in the wild are expected to occur (Schippmann et al. 2002; Nantongo and Gwali 2018). Determining the genetic structure of T. orientalis can aid in developing conservation, management, and sustainable use strategies (Frankham et al. 2002; Nantongo et al. 2016, 2020; Coates et al. 2018).
In addition to being used to make paper and poles, it has also been used in traditional medicine. Almost every part of the plant is used as medicine to treat infections in tropical areas, diseases caused by worms, and lung inflammation (Nkansa-Kyeremateng 1992; Adinortey et al. 2013). Even though they are important for medicine, not much has been written about them recently (Al-Robai et al. 2022), and there aren’t many genomic resources that can help improve and domesticate them. T. orientalis is often employed as a natural pioneer in conventional medicine to treat illnesses (Adinortey et al. 2013). Fever reduction and infection prevention are two common uses for this species. Tremetol, simiarenol, and simiarenone are important phytochemical ingredients of T. orientalis leaves; tremetol, swertianin, scopoletin, and numerous fatty acids and glycosides are found in the stem bark; sterols and fatty acids are found in the roots (Parvez et al. 2019). Plants, algae, and cyanobacteria use chloroplast organelles to perform photosynthesis. Chloroplasts also perform several crucial metabolic roles. Many amino acids, lipids, pigments, and vitamins are among these. Starch is stored, and sugar is biosynthesized as well. Plants can’t grow or develop without the energy provided by the nitrogen cycle and sulfate reduction (Neuhaus and Emes 2000; Bausher et al. 2006; Richardson and Schnell 2020). The chloroplast DNA is a typical double-stranded circular genome found in higher plants (Sugiura 1995; Odintsova and Yurina 2006; Ruhlman and Jansen 2014; Iram et al. 2019). One large single-copy (LSC) region, one short single-copy (SSC) region, and two inverted repeats (IR) sections make up the normal chloroplast genome (Zhou et al. 2016). Because of their maternal inheritance, small genome size, and low mutation rate, chloroplasts’ genomic information has been widely used to produce molecular markers for use in population genetics, genome evolution, phylogenetics, and constructing DNA barcoding markers (Sun et al. 2020; Guan et al. 2022; Chen et al. 2022; Feng et al. 2023).
As a result of their low nucleotide substitution rates, structural simplicity, and uniparental inheritance (Yang et al. 2019), chloroplast genomes are often used for species identification (Yu et al. 2021) and are excellent resources for phylogenetic investigations (Yang et al. 2019). Because of its consistent structure and wealth of genetic data may be used to investigate intricate evolutionary connections (Oldenburg and Bendich 2016). Nonetheless, DNA barcoding uses some genes, such as rbcL and matK, to identify species positively; this gives molecular marker research hope (Hollingsworth 2011; Luz et al. 2023). As chloroplast genomes carry more genetic information than gene fragments, they are used in research of plant genetic diversity and conservation (Wariss et al. 2018). Chloroplast genome data is augmented by next-generation sequencing (NGS) technology, which assembles it swiftly and affordably (Tangphatsornruang et al. 2010; Zhao et al. 2021). Thus, the current study aimed to (i) use next-generation sequencing technology to sequence, assemble, and describe the complete chloroplast genome sequences of a medicinal T. orientalis wild variant found in the Western Desert area in Saudi Arabia and (ii) study the genomic relationships among T. orientalis and its related species. This information from the chloroplast genome paves the way for investigations into the phylogenetic evaluation, practical application, and conservation genetics of T. orientalis.
Materials and methods
Sample collection and DNA extraction
Leaves of T. orientalis were collected fresh from the ground in the Jazan region of Saudi Arabia (17° 15′ N 43° 06′ E) and then air-dried before being analyzed. The studied taxa was identified according to and Tachholm (1974) and herbarium specimen was deposited in herbarium of Botany and Microbiology Department, Faculty of Science, Arish University, Egypt (Authentication number: 378). Total genomic DNA was isolated from 2 g of dried leaves using a WizPrepTM gDNA Mini Kit (Cell/Tissue; Korea) according to the manufacturer’s instructions. DNA integrity was analyzed by electrophoresis on a 1.0% agarose gel, the quality of the DNA was assessed using a Quantus™ Fluorometer (Promega, USA) at the Plant Laboratory in Botany and Microbiology Department, Faculty of Science, Arish University, Egypt. Using the standard protocol of the TruSeq library preparation kit, high-quality DNA extracts were fragmented so that a 300 bp short-insert library could be built (Illumina, San Diego, California, USA). On the Illumina HiSeq 4000 platform, the library was sequenced in pair-end mode with 150 bp reads (Novogene, China). The chloroplast genome was checked by running BLASTN against the non-redundant nucleotide database at NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi?); accessed November 18, 2022). The entire chloroplast genome was put together using the Novoplasty assembler. After removing the low-quality data, the high-quality clean reads were checked, and de novo assembly was done using the single-contig method (Magdy et al. 2019; Magdy and Ouyang 2020).
Gene annotation
The online annotation tool GeSeq (Tillich et al. 2017) was used to describe the chloroplast genome as a circular molecule. The tRNA scan-SE 2.0 search server (Lowe and Chan 2016) and their anticodon sequences and typical cloverleaf secondary structures were used to confirm that all the tRNAs were correct. The coding sequences were checked and fixed by translating them using Geneious Prime (Kearse et al. 2012). OGDRAW (version v1.2) was used to construct a map of the cpDNA for the T. orientalis strain (Al-Robai et al. 2022). The relative synonymous codon usage (RSCU) was analyzed with the help of the application CodonW (version 1.4.4) (http://codonw.sourceforge.net) (accessed on 22 November 2022).
Repeats identification
The REPuter tool4 (Kurtz et al. 2001) was used to identify repeat sequences, including forward, reverse, palindrome, and complement sequences. If the Hamming distance equals three, the maximum length of repeats is 30 base pairs, and the identity is greater than 90%. MISA’s basic repeat setting was utilized to analyze the simple sequence repeats (SSRs), as stated by Beier et al. (2017). Geneious Prime was used to detect single nucleotide polymorphisms (SNPs) and insertions/deletions (indels) in the large single copy (LSC), small single copy (SSC), and inverted repeat (IR) regions.
Comparative analysis of the chloroplast genome
The studied T. orientalis and seventeen related species were analyzed using IRscope (Amiryousefi et al. 2018) to compare and contrast their SSC, IRs, and LSC margins visually using T. orientalis sequence annotation data obtained from T. orientalis (NC_039734.1) as a reference. Ka/Ks, or the rate of nonsynonymous (Ka) to synonymous (Ks) nucleotide substitution, was also computed from the alignments using the KaKs Calculator software (v2.0).
Phylogeny analysis
The phylogenetic tree analysis utilized the commonly used CDS sequences, which were aligned using the MAFFT software for eleven Trema cpDNA genomes. We utilized the RAxML software (version 8.2.10, available at https://cme.h-its.org/exelixis/software.html; last accessed on November 22, 2022) to construct the evolutionary tree using the maximum likelihood method. The tree was generated with 1000 bootstraps and a GTRGAMMA model.
Results
Chloroplast genome characteristics and features
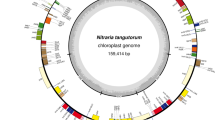
There are 157,134 bp in the assembled chloroplast genomes of T. orientalis with accession number (OQ871457). The Chloroplast genome includes a large-single copy (LSC) section that is 86,822 bp long, a small-single copy (SSC) region that is 19,320 bp long, and a pair of inverted repeat regions (IRa and IRb) that is 25,493 bp long (Fig. 1). Total GC concentration in the chloroplast genome was 36.30%, whereas GC percentages in the LSC, SSC, and IR regions were 34.0, 298.4, and 42.8%, respectively. This region’s high GC concentration (55.3%) is due to the four rRNAs’ high GC content. About 129 coding genes were found in the genome; 84 encoded proteins, 37 encoded tRNAs, and 8 encoded ribosomal RNAs. Six PCGs (rps12, rpl2, rpl23, ycf2, ndhB and rps7), seven tRNAs (trnI-GAU/CAU, trnLCAA, trnVGAC, trnAUGC, trnR-ACG and trnN-GUU), and four rRNAs (rrn16, rrn23, rrn4.5 and rrn5) were replicated. The SSC area had 12 protein-coding genes and 1 tRNA gene, whereas the LSC had 62 protein-coding genes and 22 tRNA genes. Twenty intron-containing genes (12 PCGs and six tRNA genes) and two intron-containing genes (clpP and ycf3) were found in the T. orientalis chloroplast genome. The 5′ ends of rps12 were found in the LSC area, while the duplicated 3′ ends were found in the IR region, indicating that trans-splicing occurred during its production. Of all introns containing the matK gene, the one in trnKUUU is the longest, with 2603 base pairs (Table 1). There were just 11 genes in the SSC area, whereas the LSC region had 62. Not only did IRa and IRb share four rRNA genes, but the location of those genes was also interesting (Table 2).

T. orientalis chloroplast gene map; the genes inside and outside the outer circle are transcribed clockwise and anticlockwise. The thick lines show the inverted repeats (IRa and IRb) that divide the genomes into large-single copy (LSC) and small-single copy (SSC) regions. Grey bars inside the circle represent GC content, while lighter grey indicates AT content
The cpDNA genome structure of T. orientalis and its closely related species was compared and analyzed using the CGVIEW program and the annotated cpDNA genome sequence of T. orientalis (Fig. 2). The rRNA and tRNA coding regions were shown to have significant similarities between different Trema species. Also, there were minor variations in the protein-coding areas. It was noticed that the GC content of the two IR regions is noticeably higher than that of the LSC and SSC regions (Fig. 2).

GC content of T. orientalis Chloroplast DNA; the outer five circles demonstrate similarities to related species. The inner circles indicate the CDSs of protein-coding genes (blue), rRNA operon (light purple), and tRNA information (reddish brown); the third circle shows GC contents; the fourth circle represents GC skew (green, > 0; purple, < 0); and the fifth circle represents cpDNA locations
Tandem repeats
In the present study, 735 tandem repeats were identified in the T. orientalis chloroplast genome, mostly in noncoding regions, including intergenic and introns (Table 3). The lengths of the tandem repeats ranged from 5 to 47 bp. Based on the quadripartite structure of the chloroplast genome, the most repeat sites were detected in the LSC region (549, 74.5%), followed by IR (104, 14.82%) and SSC (82, 11.15%). These repeats included 238 mononucleotides (32.38%), 45 dinucleotides (6.12%), 39 trinucleotides (5.31%), 90 tetranucleotides (12.24%), 111 pentanucleotide (15.10%), 116 hexanucleotides (15.78), 47 hepta-nucleotide (6.39%), 24 octa nucleotide (3.27%), 14 nona-nucleotide (1.90%), and 11 deca-nucleotide (1.50%). The A/T (227) mononucleotide repeats profusely existed in the T. orientalis chloroplast genome and less frequently C/G (11), and the longest repeat was one T type of 27 bp. The AT/TA motif contributed to 37 dinucleotides (82.22%), and the longest type of dinucleotides was AT type of 24 bp. The highest abundance motifs in trinucleotide repeats were AAT and TAA, AAAT motif in tetranucleotide, and AATAA in pentanucleotide repeats. The rest motifs in other repeat classes had a similar abundance ratio which ranged from 0.27 to 0.14%.
SNPs and indels
The pair-wise sequence alignment of T. orientalis chloroplast genomes revealed 147 variants, including 75 SNPs and 72 InDels in protein-coding and non-coding areas (introns and intergenic regions; Tables 4 and 5). 32 Single nucleotide polymorphism SNPs and zero indels were found in chloroplast genome protein-coding genes, with ycf1 and rpoB genes having the most SNPs. The LSC and SSC regions have more substitutions than the IRs regions, and transversion (65.33%) outnumbers transition (34.6%), as shown in Table 4. The LSC region had the most SNPs (58), followed by the SSC (13), and each IR region was the fewest (4). This study found 72 intergenic indels, 40 deletions and 32 insertions (Table 5). T. orientalis chloroplast genomes are rich in short indels, especially 1 bp indels. Deletions favored the LSC region (29 LSC, 7 in IR, and 4 in SSc). The LSC had 25 insertions, the SSC 7, and the IR area had no insertion.
Codon usage pattern analysis
The results from sequencing the chloroplast genome of T. orientalis revealed 52,378 codons (Fig. 3). One codon, either AUG or UGG, encoded the amino acids methionine and tryptophan. Two to six codons were used to encode the remaining amino acids, including the sic. codons for Arginine, leucine, and Serine. Codon usage was illustrated in Fig. 3. As an amino acid, serine was the most common among T. orientalis and appeared 4872 times. Tryptophan, on the other hand, was the rarest of the amino acids (647). Meanwhile, AGA had the highest RSCU value (1.94) of the six codons encoding Arginine, indicating that it was the most preferred and widely used. Also, 32 codons had RSCU values above 1, 25 of them ended in A or U. Most codons with RSCU values larger than 1 had A/U as the terminal codon, while those with C/G as the terminal codon often had RSCU values less than 1. In general, this suggests that codons ending in A or U are preferred by the cpDNA gene of T. orientalis. The RSCU values of T. orientalis and five closely related species were compared. The total RSCU of all the codons used to encode a single amino acid was nearly identical. Furthermore, the RSCU values of identical codons were nearly equal in these species, suggesting that their codon usage habits were more stable and rarely changed (Tables S1, S2).

Relative synonymous codon usage (RSCU) values for amino acids of T. orientalis. The colors of the histograms correspond to the colors of the codons
Analysis of the repeats
Tandem repeat sequences of tens of nucleotides are known as simple sequence repeat (SSR) markers. Each repeat unit of an SSR marker consists of a small number of nucleotides (often between one and six). The chloroplast gene of T. orientalis was analyzed, and 127 SSR sites were discovered. Ninety of the mononucleotide repetitions were A/T pairs, 33 were T repeats, and 34 were A repeats; just one was G/C. The base composition of SSRs favors AT, which is in keeping with the fact that AT is present in relatively high concentrations in the chloroplast genome. The dinucleotide sequence consisted of AT/TA repeats, appearing nine times, followed by the trinucleotide sequence ATT/TTA repeats three times, as shown in Table S3. The 105 IRSs were found; 17 were forward repeats (F), 25 were palindromic repeats (P), 5 were reverse repetitions (R), and 16 were complement repeats (C) (Table S4). IRS length was between 30 and 51 bp (Fig. 4 and Table S5). The P sequence was the longest at 149,825 base pairs. Seven repeats were found in the P-type (with a length of 31 & 32 bp), while five were found in the F-type (with a length of 35 bp). There are six types of SSRs from P1 to P6; type P1 was the highest with a value of 60, followed by P4 with 14, and compared to its related species, it showed that light difference between them as shown in Table S6. Repeat type and position 1 and two for each one with their E-value were illustrated in Table S7.

Analysis of repeat sequences in chloroplast genome of T. orientalis; Size and dispersion of the IRS. Length is an indicator of how many times this pattern will be repeated. The repetition sequence type is indicated by the variable type. In this case, the number of each type accurately reflects the total number of types. The X-axis depicts the various categories of distributed repeats, while the Y-axis indicates the total number of such elements. F for forward, P for palindrome,and R for reverse
IR regions characteristics
Using the chloroplast genomes of T. orientalis and 17 related genus from the Cannabaceae family (8 species from Trema and 9 Cetlis), we compared the junction structure to observe the change of IR borders (Fig. 3). The node genes were mostly rpl22, rps19, rpl2, ndhF, ycf1, trnh, and PsbA for all nine Trema species, except T. domingense, where ycf1 was replicated with 1103 bp before ndhf and T. orientalis (157, 174 bp), and rpl19, which was duplicated with 90 bp. In contrast to the Trema species, where the rps19 and rpl2 genes have vanished and the rps3 gene is positioned on the left (IRB/SSC) with 650 bp, Cetlis species showed variation in their IR borders. The results revealed that the genomic structure, such as gene order and number, was conserved between the four chloroplast genomes. However, some differences in the IR expansions and contractions still existed. The T. orientale had shorter IR regions compared with T. orientalis. Additionally, the length of ndhf gene in T. orientale was similar to T. orientale (157, 174 bp) and 4 bp shorter than that of T. orientalis and T. orientale (157, 192 bp), whereas and ycf1 gene was similar to T. orientale (157, 174 bp) and 2 bp longer than that of T. orientalis and T. orientale (157, 192 bp) as shown in Fig. 5.

Boundary analysis of LSC, SSC, IRa-IRb regions of chloroplast genomes of eighteen species of T. orientalis and its related species from family Cannabaceae
Synonymous and nonsynonymous mutations analysis
The correlation was calculated using KaKs analysis. To look for evidence of adaptive mutation, we calculated nonsynonymous (Ka) and synonymous (Ks) substitution rates (Table S8). These findings indicate that nine genes in T. orientalis cpDNA, psbK, petN, psbC, psaI, petG, rpl20, psbT, rpl16 and psaC, with Ka/ks ratios of > 1, were subjected to positive selection in comparisons of this species. However, the remaining genes, with Ka/Ks > 1, were subjected to negative selection, which indicates a slower rate of evolution.
Phylogenetic analysis
To investigate the family tree of the investigated plant, we sampled chloroplast DNA from T. orientalis and ten closely related species. Alignments of all 11 cpDNAs were calculated with MAFFT, and an ML tree was established with RAxML, which implemented the GTR-model with Arabidopsis thaliana as an out-group. The high bootstrap values (between 90 and 100) substantially support all associations in Fig. 6. A phylogenetic tree with strong support for most branches reveals two separate clades. T. orientalis YXing886 forms one clade, whereas other closely related species constitute the other. With a 157,192 bp sequence, the T. orientalis under study is geographically near to its closest relatives (NC_039734.1).

Phylogenetic tree of 12 complete cpDNAs constructed using the maximum likelihood (ML).
Discussion
Plant cpDNA typically contains around 120 genes, several involved in gene expression or photosynthesis (Jansen et al. 2005). There are 84 putative protein-coding genes, 46 transfer RNA (tRNA), and eight ribosomal RNA (rRNA) genes in T. orientalis’s exact cpDNAs, which are a circular molecule of 157,134 bp and exhibit a peculiar quadripartite structure (Fig. 1, Table 2). The average GC concentration of cpDNA is 36. 3%; however, the GC content of IR regions is higher than that of LSC and SSC regions (Fig. 2). T. orientalis species were similar to the GC levels found in the chloroplast genomes of other angiosperm species, ranging from 36.1 to 36.9% (Zhang et al. 2018). T. orientalis and other members of the Cannabaceae family experienced the same phenomenon (Zhang et al. 2018). The amount of GCs in a molecule is used as a proxy for its secondary structures’ stability and the local recombination rate (Meunier and Duret 2004).
Chloroplast genomes provide rich sources of phylogenetic information, and numerous investigations using chloroplast DNA sequences have been carried out during the past two decades, greatly enhancing our understanding of the evolutionary relationships among angiosperms (Jansen et al. 2007; Moore et al. 2007; Liu et al. 2020). Here we sequenced and assembled the complete chloroplast genome of T. orientalis from the Western desert in Saudi Arabia, using Illumina sequencing reads derived from the whole genome. It was possible to obtain the chloroplast genome without first separating the chloroplast DNA (Eguiluz et al. 2017). It provided sufficient genetic resources for discriminating species and phylogenetic analysis of Trema species through a comprehensive comparison of chloroplast genome sequences from Cannabaceae. The cpDNA rearrangements may proceed further, and the species’ genetic diversity may increase due to the presence of repetitive sequences. Because of their high polymorphism, low substitution rate, and codominant nature, cpSSRs are invaluable genetic tools for answering fundamental and practical concerns in plant biology (Deng et al. 2021).
Whereas previous research has shown between 118 and 140 SSRs and 30 and 101 long repeats in the cpDNAs of six Trema species (T. orientalis, T. tomentosum, T. levigatum, T. sdomingense, T. cannabinum, and T. angustifolia) (Meunier and Duret 2004), the present study found just 127 SSRs. Most of the SSRs and long repeats found in the seven species of Euonymus were mononucleotide SSRs, whereas the complement repeats made up a smaller percentage. SSR motifs may be useful as molecular identifiers for determining species, examining population genetics, and distinguishing between individuals (Pereira et al. 2013; Pezoa et al. 2021).
Interestingly, the complete chloroplast genome of T. orientale species has been previously published in the GenBank database (Accession number: MT165918.1) but recorded under the Ulmaceae family. The percentage of pairwise identity between the newly sequenced chloroplast genomes and the T. orientale was 99.8%. Although the high similarity percentage, there was a difference in the number of tRNA genes, 37 tRNAs in newly sequenced chloroplast genomes. In contrast, 36 tRNAs in the published chloroplast genome lacked trnKUUU. According to previous research, repetitive sequences plays an important role in stabilizing and rearranging chloroplast genome sequences (Weng et al. 2014; Wang et al. 2018). In addition, it is very important to note that the majority of repeats were found in regions that weren’t coding, including introns and intergenic regions, which can be taken as an indication that non-coding regions evolved faster than coding regions (Hong et al. 2017; Skuza et al. 2019). This study confirmed this result in the LSC region as this contains a large amount of intergenic sequence. Because of their analytical and highly polymorphic nature, long repetitive sequences have been used as suitable molecular markers for authentication (Choi et al. 2016), plant evolution, phylogenetics, and polymorphism research (Williams et al. 2016; Park et al. 2017). A phylogenetic tree was constructed using cpDNA sequence data from 11 species in the Trema species. The cluster of the studied Charcoal tree was closely related to the sister species of T. orientalis to the rest of the family, with a bootstrap support of 99%.
Significant evolutionary events, such as the frequent expansion and contraction of the IR region, may be responsible for the cpDNA size change (He et al. 2017). These events cause fluxes in the LSC/IR junctions, which initiate pseudogenes, gene duplication, or the reversion of duplicated genes to a single copy. Earlier research demonstrated that the expansion and contraction of IRs alter the evolution of protein-coding genes in the Cannabaceae family (Zhang et al. 2018). The cpDNAs of 18 different species from the family Cannabaceae were compared in this study. When comparing cpDNAs from different angiosperm species, there is a high level of conservation at the LSC/SSC and IR region boundaries (Palmer 1985). Trema species differed in IR contraction and expansion from Celtis species. The findings add to our existing understanding of evolutionary trends in angiosperms. Plants rely on genetic variation to maintain their evolution potential to adapt to ever-changing environmental conditions (Livingston 1996). It is believed that nucleotide substitutions and microstructural mutations, such as insertions and deletion inversions, are a major driving force in sequence evolution despite the remarkable conservation of chloroplast genomes relative to gene content (Britten et al. 2003). Natural mutations and point mutations were more common than frameshifts (Raes and Van de Peer 2005). As expected, more SNPs were found in the T. orientalis chloroplast genomes than in Indel. Most occurred in intergenic regions, consistent with the hypothesis that non-coding sequences evolved more slowly than CDS (Wu et al. 2023).
Conclusion
Finally, we evaluated repeat sequencing, codon preferences, and nucleic acid diversity after assembling the chloroplast genome of T. orientalis (charcoal tree), providing information for the cpDNA genome, evolution and phylogenetic relationship of related species of Trema. The chloroplast genome of T. orientalis is 157,134 bp long, and it differs from the chloroplast genomes of other T. orientalis species in a few base positions. The phylogenetic study shows that T. orientalis is closely related to T. orientalis reference (NC_039734.1). Intriguingly, we identified four candidate target sites in the IR, LSC, and SSC that may serve as molecular markers: rpl22, rps19, ycf1, ndhF, psbT, and rpl2. Nearly 735 tandem repeats have been identified, which can be used for population genetics research within Trema species. Our results enrich the data on the chloroplast genomes of the T. orientalis species, lay an essential foundation for accurate molecular identification, and give insight into the evolutionary pattern of these species. These molecular markers can differentiate across Trema species and its related genera and provide a theoretical foundation for future research into germplasm resources and genetic breeding methods. They have also been used to examine DNA sequence variations among plant species.
Data availability
The complete chloroplast genome sequence of T. orientalis was deposited at NCBI GenBank (accession number OQ871457). All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Abbreviations
- IRs:
-
Inverted repeats
- LSC:
-
Large single-copy
- SSC:
-
Short single-copy
- NGS:
-
Next-generation sequencing
- cpDNA:
-
Chloroplast DNA
- RSCU:
-
Relative synonymous codon usage
- SSRs:
-
Simple sequence repeats
- SNP:
-
Single nucleotide polymorphism
References
Adinortey MB, Galyuon IK, Asamoah NO (2013) Trema orientalis Linn. Blume: a potential for prospecting for drugs for various uses. Pharmacogn Rev 7:67
Al-Robai SA, Zabin SA, Ahmed AA et al (2022) Phenolic contents, anticancer, antioxidant, and antimicrobial capacities of MeOH extract from the aerial parts of Trema orientalis plant. Open Chem 20:666–678
Amiryousefi A, Hyvönen J, Poczai P (2018) IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34:3030–3031
Bausher MG, Singh ND, Lee SB (2006) The complete chloroplast genome sequence of Citrus Sinensis (L.) Osbeck Var’Ridge pineapple’: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol 6:1–11
Beier S, Thiel T, Münch T et al (2017) MISA-Web: a web server for microsatellite prediction. Bioinformatics 33:2583–2585
Bell CD, Soltis DE, Soltis PS (2010) The age and diversification of the angiosperms re-revisited. Am J Bot 97:1296–1303
Britten RJ, Rowen L, Williams J (2003) Majority of divergence between closely related DNA samples is due to Indels. PNAS 100:4661–4665
Byng JW, Chase MW, Christenhusz MJM (2016) An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc 181:1–20
Chen D, Zhang H, Chang L et al (2022) A molecular identification of medicinal Rheum species cultivated germplasm from the northwest of China using DNA barcoding. Genet Resour Crop Evol 69:997–1008. https://doi.org/10.1007/s10722-021-01276-4
Choi KS, Chung MG, Park S (2016) The complete chloroplast genome sequences of three veroniceae species (Plantaginaceae): comparative analysis and highly divergent regions. Front Plant Sci 7:355
Coates DJ, Byrne M, Moritz C (2018) Genetic diversity and conservation units: dealing with the species population continuum in the age of genomics. Front Ecol Evol 6:165
Deng G, Yang M, Zhao K, Yang Y, Huang X, Cheng X (2021) The complete chloroplast genome of Cannabis sativa variety Yunma 7. Mitochondrial DNA B Resour 6(2):531–532
Eguiluz M, Rodrigues NF, Guzman F et al (2017) The chloroplast genome sequence from Eugenia uniflora, a Myrtaceae from neotropics. Plant Syst Evol 303:1199–1212
Farzana M, Rahman MM, Ferdous T (2022) Review on Trema orientalis as a potential bioresource in tropical countries. Trees 36:1169–1177
Feng J, Xiong Y, Su X et al (2023) Analysis of complete chloroplast genome: structure, phylogenetic relationships of Galega orientalis and evolutionary inference of Galegeae. Genes 14:176
Frankham R, Ballou SEJD, Briscoe DA et al (2002) Introduction to conservation genetics. Cambridge University Press
Franzel S, Carsan S, Lukuyu B et al (2014) Fodder trees for improving livestock productivity and smallholder livelihoods in Africa. Curr Opin Environ Sustain 6:98–91
Goodale UM, Berlyn GP, Gregoire TG et al (2014) Differences in survival and growth among tropical rain forest pioneer tree seedlings in relation to canopy openness and herbivory. Biotropica 46(2):183–193
Guan YH, Liu WW, Duan BZ (2022) The first complete chloroplast genome of Vicatia thibetica de Boiss.: genome features, comparative analysis, and phylogenetic relationships. Physiol Mol Biol Plants 28:439–454
He L, Qian J, Li X et al (2017) Complete chloroplast genome of medicinal plant Lonicera Japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 22:249
Hollingsworth PM (2011) Refining the DNA barcode for land plants. Proc Natl Acad Sci USA 108:19451–19452
Hong SY, Cheon KS, Yoo KO (2017) Complete chloroplast genome sequences and comparative analysis of Chenopodium quinoa and C. album. Front Plant Sci 8:1696
Iram S, Hayat MQ, Tahir M (2019) Chloroplast genome sequence of Artemisia scoparia: comparative analyses and screening of mutational hotspots. Plants 8:476
Jansen RK, Raubeson LA, Boore JL et al (2005) Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol 395:348–384
Jansen RK, Cai Z, Raubeson LA et al (2007) Analysis of 81 genes from 64 plastid genomes resolves relationships in angio-sperms and identifies genome-scale evolutionary patterns. PNAS 104:19369–19374
Kearse M, Moir R, Wilson A et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kurtz S, Choudhuri JV, Ohlebusch E et al (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29:4633–4642
Liu H, Hu H, Zhang S et al (2020) The complete chloroplast genome of the rare species Epimedium tianmenshanensis and comparative analysis with related species. Physiol Mol Biol Plants 26:2075–2083
Livingston K (1996) Conservation genetics. In: Avise JC, Hamrick JL (eds) Case histories from nature. Chapman and Hall, New York
Lowe TM, Chan PP (2016) TRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 44:W54–W57
Luz TZ, Cunha-Machado AS, da Silva Batista J (2023) First DNA barcode efficiency assessment for an important ingredient in the Amazonian ayahuasca tea: mariri/jagube, Banisteriopsis (Malpighiaceae). Genet Resour Crop Evol 70:1605–1616. https://doi.org/10.1007/s10722-022-01522-3
Magdy M, Ouyang B (2020) The complete mitochondrial genome of the chiltepin pepper (Capsicum Annuum Var. Glabriusculum), the wild progenitor of Capsicum Annuum L. Mitochondrial DNA Part B 5:683–684
Magdy M, Ou L, Yu H et al (2019) Pan-plastome approach empowers the assessment of genetic variation in cultivated capsicum species. Hortic Res 6:108
Meunier J, Duret L (2004) Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol 21:984–990
Moore MJ, Bel CD, Soltis PS et al (2007) Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. PNAS 104:19363–19368
Nantongo JS, Gwali S (2018) Long-term viability of populations of Prunus africana ((hook. f.) kalm.) in Mabira forest: implications for in situ conservation. Afr J Ecol 56(1):136–139
Nantongo JS, Eilu G, Geburek T et al (2016) Detection of self-incompatibility genotypes in Prunus africana: characterization, evolution and spatial analysis. PLoS ONE 11(6):e0155638
Nantongo JS, Potts BM, Hugh F et al (2020) Quantitative genetic variation in bark stripping of Pinus radiata. Forests 11(12):1356
Neuhaus H, Emes M (2000) Nonphotosynthetic metabolism in plastids. Annu Rev Plant Biol 51:111
Nkansa-Kyeremateng K (1992) Ghana herbal pharmacopoeia. Policy Research and Strategic Planning Institute (PORSPI), Accra
Odintsova MS, Yurina NP (2006) Chloroplast genomics of land plants and algae. In: Giardi MT, Piletska EV (eds) Biotechnological applications of photosynthetic proteins: biochips, biosensors and biodevices; biotechnology intelligence unit. Springer, Boston, pp 57–72
Oldenburg DJ, Bendich AJ (2016) The linear plastid chromosomes of maize: terminal sequences, structures, and implications for DNA replication. Curr Genet 62:431–442
Orwa C, Mutua A, Kindt R (2009) Agroforestree database: a tree reference and selection guide. Version 4
Palmer JD (1985) Comparative organization of chloroplast genomes. Annu Rev Genet 19:325–354
Park I, Yang S, Choi et al (2017) The complete chloroplast genome sequences of Aconitum pseudolaeve and Aconitum longecassidatum, and development of molecular markers for distinguishing species in the Aconitum Subgenus Lycoctonum. Molecules 22:2012
Parvez A, Azad AK, Islam MZ et al (2019) A phytochemical and pharmacological review on Trema oreintalis: a potential medicinal plant. Pharmacologyonline 3:103–119
Pereira GS, Nunes ES, Laperuta LDC et al (2013) Molecular polymorphism and linkage analysis in sweet passion fruit, an outcrossing species. Ann Appl Biol 162:347–361
Pezoa I, Villacreses J, Rubilar M et al (2021) Generation of chloroplast molecular markers to differentiate Sophora toromiro and its hybrids as a first approach to its reintroduction in rapa nui (Easter Island). Plants 10:342
Raes J, Van de Peer Y (2005) Functional divergence of proteins through frameshift mutations. Trends Genet 21:428–431
Richardson LGL, Schnell DJ (2020) Origins, function, and regulation of the TOC–TIC general protein import machinery of plastids. J Exp Bot 71(4):1226–1238
Ruhlman TA, Jansen RK (2014) The plastid genomes of flowering plants. In: Maliga P (ed) Chloroplast biotechnology methods in molecular biology, vol 1132. Humana Press, Totowa, pp 3–38
Schippmann U, Leaman DJ, Cunningham A (2002) Impact of cultivation and gathering of medicinal plants on biodiversity: global trends and issues. Biodiversity and the ecosystem approach in agriculture, forestry and fisheries Satellite event on the occasion of the Ninth regular session of the commission on genetic resources for food and agriculture Rome. Inter-departmental Working Group on Biological Diversity for Food and Agriculture, Rome. FAO2002
Skuza L, Szućko I, Filip E (2019) Genetic diversity and relationship between cultivated, weedy and wild rye species as revealed by chloroplast and mitochondrial DNA non-coding regions analysis. PLoS ONE 14:e0213023
Smith C (1966) Common names of South African plants. Pretoria: Department of Agricultural Technical Services. Botanical Research Institute. Botanical Survey Memoir, vol 35
Sugiura M (1995) The chloroplast genome. Essays Biochem 30:49–57
Sun JL, Han Y, Cui XM (2020) Development and application of chloroplast molecular markers in Panax notoginseng. Zhong Yao Cai 45:1342–1349
Sytsma KJ, Morawetz J, Pires JC (2002) Urticalean rosids: circumscription, rosid ancestry, and phylogenetics based on rbcL, trnL-trnF, and ndhF sequences. Am J Bot 89:1531–1546
Tangphatsornruang S, Sangsrakru D, Chanprasert J et al (2010) The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: structural organization and phylogenetic relationships. DNA Res 17:11–22
Tillich M, Lehwark P, Pellizzer T et al (2017) GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res 45:W6–W11
Wang X, Zhou T, Bai G (2018) Complete chloroplast genome sequence of Fagopyrum Dibotrys: genome features, comparative analysis and phylogenetic relationships. Sci Rep 8:1–12
Wariss HM, Yi TS, Wang H et al (2018) The chloroplast genome of a rare and an endangered species Salweenia bouffordiana (Leguminosae) in China. Conserv Genet Resour 10:405–407
Weng ML, Blazier JC, Govindu M (2014) Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol Biol Evol 31:645–659
Williams AV, Miller JT, Small I (2016) Integration of complete chloroplast genome sequences with small amplicon datasets improves phylogenetic resolution in Acacia. Mol Phylogenet Evol 96:1–8
Wu ML, Yan RR, Xu X et al (2023) Characterization of the plastid genome of the vulnerable endemic Indosasa lipoensis and phylogenetic analysis. Diversity 15:197
Yang MQ, van Velzen R, Bakker TF (2013) Molecular phylogenetics and character evolution of Cannabaceae. Taxon 62:473–485
Yang Z, Wang G, Ma Q et al (2019) The complete chloroplast genomes of three Betulaceae species: implications for molecular phylogeny and historical biogeography. PeerJ 7:e6320
Yu X, Wang W, Yang H et al (2021) Transcriptome and comparative chloroplast genome analysis of Vincetoxicum versicolor: insights into molecular evolution and phylogenetic implication. Front Genet 12:602528
Zhang H, Jin J, Moore MJ et al (2018) Plastome characteristics of Cannabaceae. Plant Divers 23 40(3):127–137
Zhao Z, Gao A, Huang J et al (2021) Screening of sweet wampee [Clausena lansium (Lour.) Skeels] progenies in the early growth stage based on chloroplast genome analysis. Genet Resour Crop Evol 68:1747–1750. https://doi.org/10.1007/s10722-021-01134-3
Zhou T, Zhao J, Chen C (2016) Characterization of the complete chloroplast genome sequence of Primula Veris (Ericales: Primulaceae). Conserv Genet Resour 8:455–458
Acknowledgements
All authors thank Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2023R402), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Funding
Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R402), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
AAI and DA conceived and designed the experiments; AAI Analyzed the data; AAI summarized the data; AAI and DA wrote the manuscript; All authors revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ibrahim, A.A., Alwutayd, K.M., Safhi, F.A. et al. Characterization and comparative genomic analyses of complete chloroplast genome on Trema orientalis L.. Genet Resour Crop Evol 71, 1085–1099 (2024). https://doi.org/10.1007/s10722-023-01678-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-023-01678-6