
Abstract
Soybean mosaic virus (SMV) is a member of genus Potyvirus, which causes worldwide soybean [Glycine max (L.) Merr.] yield loss and seed quality deterioration. It is of great significance to find new resistance loci and genes for cultivation of soybean variety. In the present study, a recombinant inbred line (RIL) population and a genome-wide association study (GWAS) panel, which contained 193 lines and 379 germplasms, respectively, were used for QTL mapping of resistance to SMV. Linkage mapping identified a major QTL, qSMV13, on chromosome 13, conferring resistance to SMV SC3 and SC7 strains, explaining phenotypic variations 71.21 and 76.59 %, respectively. The QTL qSMV13 was located close to the known SMV resistance loci Rsv1-h. GWAS analysis revealed five single nucleotide polymorphisms (SNPs) significantly associated with resistance to SC3 on chromosomes 2, 11, 13, 14 and 16. One of the SNP markers, ss715614844, was the right flanking marker of qSMV13. Combining linkage mapping and GWAS analysis enabled us to delimit qSMV13 in a 97.2-kb genomic region containing seven genes. A LRR-RLK protein was proposed as the candidate gene of qSMV13. These results provided selection markers and candidate genes for SMV resistance in soybean molecular breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soybean [Glycine max (L.) Merr.] is an important protein and oil crop in the world. Soybean mosaic virus (SMV) belongs to the genus Potyvirus in the family of Potyviridae (Adams et al. 2005; Lin et al. 2020b). Plant infected with SMV not only presents typical symptoms such as leaf shrinkage, necrosis, plant dwarfing and seed mottling, but also leads to a serious reduction in seed yield and deterioration in seed quality (Wrather et al. 2001). The host defenses to the SMV could be divided into two broad categories, resistance and tolerance, based on the responses to viral infection (Lin et al. 2020b). In the USA, according to the disease reactions of two sensitive and six resistant soybean cultivars, 98 SMV isolates were classified into seven strains, namely G1 to G7 (Cho and Goodman 1979, 1982). In China, based on SMV isolates reaction of ten soybean differentials, SMV isolates collected from 2003 to 2010 were grouped into 22 strains (SC1 to SC22) (Guo et al. 2005; Li et al. 2010; Wang et al. 2003; Zhan et al. 2006). Among these strains, SC3 is a prime virus strain, and SC7 is an avirulent strain in Huang-Huai-Hai eco-region in China (Wang et al. 2003).
Up to now, there is no effective chemical method to control virus disease and the most cost-effective and eco-friendly strategy is to cultivate resistant varieties. Many resistance loci to SMV different strains have been reported. In America, three distinct SMV resistance loci (Rsv1, Rsv3 and Rsv4) have been identified and mapped on soybean chromosomes 13, 14, and 2, respectively (Gore et al. 2002; Hayes et al. 2000; Hayes and SaghaiMaroof 2000; Jeong and SaghaiMaroof 2004; SaghaiMaroof et al. 2010; Yu et al. 1994). Recently, a new Rsv5 locus was designated in soybean germplasm “York” as the resistance gene to replace the original Rsv1-y allele allocation on chromosome 13 (Klepadlo et al. 2017). In China, most SMV resistance loci have been positioned on chromosomes 2, 13, 14, and 6 (Fu et al. 2006; Li et al. 2006a; Ma et al. 2011; Wang et al. 2011a, b; Yang and Gai 2011; Yang et al. 2013). Karthikeyan et al. (2017, 2018) reported resistance loci to SMV strains SC5 and SC20 on chromosome 2 and chromosome 13, respectively. Yang et al. (2013) mapped resistance loci to SMV strains SC3, SC6 and SC7 in the neighbor of Rsv1 locus. Wang et al. (2011a) identified resistance loci to SC8 near Rsv4 locus. Yan et al. (2015) described resistance loci to SC7 on chromosome 2 in Kefeng 1. Meanwhile, Wang et al. (2018) summarized the QTL related to SMV resistance in soybean, and found that the resistance loci of different SMV strains were mainly located on five chromosomes (Gm02, Gm06, Gm10, Gm13 and Gm14). Further analysis revealed that there were ten loci (Rsv1, RSC3Q and RSC12, etc.) on chromosome 13, eight loci (Rsv4, RSC6 and RSC8, etc.) on chromosome 2, two loci (RSC15 and RSC18) on chromosome 6 and two loci (Rsv3 and RSC4) on chromosome 14.
In contrast to specific resistance to SMV, horizontal resistance (often referred to as tolerance) was also reported (Zhi et al. 2005), which was typically more durable and achieved by multiple genes. Zhi et al. (2005) revealed one additive major gene plus additive-dominant polygenes responsible for the horizontal resistance to Sa strain. Lin et al. (2020b) detected one QTL (qTsmv-13) for resistance and two QTL (qTsmv-2 and qTsmv-3) for tolerance to one recombinant SMV strain. Besides the linkage mapping strategy to mine the genetic loci for SMV, GWAS is also an important approach that has been successfully applied to identify loci associated with SMV. Yan et al. (2015) identified 19 SNPs associated with disease rate of SC7 in a natural population. Che et al. (2017) reported 104 SNPs significantly associated with resistance to SC7 in a soybean mutant panel. Che et al. (2020) also found 24 SNPs significantly associated with resistance to SC3 in a natural population.
A lot of work has been done on the detecting of SMV resistance loci and the prediction of candidate genes. However, only a few SMV resistance related loci and genes have been applied to date. The reason is partially due to the diversity of SMV strains in soybean production and the complicated infection of various SMV strains. So, more resistance loci and genes should be explored in soybean breeding program. In the present study, QTL mapping for SMV resistance was performed using a population consisting 193 RIL lines with SoySNP6K array genotyping data and phenotyping results of disease index (DI) inoculated with SMV strains SC3 and SC7. Meanwhile, GWAS was also performed using a natural population of 379 germplasms genotyped with the same SNP array and phenotyped with DI of SC3. The objectives were to utilize the linkage and genome-wide association mapping strategies to identify genetic loci and candidate genes that were associated with resistance to SC3 and SC7. The results will enrich useful SC3 and SC7 resistance genes and offer markers for soybean molecular breeding.
Materials and methods
Soybean materials and SMV strains
For QTL mapping, one recombinant inbred line (RIL) population was used in this study. The RIL population was derived from a cross between “C813” × “Kennong7” and consisted of 193 F6:8 RILs. “C813” is susceptible to SC3 and SC7 with DI 83.6 and 69.9, respectively, while “Kennong7” is resistant to SC3 and SC7 with DI 3.6 and 14.8, respectively (Table 1).
The GWAS panel consists of 379 diverse soybean germplasms mainly from Huang-Huai-Hai eco-region in China, including 302 cultivars and 77 landraces (with “Kennong7” and “C813” included). Detailed information of the GWAS panel was given in Appendix A.
SMV SC3 and SC7 strains were provided by the National Center for Soybean Improvement, Nanjing Agricultural University, China, and were cultured on SMV-susceptible host Nannong 1138-2.
Methods
Population planting and SMV inoculation
The seedlings of the RIL population and the GWAS panel were planted in round plastic pots (diameter × depth: 30 cm × 25 cm) which were filled with vermiculite and nutrient soil (3 : 1) in an aphid-free greenhouse with 14 h of supplemental light (~ 10,000 lx) per day, and room temperature 26 °C ± 1 °C at Hebei Agricultural University. For each line, 30 seedlings were used for each replicate. Three biological replicates were conducted for the RIL population inoculated with SC3 and SC7, and two biological replicates were used for GWAS panel inoculated with SC3.
The Nannong 1138-2 fresh leaves infected by SC3 or SC7 were grinded in 0.01 mol/L sodium phosphate buffer (approx.10mL per g leaf tissue, pH 7.4), using mortars and pestles. Seedlings were manually inoculated with the inoculum by rubbing unfolded primary leaves at the V1 stage and were inoculated again on the first trifoliate leaf at the V2 stage.
Resistance evaluation to SC3 and SC7 strains
Resistances to SC3 and SC7 strains were evaluated by scoring disease index one month after inoculation as described in Zhi et al. (2005). Disease index under 35 was defined as resistant accession and disease index over 35 was defined as susceptible accession. The statistical descriptive analysis was performed using SPSS V19.0 software. The disease index was transformed via the arcsine function, and the analysis of variance was conducted by using the transformed data via the SPSS V19.0 software. The broad-sense heritability was estimated via the transformed data according to the method described by Nyquist and Baker (1991).
where Si was disease severity; fi was the number of plants with the Si; Smax was the highest Si; n was total number of surveyed plants.
Construction of linkage map and QTL mapping
The RIL population used in this study was genotyped by SoySNP6K array (the core subset selected from the SoySNP50K Illumina Bead Chip array), which contained 5403 SNPs and could represent major linkage disequilibrium (LD) blocks across the soybean genome (Akond et al. 2013, 2015; Wen et al. 2014).
The polymorphic SNP markers were imported into Joinmap software (v4.0) for construction of linkage map according to the previously described procedures (Ooijen 2006). Maximum likelihood algorithm and an independence LOD = 3.0 was used to calculate the distance between markers.
IciMapping version 4.0 (Wang et al. 2014) was used for inclusive composite interval mapping of additive (ICIM-ADD). The threshold of LOD score was 4.0 for statistical significance of QTL effects. The MapChart 2.3 software (Voorrips 2002) was used to graph the linkage map and analyze QTLs.
Population genetic analysis of GWAS panel
The natural population was genotyped by SoySNP6K array as above. The GWAS panel structure was analyzed by STRUCTURE 2.3.4 software (Evanno et al. 2005). Three independent runs were conducted and the K value (number of sub-groups) was from 1 to 12. Burn-in time and MCMC (Markov chain Monte Carlo) replication number were set both to 100,000 according to the previously described procedures (Sonah et al. 2015; Sun et al. 2017). Principal component and neighbor-joining tree analysis were performed using TASSEL 5.0 based on 3863 SNPs with minor allele frequency (MAF) > 20 %. TASSEL 5.0 was used to analyze linkage disequilibrium. The extent of LD decay was chosen by using the method of decreasing r2 to half of the maximum value.
Genome-wide association analysis
Mixed linear model was used to test associations between the SNPs (MAF > 5 %) and DI to SC3. In addition to the SNP and PCA matrix being tested, relative kinship matrix (K) was included as fixed and random effects, respectively. Top five principal components were used to build the P matrix for population structure correction. Analysis was performed with the software TASSEL 5.0. False discovery rate (FDR) ≤ 0.05 was used to identify significant associations between the SNP and DI.
Expression analysis of candidate genes in associated regions
To further verify the genetic loci and candidate genes found in this study controlling SMV resistance, the annotation of the candidate genes were analyzed. Gene annotation and candidate gene identification were based on the GlymaWm82.a2.v1 soybean sequence annotation database (https://www.soybase.org). The expressions of candidate genes were analyzed by using the transcriptome sequencing data of two soybean varieties (Zheng 92116 and Qihuang 30) at 0, 3, 10, 24, 120 and 240 h post inoculation (hpi) of the SC7 strain, which was described in our previous study (Chu et al. 2021).
Meanwhile, the expression analysis of candidate genes at 0, 4, 8, 12, 24, 48, 72 h post-inoculation (inoculated with SC3, mock inoculated with sodium phosphate buffer as control) was performed in resistant accessions (Kennong7, Han5408chun, Yudou12) and susceptible accessions (C813, Huangdou, Weidou267) via real-time qPCR (qRT-PCR) method. The qRT-PCR primers of the candidate genes and housekeeping gene were listed in Appendix B, and methods were similar as our previous study (Kong et al. 2018), with three technical replicates for each sample.
The virus contents were analyzed by detecting the relative expressions of CP gene of SMV as the description in Yuan et al. (2020).
Sequence analysis of candidate gene Glyma.13G184200
The amplification primers of Glyma.13G184200 were designed via the Primer Premier 5.0 software (Premier Biosoft International, Palo Alto, CA) (Appendix B). The coding sequences (CDS) were amplified by using the leaf cDNAs of Kennong7 and C813. PCR products were purified and ligated into the pMD19-T vector (Takara) according to the manufacturer’s protocol. The ligated product was transformed into DH5α competent cells, and the positive clones were sequenced by GENEWIZ (Tianjin, China). The CDS sequences and deduced amino acid sequences were aligned via the DNAMAN software.
Results
Genetic variation of resistance to SC3 and SC7 in the RIL population
The RIL population showed relative wide genetic variations to SC3 and SC7 with the range of DI 0 ~ 97.9 and 0 ~ 100, and the variation coefficient was up to 70.6 and 77.0 %, respectively (Table 1). Histogram analysis showed that the segregation of DI to SC3 and SC7 in the RIL population were both bi-modal distribution, suggesting that the resistance to SC3 and SC7 might be controlled by a major effect gene/QTL (Appendix C). In addition, the broad sense heritability of the resistance to SC3 and SC7 in the RIL population were 84.14 and 79.90 %, respectively (Table 1).
Genetic linkage map construction of the RIL population
The high density molecular genetic linkage map was constructed by the polymorphic SNP markers, which contained a total of 2,234 markers, and were grouped into 23 linkage groups (Appendix D). Chromosome 13 showed the most quantity of markers (193 SNPs) in the linkage groups. The whole map covered a total of 4,229.01 cM genetic distance and the average distance of the individual chromosome ranged from 1.21 cM (chromosome 3) to 3.38 cM (chromosome 4) and the average distance of the whole genetic map was 1.89 cM (Appendix E).
QTL mapping of disease index to SC3 and SC7 in the RIL population
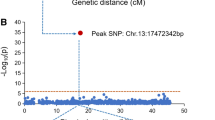
After QTLs analysis basing on disease index, a co-located QTL with LOD values of 51.89 and 64.73 for SC3 and SC7, respectively, designated as qSMV13, was mapped in a narrow region between 142.39 ~ 142.74 cM on chromosome 13. The QTL qSMV13 accounted for 71.21 and 76.59 % of phenotypic variations to SC3 and SC7, respectively. The desirable allele of the QTL for SC3 and SC7 were both derived from the resistant parent “Kennong7”.
Further analysis revealed that qSMV13 were flanked by markers ss715614844 and ss715614864. Marker ss715614844 was nearby the reported SMV resistance genetic loci Rsv1-h (Ma et al. 2016), and marker ss715614864 was just within the Rsv1-h locus (Fig. 1).

QTL qSMV13 and candidate genes detected on chromosome 13 for resistance to SC3 and SC7 strains A, Associated marker on chromosome 13 for resistance to SC3 in GWAS panel B, QTL qSMV13 on chromosome 13 for resistance to SC3 and SC7 in RIL population C, Expressions of the candidate genes in the qSMV13 region and coat protein gene of SMV between resistant and sensitive varieties after inoculation with SC7
Haplotype analysis of the qSMV13 flanking markers ss715614844 and ss715614864
To further verify the QTL obtained in the present study, the haplotype of the qSMV13 flanking markers ss715614844 and ss715614864 were analyzed. The RIL population could be divided into two haplotype groups, CT and TG, according to the SNP calls of the linkage markers. The disease index of CT-group (same genotype as resistance accession “Kennong7”) were 11.8 and 7.9, which were resistance to SC3 and SC7, while the TG-group (genotype as susceptible accession “C813”) were 71.0 and 63.1, which showed sensitive to SC3 and SC7, respectively. These results not only verified the QTL mapping results in this study, but also offered effective favorite allelic variation for marker-assisted selection of anti-SMV in soybean.
Genetic structure and linkage disequilibrium analysis of GWAS panel
The GWAS panel was genotyped by SoySNP6K beadchip as indicated above, which contained 5,403 SNPs. SNPs with MAF of > 0.05 and call rate > 80 % were selected for further analysis and 5,046 high-performing SNPs were obtained.
The GWAS panel was divided into two major subgroups (group1 and group2) according to the STRUCTURE analysis, which was verified by phylogenetic (NJ tree) analysis and PCA analysis (Appendix F). Among the two groups, group 1 contained 308 accessions, and nearly all were attributed to the cultivars. The group 2 contained 71 accessions, and most of those belonged to the landraces (Appendix A).
The average extent of genome-wide LD decay distance in the panel was approximately 210 kb, where the r2 drops to half its maximum value (0.17) (Appendix G), and offered the select region of candidate genes. Linkage disequilibrium decay distance were different between chromosomes, with 320 kb in chromosome 17, 100–200 kb in chromosomes 4, 5, 6, 7, 9, 10, 12, 13 and 15, and 200–300 kb in the remaining 10 chromosomes in the panel.
SNP associated with SC3 resistance in GWAS panel
As shown in the quantile-quantile (QQ) plots, lower inflation of nominal P-value was observed (Appendix H). Using this model, five SNP markers, ss715583175, ss715608741, ss715614844, ss715617664 and ss715625254 were significantly associated (FDR < 0.05) with resistance to SC3 on chromosomes 2, 11, 13, 14 and 16, and explained 6.0–19.0 % of phenotypic variations (Appendix H, Table 2).
Further analysis found that the associated marker ss715614844 was also the flanking marker of qSMV13 on chromosome 13 with PVE 71.21 % in the RIL population and explained 8.1 % of phenotypic variation in the natural population (GWAS panel), indicating consistency of the two mapping strategies.
Identification of resistance genes to SC3 and SC7 strains in qSMV13 region
To predict the candidate genes associated with resistance to SC3 and SC7 strains, the linkage mapping and GWAS analysis results were comprehensively analyzed. The qSMV13 genetic region defined by markers ss715614844 and ss715614864 spanned a 97.2-kb genomic region in the Williams 82 reference genome (Glyma.Wm82.a2.v1) containing seven annotated genes. Five of these genes, Glyma.13G183800, Glyma.13G184000, Glyma.13G184300, Glyma.13G184500 and Glyma.13G184600 showed relatively high expression levels in the resistance variety “Qihuang30” after the SC7 strain inoculation via the transcriptome analysis (Fig. 1). Meanwhile, the virus contents in resistant variety Qihuang30 kept decreasing and relative lower contents than that in sensitive variety Zheng92116 after 3 h post inoculation (Fig. 1).
Meanwhile, the expressions of other two genes, Glyma13g184200 and Glyma13g183900, were analyzed via qRT-PCR method after SC3 strain inoculation. Glyma13g184200 encodes a LRR receptor-like protein kinase and showed relative higher expressions at many time-points in resistant varieties (Kennong7, Han5408chun and Yudou12), but presented relatively higher expression at only one or two time-points in the sensitive varieties (C813, Huangdou and Weidou267). Another gene Glyma13g183900, encoding a zinc finger protein, showed higher expressions at more time-points in sensitive varieties, and presented relatively higher expressions only at 72 hpi in the resistance varieties (Fig. 2).

Expression analysis of the candidate genes to SC3 by qRT-PCR. Y-axes indicated the relative expression ratio of mRNA between samples inoculated with SC3 and phosphate buffer at different time-points (0–72 hpi). Asterisks represented statistical significance (*P < 0.05; **P < 0.01)
In addition, to further analyze the sequence differences of LRR receptor-like (LRR-RLK) protein kinase gene Glyma13g184200 in resistant and susceptible varieties, the parental lines “C813” and “Kennong7” were used to amplify the coding sequences (CDS) of Glyma13g184200. The results showed that there existed a 72-bp insertion in the CDS of C813 compared with Kennong7, which was located in the LRR-RLK domain (Appendix I). Thus, we considered that Glyma13g184200 might be the most likely candidate gene for the SMV resistance in the qSMV13 region on chromosome 13.
Discussion
SMV disease causes yield decrease and quality deterioration and results in 35–50 % of yield loss when seriously occurred in soybean field (Wrather et al. 2001). QTL analysis is used to detect traits associated genetic loci by linkage mapping and genome-wide association analysis. However, the linkage mapping method can only investigate very limited variations from the two parents in the genetic population, and the GWAS results usually are affected by the Q value and individual relationships. A combination of both methods could provide a much higher power and resolution to detect QTL or genes that can hardly be detected by either method alone (Goddard 2005). Combination of the two strategies has been successfully applied to analyze complex traits, such as drought tolerance, Pythium resistance, downy mildew resistance, flowering time, seed size and shape (Lin 2020a; Lu et al. 2010; Hu et al. 2013; Nemri et al. 2010; Zhang et al. 2012). But, to date, the combination of both methods to detect the QTL of SMV resistance has not been reported. In this study, one RIL population and one natural population were used to detect the resistance sites of soybean mosaic virus SC7 and SC3 strains, and same marker ss715614844 was detected in the two populations, and the resistance site was further validated through the analysis of annotated genes in the mapping interval using transcriptome analysis and qRT-PCR technology (Figs. 1 and 2).
High marker densities are critical to improve the accuracy of QTL mapping (Song et al. 2016; Cao et al. 2017). By comparison, SNPs have a more abundant DNA variation than traditional markers. In our study, BARCSoySNP6K iSelect BeadChip was used to genotype the RIL population and GWAS panel. BARCSoySNP6K iSelect BeadChip was the core subset selected from the SoySNP50K Illumina Bead Chip array. Based on this BeadChip, Lin et al. (2020a, 2021) identified five QTL to Pythium sylvaticum and two QTL to Phytophthora sansomeana, and 20 SNP loci significantly associated with symbiotic nitrogen fixation-related characteristics were identified in a soybean natural population by GWAS (Huo et al. 2019). Previous studies have shown that the LD of self-pollinated species is higher than the cross-pollinated species, as the maize and rice LD decays less than 1 kb, Arabidopsis thaliana about 3–4 kb, soybean more than 150 kb (Gore et al. 2009; Hao et al. 2012; Lam et al. 2010; Wen et al. 2014; Zhu et al. 2007). In the present study, the average extent of genome-wide LD decay distance in the GWAS panel was approximately 210 kb. Since the soybean genome is known to extend slightly over 1000 Mb, the estimates of LD decay herein suggested at least 4762 (1000 Mb/210 kb) markers will be needed for whole genome scanning in soybean. Thus, we considered that the 5,046 SNPs in BARCSoySNP6K were sufficient for QTL analysis in soybean.
Recently, several SC3 and SC7 resistance loci have been mapped. Yang et al. (2013) reported that the resistance gene for SC3 was positioned between BARCSOYSSR_13_1128 and BARCSOYSSR_13_1136, while the resistance gene for SC7 was mapped between BARCSOYSS R_13_1140 and BARCSOYSSR_13_1155. Li et al. (2017) reported that four candidate genes for resistance to SC3 on chromosome 13 nearby Rsv1 using fine mapping and transcriptome analysis. Che et al. (2017) found SNPs associated with the resistance to SC7 on chromosomes 1, 3, 5, 9, 10, 12, 13, and 15 by GWAS. In our present study, one major QTL, qSMV13, was identified to associate with SC3 and SC7 resistance in the RIL population, and a qSMV13 flanking SNP marker was found to associate with SC3 strain resistance in the natural population (Fig. 1). Furthermore, the qSMV13 is about 126 kb away from ss715614889, a SNP marker that was associated with resistance to SMV G1, G2 and G3 strains (Chang et al. 2016), suggesting qSMV13 may also confer SMV G1, G2 and G3 resistance. In addition, the Chi-square fitness test of the qSMV13 locus based on the symptomless (resistant) and symptomatic (susceptible) of RIL lines (Karthikeyan et al. 2018) revealed a deviation of the 1 : 1 ratio of resistant and susceptible (χ2 = 16.9, 7.4, P < 0.05), indicating that the qSMV13 is a major resistance gene with much higher PVE to SMV and minor effects QTL are also available for the SMV resistance.
More importantly, seven annotated genes were found in the QTL qSMV13 genomic interval. Two of those genes, Glyma.13g184500 encoding bromodomain-containing protein and Glyma.13g184600 encoding eukaryotic membrane family protein, were located within the overlapping region of qSMV13 and Rsv1-h. Another gene, Glyma.13g184200 was predicted to encode a leucine rich repeat receptor-like protein kinase (LRR-RLK). LRR-RLK genes in soybean, such as GsLRPK and GmSARK, were participated in the immune response to pathogen, and also played an important role in the anti-cold and anti-aging (Li et al. 2006b; Yang et al. 2014). Glyma.13g183900 encoded a C3HC4 zinc finger protein that reported to play an important regulatory role in the basic immunity of Arabidopsis to pathogens (Wang et al. 2019). Glyma.13g183800 was predicted to encode esterase/lipase/thioesterase family protein. Lippold et al. (2012) reported esterase/lipase/thioesterase genes, PES1 and PES2, with higher expressions under low nitrogen stress and senescence, and could convert leaf alcohol and fatty acid into phytate to maintain the integrity of photosynthetic membrane in Arabidopsis thaliana. Glyma.13g184300 encoded the transducin/WD40 repeat-like superfamily protein, and this superfamily presented vital functions in plant development and stress signal transduction (Gachomo et al. 2014). Glyma.13g184000 was found to encode integral component of membrane.
Further analysis of these annotated genes showed that five of them were highly induced in resistance variety “Qihuang30” after SC7 inoculation based on the transcriptome analysis (Fig. 1), and the other two, Glyma.13g184200 and Glyma.13g183900, showed different expressions between the resistance varieties (Kennong7, Han5408chun and Yudou128) and sensitive varieties (C813, Huangdou and Weidou267) after SC3 inoculation based on the qRT-PCR analysis (Fig. 2). Moreover, a 72-bp insertion/deletion difference at the LRR-RLK domain sequences of Glyma.13g184200 was detected between C813 and Kennong7. Karthikeyan et al. (2018) reported three insertion/deletion variations and 63 SNPs in Glyma.13G194700, and identified one insertion/deletion variation and 17 SNPs in Glyma.13G195100, which located in the TIR, NB and LRR domains of TIR-NBS-LRR proteins, between the SMV resistant and sensitive soybean varieties. Because the LRR domains are required for the resistance response to pathogens in plants, amino acid variations may cause the function change of a disease resistant protein (Karthikeyan et al. 2018). Collectively, these candidate genes, especially the LRR receptor-like protein kinase Glyma13g184200, worth further functional validation in the future.
Conclusions
In this study, we identified a major QTL qSMV13 conferring resistance to SC3 and SC7 strains and explaining phenotypic variations of 71.21 and 76.59 %, respectively. qSMV13 was defined to a genomic interval of approximately 97.2 kb containing 7 annotated genes on chromosome 13 by linkage mapping, and the flanking marker ss715614844 was further verified for association with SC3 resistance via the genome-wide association analysis. These results offered useful selection markers and candidate genes with resistance to SMV in soybean molecular breeding.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Adams MJ, Antoniw JF, Fauquet CM (2005) Molecular criteria for genus and species discrimination within the family Potyviridae. Arch Virol 150:459–479
Akond M, Liu SM, Schoener L, Anderson J, Kantartzi SK, Meksem K, Song QJ, Wang DC, Wen ZX, Lightfoot DA, Kassem MA (2013) A SNP-based genetic linkage map of soybean using the SoySNP6K Illumina Infinium BeadChip genotyping array. Plant Genet Genom Biotechnol 1:80–89
Akond M, Liu SM, Kantartzi SK, Meksem K, Bellaloui N, Lightfoot DA, Yuan JZ, Wang DC, Anderson J, Kassem MA (2015) A SNP genetic linkage map based on the ‘Hamilton’ by ‘Spencer’ recombinant inbred line population identified QTL for seed isoflavone contents in soybean. Plant Breed 5:580–588
Cao YC, Li SG, He XH, Chang FG, Kong JJ, Gai JY, Zhao TJ (2017) Mapping QTLs for plant height and flowering time in a Chinese summer planting soybean RIL population. Euphytica 213:39
Chang HX, Lipka AE, Domier LL, Hartman GL (2016) Characterization of disease resistance loci in the USDA soybean germplasm collection using Genome-wide association studies. Phytopathology 10:1139–1151
Che ZJ, Liu HL, Yi FL, Cheng H, Yang YM, Wang L, Du JY, Zhang PP, Wang J, Yu DY (2017) Genome-wide association study reveals novel loci for SC7 resistance in a soybean mutant panel. Front Plant Sci 8:1771
Che ZJ, Yan HL, Liu HL, Yang H, Du HP, Yang YM, Liu BH, Yu DY (2020) Genome-wide association study for soybean mosaic virus SC3 resistance in soybean. Mol Breed 40:69
Cho ER, Goodman RM (1979) Strains of soybean mosaic virus: classification based on virulence in resistant soybean cultivars. Phytopathology 69:467–470
Cho ER, Goodman RM (1982) Evaluation of resistance in soybeans to soybean mosaic virus strains. Crop Sci 22:1133–1136
Chu JH, Li WL, Piao DR, Kong YB, Du H, Lin F, Li XH, Zhang CY (2021) Mining of a major QTL and novel genes conferring resistance to SC3 and SC7 strains in soybean. Plant Breed 00:1–9
Evanno GS, Regnaut SJ, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 8:2611–2620
Fu SX, Zhan Y, Zhi HJ, Gai JY, Yu DY (2006) Mapping of SMV resistance gene Rsc-7 by SSR markers in soybean. Genetica 128:63–69
Gachomo EW, Jimenez-Lopez JC, Baptiste L (2014) GIGANTUS1 (GTS1), a member of Transducin/WD40 protein superfamily, controls seed germination, growth and biomass accumulation through ribosome-biogenesis protein interactions in Arabidopsis thaliana. BMC Plant Biol 1:37–37
Goddard M (2005) The use of association to map quantitative trait loci. Aust J Exp Agric 45:837–845
Gore MA, Chia JM, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, Grills GS, Ross-Ibarra J, Ware DH, Buckler ES (2009) A first-generation haplotype map of maize. Science 326:1115–1117
Gore MA, Hayes AJ, Jeong SC, Yue YG, Buss GR, SaghaiMaroof MA (2002) Mapping tightly linked genes controlling potyvirus infection at the Rsv1 and Rpv1 region in soybean. Genome 45:592–599
Guo DQ, Zhi HJ, Wang YW, Gai JY, Zhou XA, Yang CL, Li K, Li HC (2005) Identification and distribution of soybean mosaic virus strains in Middle and Northern Huang Huai Region of China. Chinese Journal of Oil Crop Sciences 27:64–68. (in Chinese)
Hao DR, Chao MN, Yin ZT, Yu DY (2012) Genome-wide association analysis detecting significant single nucleotide polymorphisms for chlorophyll and chlorophyll fluorescence parameters in soybean (Glycine max) landraces. Euphytica 186:919–931
Hayes AJ, Ma GR, Buss GR, SaghaiMaroof MA (2000) Molecular marker mapping of Rsv4, a gene conferring resistance to all known strains of soybean mosaic virus. Crop Sci 40:1434–1437
Hayes AJ, SaghaiMaroof MA (2000) Targeted resistance gene mapping in soybean using modified AFLPs. Theor Appl Genet 100:1279–1283
Hu ZB, Zhang HR, Kan GZ, Ma DY, Zhang D, Shi GX, Hong DL, Zhang GZ, Yu DY (2013) Determination of the genetic architecture of seed size and shape via linkage and association analysis in soybean (Glycine max L. Merr.). Genetica 141:247–225
Huo XB, Li XH, Du H, Kong YB, Tian R, Li WL, Zhang CY (2019) Genetic loci and candidate genes of symbiotic nitrogen fixation–related characteristics revealed by a genome-wide association study in soybean. Mol Breeding 39:127
Jeong SC, SaghaiMaroof MA (2004) Detection and genotyping of SNPs tightly linked to two disease resistance loci, Rsv1 and Rsv3, of soybean. Plant Breed 123:305–310
Karthikeyan A, Li K, Jiang H, Ren R, Li C, Zhi HJ, Chen SY, Gai JY (2017) Inheritance, fine-mapping, and candidate gene analyses of resistance to soybean mosaic virus strain SC5 in soybean. Mol Genet Genom 292:811–822
Karthikeyan A, Li K, Li C, Yin JL, Li N, Wang YH, Song YP, Ren R, Zhi HJ, Gai JY (2018) Fine–mapping and identifying candidate genes conferring resistance to Soybean mosaic virus strain SC20 in soybean. Theor Appl Genet 131:461–476
Klepadlo M, Chen P, Shi A, Mason RE, Korth KL, Srivastava V, Wu C (2017) Two tightly linked genes for soybean mosaic virus resistance in soybean. Crop Sci 57:1844–1853
Kong YB, Li XH, Wang B, Li WL, Du H, Zhang CY (2018) The soybean purple acid phosphatase GmPAP14 predominantly enhances external phytate utilization in plants. Front Plant Sci 9:292
Lam HM, Xu X, Liu X, Chen WB, Yang GH, Wong FL, Li MW, He WM, Qin N, Wang B, Li J, Jian M, Wang J, Shao GH, Wang J, Sun SSM, Zhang GY (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42:1053–1059
Li C, Adhimoolam K, Yuan Y, Yin JL, Ren R, Yang YQ, Zhi HJ (2017) Identification of candidate genes for resistance to Soybean mosaic virus strain SC3 by using fine mapping and transcriptome analyses. Crop Pasture Sci 68:156–166
Li HC, Zhi HJ, Gai JY, Guo DQ, Wang YW, Li K, Bai L, Yang H (2006a) Inheritance and gene mapping of resistance to soybean mosaic virus strain SC14 in soybean. J Integr Plant Biol 48:1466–1472
Li K, Yang QH, Zhi HJ, Gai JY (2010) Identification and distribution of soybean mosaic virus strains in Southern China. Plant Dis 94:351–357
Li XP, Gan R, Li PL, Ma YY, Zhang LW, Zhang R (2006b) Identification and functional characterization of a leucine-rich repeat receptor-like kinase gene that is involved in regulation of soybean leaf senescence. Plant Mol Biol 61:829–844
Lin F, Li WL, McCoy AG, Gao X, Collins PJ, Zhang N, Wen ZX, Cao SZ, Wani SH, Gu CH, Chilvers MI, Wang DC (2021) Molecular mapping of quantitative disease resistance loci for soybean partial resistance to Phytophthora sansomeana. Theor Appl Genet 134:1977–1987
Lin F, Wani SH, Collins PJ, Wen ZX, Li WL, Zhang N, McCoy AG, Bi YD, Tan RJ, Zhang SC, Gu CH, Chilvers MI, Wang DC (2020a) QTL mapping and GWAS for identification of loci conferring partial resistance to Pythium sylvaticum in soybean (Glycine max (L.) Merr). Mol Breed 40:54
Lin J, Lan ZJ, Hou WH, Yang CY, Wang DG, Zhang MC, Zhi HJ (2020b) Identification and fine-mapping of a genetic locus underlying soybean tolerance to SMV infections. Plant Sci 292:110367
Lu Y, Zhang S, Shah T, Xie C, Hao Z, Li X, Farkhari M, Ribaut JM, Cao M, Rong T, Xu Y (2010) Joint linkage-linkage disequilibrium mapping is a powerful approach to detecting quantitative trait loci underlying drought tolerance in maize. Proc Natl Acad Sci USA 107:19585–19590
Ma FF, Wu XY, Chen YX, Liu YN, Shao ZQ, Wu P, Wu M, Liu CC, ·WuWP,·Yang JY, ·Li DX, ·Chen JQ, ·Wang B (2016) Fine mapping of the Rsv1–h gene in the soybean cultivar Suweon 97 that confers resistance to two Chinese strains of the soybean mosaic virus. Theor Appl Genet 129:2227–2236
Ma Y, Wang DG, Li HC, Zheng GJ, Yang YQ, Li HW, Zhi HJ (2011) Fine mapping of the Rsc14Q locus for resistance to soybean mosaic virus in soybean. Euphytica 181:127–135
Maroof MAS, Tucker DM, Skoneczka JA, Bowman BC, Tolin SA (2010) Fine mapping and candidate gene discovery of the soybean mosaic virus resistance gene, Rsv4. Plant Genome 3:14–22
Nemri A, Atwell S, Tarone AM, Huang YS, Zhao K, Studholme DJ, Nordborg M, Jones JD (2010) Genome-wide survey of Arabidopsis natural variation in downy mildew resistance using combined association and linkage mapping. Proc Natl Acad Sci USA 107:10302–10307
Nyquist WE, Baker RJ (1991) Estimation of heritability and prediction of selection response in plant populations. Critical Reviews in Plant Science 10:235–322
Ooijen JV (2006) JoinMap4.0 software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Sonah H, O’Donoughue L, Cober E, Rajcan I, Belzile F (2015) Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol J 2:211–221
Song QJ, Jenkins J, Jia GF, Hyten DL, Pantalone V, Jackson SA, Schmutz J, Cregan PB (2016) Construction of high resolution genetic linkage maps to improve the soybean genome sequence assembly Glyma1.01. BMC Genom 17:33
Sun ZW, Wang XF, Liu ZW, Gu QS, Zhang Y, Li ZK, Ke HF, Yang J, Wu JH, Wu LQ, Zhang GY, Zhang CY, Ma ZY (2017) Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol J 8:982–996
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang XQ, Gai JY, Pu ZQ (2003) Classification and distribution of strain groups of soybean mosaic virus in middle and lower Huang-Huai and Changjiang valleys. Soybean Science 22:102–107. (in Chinese)
Wang DG, Ma Y, Yang YQ, Liu N, Li C, Song YP, Zhi HJ (2011a) Fine mapping and analyses of RSC8 resistance candidate genes to soybean mosaic virus in soybean. Theor Appl Genet 122:555–565
Wang DG, Ma Y, Liu N, Yang ZL, Zheng GJ, Zhi HJ (2011b) Fine mapping of resistance to soybean mosaic virus strain SC4 in soybean based on genomic-SSR markers. Plant Breeding 130:653–659
Wang JK, Li HH, Zhang Y, Meng L (2014) Users’ Manual of QTL IciMapping v4.0. Institute of Crop Science, CAAS, Beijing
Wang DG, Li K, Zhi HJ (2018) Progresses of resistance on soybean mosaic virus in soybean. Scientia Agricultura Sinica 51:3040–3059. (in Chinese)
Wang YZ, Li YS, Rosas-Diaz T, Caceres-Moreno C, Lozano-Duran R, Macho AP (2019) The IMMUNE-ASSOCIATED NUCLEOTIDE-BINDING 9 protein is a regulator of basal immunity in Arabidopsis thaliana. Molecular Plant-Microbe Interactions 32:65-75
Wen ZX, Tan RJ, Yuan JZ, Bales C, Du WY, Zhang SC, Chilvers MI, Schmidt C, Song QJ, Cregan PB, Wang DC (2014) Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genom 15:809
Wrather JA, Anderson TR, Arsyad DM, Tan Y, Ploper LD, Porta-Puglia A, Ram HH, Yorinori JT (2001) Soybean disease loss estimates for the top ten soybean-producing countries in 1998. Can J Plant Path 23:115–121
Yan HL, Wang H, Cheng H, Hu ZB, Chu SS, Zhang GZ, Yu DY (2015) Detection and fine-mapping of SC7 resistance genes via linkage and association analysis in soybean. J Integr Plant Biol 57:722–729
Yang L, Wu KC, Gao P, Liu XJ, Li GP, Wu ZJ (2014) GsLRPK, a novel cold activated leucine-rich repeat receptor-like protein kinase from Glycine soja, is a positive regulator to cold stress tolerance. Plant Sci 215:19–28
Yang QH, Gai JY (2011) Identification, inheritance and gene mapping of resistance to a virulent Soybean mosaic virus strain SC15 in soybean. Plant Breed 130:128–132
Yang Y, Zheng G, Han L, Wang DG, Yang X, Yuan Y, Huang S, Zhi H (2013) Genetic analysis and mapping of genes for resistance to multiple strains of Soybean mosaic virus in a single resistant soybean accession PI 96983. Theor Appl Genet 126:1783–1791
Yu YG, SaghaiMaroof MA, Buss GR, Maughan PJ, Tolin SA (1994) RFLP and micro satellite mapping of a gene for soybean mosaic virus resistance. Phytopathology 84:60–64
Yuan Y, Yang YQ, Shen YC, Yu KS, Wang LQ, Ren R, Yin JL, Zhi HJ (2020) Mapping and functional analysis of candidate genes involved in resistance to soybean (Glycine max) mosaic virus strain SC3. Plant Breeding 139:618-625
Zhan Y, Zhi HJ, Yu DY, Gai JY (2006) Identification and distribution of SMV strains in Huang-Huai valleys. Scientia Agricultura Sinica 39:2009–2015. (in Chinese)
Zhang D, Cheng H, Hu ZB, Wang H, Kan GZ, Liu CY, Yu DY (2012) Fine mapping of a major flowering time QTL on soybean chromosome 6 combining linkage and association analysis. Euphytica 191:23–33
Zhi HJ, Gai JY, He XH (2005) Inheritance of resistance in infection and resistance in development to soybean mosaic virus in soybeans. Acta Agronomica Sinica 31:1260–1264. (in Chinese)
Zhu QH, Zheng XM, Luo JC, Gaut BS, Ge S (2007) Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: severe bottleneck during domestication of rice. Mol Biol Evol 24:875–888
Funding
This study was supported by the Project of Hebei province Science and Technology Support Program (17927670 H, 16227516D-1) and the Natural Science Foundation of Hebei Province, China (C2020204042).
Author information
Authors and Affiliations
Contributions
CZ designed the research; JC, WL, DP, XH, HZ, HD, YK, YJ, XL performed experiments; JC, WL and FL analyzed data; JC and WL wrote the manuscript; CZ and XL revised the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
The experiments were performed in compliance with the current laws of China.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chu, J., Li, W., Piao, D. et al. Identification of a major QTL related to resistance to soybean mosaic virus in diverse soybean genetic populations. Euphytica 217, 176 (2021). https://doi.org/10.1007/s10681-021-02907-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-021-02907-8