
Abstract
This study presents a new method for the determination of nickel in aqueous samples by slotted quartz tube-flame atomic absorption spectrometry (SQT-FAAS) after a dispersive assisted simultaneous complexation and extraction (DASCE) process. Synthesized ligand was directly dissolved in the extraction solvent to eliminate the complex formation step prior to the extraction. All parameters of the SQT-FAAS and DASCE method were systematically optimized to improve the detection power of nickel for trace determinations. Under the optimum experimental conditions, the optimized method (DASCE-SQT-FAAS) recorded 137-fold enhancement in detection power over the conventional FAAS. The limits of detection and quantification were determined to be 1.6 μg/L and 5.2 μg/L, respectively. The calibration plot was linear over a wide concentration range and the precision for replicate measurements was appreciably high. Nickel was not detected in five different water samples but spiked recovery tests for three samples yielded results that were close to 100%, confirming the method’s accuracy and applicability to the matrices tested.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nickel is known as a transition element which forms alloys with considerable strength and ductility and shows resistance to heat and corrosion (Haudet et al. 2015). It is therefore mostly used for computer components, dental and surgical prosthesis, electroplating, magnetic tapes, and Ni-Cd batteries. Ni accumulates in the environment through processes such as volcano eruptions, soil and rock dissolution, biological cycles, atmospheric fallout from industrial plants, and waste disposal (Hol et al. 2014). The main modes of exposure to Ni are dermal contact, ingestion, and inhalation, where cigarettes and foods are the highest exposure routes to humans with typical concentrations ranging between 0.64–1.2 μg/g and 0.02–13 μg/g, respectively (Reclo et al. 2017; Zambelli et al. 2016). Dermatitis (chronic skin disorder) is the most common health complication associated with Ni but carcinogenesis has also been reported (Büyükpınar et al. 2017). The World Health Organization (WHO) has set an allowable limit of Ni concentration as 0.07 mg/L in water samples. This element is usually detected in most soils, animal tissues, plants, and water at trace levels and, therefore, accurate and precise analytical methods for its determination are required (Weldeabzgi et al. 2017).
In literature, there are many determination methods for the determination of this element. Accuracy and precision are the main factors for the selection of proper instrument for variety of matrices. Spectroscopic methods working on the principles of atomic absorption and atomic emission are widely used for the determination of this element and other metals (IUPAC 1998). Inductively coupled plasma (ICP) is a very useful source for atomization, ionization, and excitation of elements for mass spectrometry (ICP-MS) and optical emission spectrometry (ICP-OES) systems (Dos Anjos et al. 2018; Peeters et al. 2017). These plasma techniques exhibit high sensitivity for most elements and are capable of simultaneous determination of multi elements (Yang et al. 2011). Atomic absorption spectrometry methods based on electrothermal atomization (ETAAS) and photochemical vapor generation (PVG) also provide sensitive determinations but they are only capable of determining only one element at a time (Bidabadi et al. 2009; Büyükpınar et al. 2017). Besides spectroscopic methods, electroanalytical methods such as anodic or cathodic adsorptive stripping voltammetry offer high selectivity and sensitivity for metal determinations (Gonzalez 2002; Mettakoonpitak et al. 2017). For routine laboratory analysis of metals in high concentrations, flame atomic absorption spectrometry (FAAS) is a preferred technique due to its simplicity, robustness, and low operational cost (Özzeybek et al. 2017). However, due to the low sample nebulization efficiency and the relatively low residence time of atoms in the optical path, it suffers from low sensitivity for trace determination of most elements in comparison to the techniques mentioned above. In order to overcome this drawback, preconcentration methods have been studied. Slotted quartz tubes (SQTs) are basic components that can be attached to the flame unit of FAAS to improve the detection power. Under fuel-lean conditions (low flame temperature), atoms adsorb onto the inner walls of the quartz tube and the amount of adsorbed atoms increase until the surface is saturated (Özzeybek et al. 2017). A sudden increase in flame temperature using low amounts of organic solvents or the use of hydrogen gas to provide a reducing environment leads to the release of trapped atoms to obtain a sharp analytical signal (Uslu et al. 2018). In addition to atom trap studies, SQTs with both entry and exit slots are used to increase the residence time of atoms in the flame, thereby increasing their interaction with analyte radiation coming from source. Depending on the type of element, the absorbance signal can be increased by two- to fivefolds using this simple component (Özzeybek et al. 2017). There are several microextraction methods developed for separation of analytes from sample matrix and significantly building up analyte amount for trace and ultratrace determinations. Some of these include solid-liquid-solid dispersive extraction ionic liquid-based dispersive liquid-liquid microextraction (SLSDE-IL-DLLME), cloud point extraction (CPE), solidified floating organic drop microextraction (SFODME), solid-phase microextraction (SPME), and single drop microextraction (SDME) (Jalbani and Soylak 2015b; Kocot et al. 2016; Viñas et al. 2015; Wang et al. 2013). Dispersive liquid-liquid microextraction (DLLME) is a relatively new microextraction method which is widely used to extract both organic and inorganic analytes from a variety of aqueous matrices (Kocot et al. 2016; Rezaee et al. 2006). In DLLME, an organic solvent miscible with both extractant and aqueous solution is used to increase the surface area by dispersing the extractant as fine droplets throughout the sample solution (Chormey et al. 2017). This results in high extraction efficiencies and the extraction is completed within a few seconds after injecting the dispersive and extractant mixture. There are very few ligandless extraction methods in literature for metal determinations (Alothman et al. 2012; Reclo et al. 2017). Extraction procedures are generally preceded by metal complexation with an appropriate ligand. In this study, a laboratory synthesized ligand was dissolved in chloroform with the aim of performing simultaneous complexation and extraction. The method namely dispersive assisted simultaneous complexation and extraction (DASCE) was comprehensively optimized and coupled to SQT-FAAS for trace determination of nickel in aqueous matrices.
Material and methods
Instruments
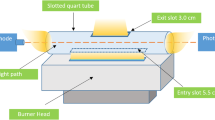
In the determination of nickel, an Analytik Jena NovAA 300 model atomic absorption spectrometer with a flame unit was employed. A 13-cm long quartz tube with 18-mm i.d and 20-mm o.d was attached to the flame burner head. A rotary cutting tool was used to make an entry slot of 5.5 cm to fit the flame length of 5.0 cm, and a 3.0-cm exit slot was cut at 180° to the entry slot. The 232-nm wavelength of Ni was selected from a multi-element hollow cathode lamp (Fe, Co, Ni, Mn, Cu, Cr) operating at 10 mA and a monochromator spectral bandpass of 0.50 nm. Deuterium hollow cathode lamp (D2) was used for background correction. A schematic diagram of an SQT attached flame burner head system is presented in Fig. 1.

Schematic diagram of SQT attached flame burner head (Erarpat et al. 2017)
Reagents
Unless stated otherwise, all chemicals and reagents used in this study were of analytical grade. Working and calibration standard solutions were prepared from a 1000 mg L−1 stock standard solution of nickel purchased from High Purity Standards. Chloroform, sodium chloride, nitric acid (65%), 2-propanol, ethanol, methanol, hydrochloric acid (37%), sodium hydroxide, potassium chloride, 5-bromosalisiladehyde, p-toluidine, para-toluene sulphonic acid, and potassium nitrate were purchased from Merck (Darmstadt, Germany). Ultrapure water (resistivity 18.2 Ω cm) generated by a Milli-Q Reference Water Purification System at the Central Laboratory of Yıldız Technical University was used for aqueous standard preparations and all cleanup purposes.
Ligand synthesis
The ligand used in this study, (Z)-3-bromo-5-((p-tolylimino)methyl)phenol (S8) (Schiff base ligand) was synthesized by adding p-toluenesulfonic acid as a catalyzer to 10 mmol of 5-bromosalisiladehyde (dissolved in 25 mL ethyl alcohol) in a 200-mL flat bottom flask. After reaching 60 °C, 20 mmol of p-toluidine (dissolved in ethanol) was added drop by drop to into the solution and kept under reflux at 46–47 °C. The dropwise addition was continued until an orange color accompanied by sedimentation was observed. The supernatant was filtered through a 125-mm filter paper and the residue was washed with ethanol. The residue was left to dry under ambient conditions. FTIR and 1H-NMR measurements proved the formation of the ligand after the reaction.
Extraction procedure
A mixture of 2-propanol (2.0 mL) and the ligand ((Z)-3-bromo-5-((p-tolylimino)methyl)phenol) (350 μL) dissolved in chloroform was withdrawn with a syringe and solvent mixture was rapidly injected into a 15-mL centrifuge tube containing 8.0 mL of aqueous sample/standard solution and 2.0 mL buffer solution (pH 10). This resulting solution was vortexed for 1.0 min and centrifuged at 3461g for 2.0 min. The supernatant aqueous phase was carefully discarded and the extracted sediment was transferred into clean tube and kept in a water bath (100 °C). After complete evaporation, 150 μL concentrated nitric acid was added to dissolve the residue and break the Ni-S8 bonds. Vortexing was employed to facilitate dissolution and centrifugation was used to recollect the acidified extract at the bottom of the tube before sending to the SQT-FAAS system.
Samples
Three Aegean seawater samples were collected from the shores of Bodrum, Patmos, and Kalavrai and one Marmara seawater sample was taken from the shores of Beşiktaş. The samples were used to rinse the plastic bottles several times before filling to the brim without leaving airspaces. Tap water was sampled from the laboratory faucet and analyzed at the same day.
Results and discussions
Optimization of experimental parameters was started with proper concentration of nickel. In the stepwise optimization process, different variables were tested for one parameter while other parameters were kept at constant values. The highest mean value from triplicate readings was selected as optimum one.
SQT-FAAS optimizations
In order to increase the amount of sample volume reaching the flame and atomization efficiency for Ni, sample flow rate and acetylene flow rates were respectively optimized for both FAAS and SQT-FAAS systems. A rotating knob on the nebulizer unit was adjusted at different positions while taking absorbance measurements. The flow rate of each knob position was determined with 2.0 mL of 5.0 mg/L and 2.0 mg/L aqueous standard solutions for FAAS and SQT-FAAS, respectively. Optimum sample flow rate determined for FAAS was found to be 5.88 mL/min while the optimum sample flow rate for SQT-FAAS increased slightly to 6.06 mL/min. Optimization of flame temperature is essential to get efficient atomization. Fuel flow rate was conveniently adjusted at 5.0 L/h intervals with the instruments software. Optimum fuel flow rate for FAAS was determined as 40 L/h after testing flow rates between 40 and 70 L/h. The same optimum fuel flow rate was obtained for SQT-FAAS but up to 60 L/h, there was the occurrence of horn-shaped flame at the open ends of SQT. The closeness of these flames to electrical systems of the instrument prevented the testing of higher fuel flow rates. The position of SQT on burner head determines the entrance and exit rate of aspirated sample and also the path of hollow cathode lamp radiation through the flame. At 0.0-mm height, the SQT was directly placed onto the burner head and triplicate measurements were performed. The average absorbance value obtained was compared to the average absorbances obtained at 1.0-mm and 2.0-mm SQT heights, for which 1.0 mm was selected as optimum SQT height.
Complex formation optimization
With the exception of a few ligandless extractions, conventional extraction methods for metals are preceded by forming coordinate covalent complexes with ligands. In manual procedures, multiple experimental steps are susceptible to high error margins. Cutting down the number of steps in effect lowers the errors associated with the entire method. This study tested for the first time simultaneous complexation and extraction of Ni using the SQT-FAAS determination. Chloroform is a commonly used solvent for metal complex extractions and with this knowledge, the solubility of the laboratory synthesized ligand was tested by dissolving a small amount in chloroform. Upon complete dissolution, a test run was performed using methanol as dispersive solvent and a significantly high absorbance signal was obtained. This simultaneous process signifies a reduction in types and volume of solvents, a rapid and environmentally friendly method. Optimization of complexing conditions was then performed, starting with the pH of buffer solution. Buffer solutions in the pH range of 3.0 and 13 were tested for this purpose but no absorbance signals were recorded in the acidic region (below pH 7). A significant signal was recorded at pH 9 and this increased at pH 10 before decreasing marginally up to pH 13. Hence, pH 10 was selected and its optimum amount was then determined by testing 1.0 mL, 2.0 mL, and 3.0 mL and a fourth standard prepared solely at pH 10. The absorbance signal increased sharply from 1.0 to 2.0 mL, after which it increased slightly as shown in Fig. 2. Two milliliters of buffer solution was selected for recording not so different absorbance values and to cut down the amount of chemical usage.

A plot of pH 10 buffer solution volumes against absorbance
Degradation of ligand by light or sunlight was prevented by keeping the ligand solution in a dark, cool, and dry cabinet. The optimum amount of ligand concentration was investigated by testing 0.33, 0.66, 1.0, and 2.0% (mass/mass) concentrations. Absorbance values recorded were very close to each other and testing lower concentrations did not result in any significant change either. 0.33% was therefore selected as the optimum ligand concentration for recording lower standard deviations for a relatively high average absorbance. Determining the optimum amount of ligand solution was very significant in this study not only because of its effect on complexation, but also because it determined the optimum amount of extraction solvent. For this reason, 0.20-, 0.25-, 0.30-, 0.35-, and 0.40-mL volumes were tested for their complexation and extraction efficiencies. The highest absorbance value was recorded for 0.35 mL, indicating that the volume was sufficient to form and extract appreciable amounts of Ni-S8 complex from solution. Mixing by vortex was performed to facilitate a uniform distribution of both ligand and chloroform and the mass transfer of analytes into the extraction solvent. The highest average absorbance was recorded for 60 s after testing 15-, 30-, 60-, 120-, and 300-s periods of vortex.
Effect of type and volume of dispersive solvent
The most important feature of dispersive solvents is their capability to disperse extraction solvents as very fine droplets throughout aqueous solution leading to increased surface area for effective extraction. The dispersion efficiencies of ethanol, methanol, and 2-propanol were tested by mixing 2.0 mL of each with chloroform in separate extractions. Methanol and ethanol yielded absorbance values almost half of that recorded by 2-propanol. The optimum amount of 2-propanol was then determined by comparing the 2.0-mL volume extraction result to 0.50, 1.0, and 3.0-mL volumes. The absorbance signals increased from 0.50 to 2.0 mL and declined marginally at 3.0 mL. It could be stated that the volumes lower than 2.0 mL were insufficient for an effective dispersion and the 3.0-mL volume could have resulted in an increase in chloroform solubility in aqueous solution, leading to reduced extracted phase volume and low absorbance values. The optimum volume of 2-propanol was therefore selected as 2.0 mL for subsequent optimizations.
Effect of ionic strength
Ionic strength is an important factor affecting the extraction recovery of analyte(s). When the ionic strength of the aqueous solution is increased by salt addition, the solubility of the analyte could be reduced. Salting out effect was therefore examined on the method by testing 1.0 g each of sodium chloride, potassium nitrate, and potassium iodide. The average absorbance values obtained for these salt added extractions were lower when compared to a saltless extraction average absorbance. Hence, no addition of salt was applied for further studies.
Analytical figures of merit
The results of the entire optimization process (complexation, extraction, and SQT) are presented in Table 1. Under the optimum conditions, the analytical performance of the different systems (Table 2) was determined by developing calibration plots with aqueous standard solutions. Linear dynamic ranges were determined from calibration plots with high regression coefficients (0.999>), and relative standard deviations were calculated from six replicate measurements of the lowest concentration in calibration plot. The %RSD results obtained were lower than 10% for all the systems studied and this established high precision (repeatability) for both extraction process and instrumental measurements. The limits of detection and quantification of the FAAS were significantly increased by the methods employed. The orders of increase in detection power in comparison to FAAS were 3.3-, 32-, and 137-folds for SQT-FAAS, DASCE-FAAS, and DASCE-SQT-FAAS, respectively.
Real samples analysis and recovery
This method was developed with aqueous standards prepared in ultrapure deionized water and was therefore subjected to little or no interferences. However, real samples of environmental and biological origins tend to have complex matrices that could impede the extraction and determination of analyte. Four seawater samples taken from the Aegean and Marmara seas and tap water from the laboratory faucet were analyzed under the optimum conditions. There was no significant absorbance signal recorded for the samples tested to suggest nickel was present within the detection limit of the method. To determine the method’s accuracy and applicability for these samples, spiked recovery tests were performed on two seawater samples and the tap water sample. The samples were spiked to a final concentration of 14 μg/L and their respective concentrations determined with the linear calibration plot. The percent recoveries obtained ranged between 92 and 99% as given in Table 3. These results suggest that Ni when present in these matrices can be determined and accurately quantified with appreciable precision.
Conclusions
This study presents a novel analytical method for nickel determination by SQT-FAAS based on simultaneous complexation and extraction of Ni-S8 complex. Dispersive assisted simultaneous complexation and extraction process was developed in this study. To the best of our knowledge, this is the first study in literature where DASCE-SQT-FAAS is used for the determination of Ni at trace levels. Stepwise optimization of experimental parameters led to 137-fold enhancement in the detection power of the conventional FAAS. This increase correlates to a detection limit of 1.6 μg/L and a wide linear calibration range. In the recovery studies, Ni was not determined in the five aqueous samples analyzed but spiked recovery tests with close to 100% recovery established the accuracy and applicability of the developed method to these sample matrices. The developed analytical method is simple, rapid, high-yielding, and eco-friendly.
References
Alothman, Z. A., Habila, M., Yilmaz, E., & Soylak, M. (2012). Solid phase extraction of Cd(II), Pb(II), Zn(II) and Ni(II) from food samples using multiwalled carbon nanotubes impregnated with 4-(2-thiazolylazo)resorcinol. Microchimica Acta, 177(3–4), 397–403. https://doi.org/10.1007/s00604-012-0789-2.
Bidabadi, M. S., Dadfarnia, S., & Shabani, A. M. (2009). Solidified floating organic drop microextraction (SFODME) for simultaneous separation/preconcentration and determination of cobalt and nickel by graphite furnace atomic absorption spectrometry (GFAAS). Journal of Hazardous Materials, 166(1), 291–296. https://doi.org/10.1016/j.jhazmat.2008.11.052.
Büyükpınar, Ç., Maltepe, E., Chormey, D. S., San, N., & Bakırdere, S. (2017). Determination of nickel in water and soil samples at trace levels using photochemical vapor generation-batch type ultrasonication assisted gas liquid separator-atomic absorption spectrometry. Microchemical Journal, 132, 167–171. https://doi.org/10.1016/j.microc.2017.01.024.
Chormey, D. S., Buyukpinar, C., Turak, F., Komesli, O. T., & Bakirdere, S. (2017). Simultaneous determination of selected hormones, endocrine disruptor compounds, and pesticides in water medium at trace levels by GC-MS after dispersive liquid-liquid microextraction. Environmental Monitoring and Assessment, 189(6), 277. https://doi.org/10.1007/s10661-017-6003-6.
Deng, Q., Chen, M., Kong, L., Zhao, X., Guo, J., & Wen, X. (2013). Novel coupling of surfactant assisted emulsification dispersive liquid-liquid microextraction with spectrophotometric determination for ultra trace nickel. Spectrochimica Acta. Part A, Molecular and Biomolecular Spectroscopy, 104, 64–69. https://doi.org/10.1016/j.saa.2012.10.080.
Dos Anjos, S. L., Alves, J. C., Rocha Soares, S. A., Araujo, R. G. O., de Oliveira, O. M. C., Queiroz, A. F. S., et al. (2018). Multivariate optimization of a procedure employing microwave-assisted digestion for the determination of nickel and vanadium in crude oil by ICP OES. Talanta, 178, 842–846. https://doi.org/10.1016/j.talanta.2017.10.010.
Erarpat, S., Ozzeybek, G., Chormey, D. S., & Bakirdere, S. (2017). Determination of lead at trace levels in mussel and sea water samples using vortex assisted dispersive liquid-liquid microextraction-slotted quartz tube-flame atomic absorption spectrometry. Chemosphere, 189, 180–185. https://doi.org/10.1016/j.chemosphere.2017.09.072.
Escudero, L. A., Blanchet, A. J., Sombra, L. L., Salonia, J. A., & Gasquez, J. A. (2014). Determination of the total and extractable fraction of Ni in lake sediments and natural waters of San Luis (Argentina) by FAAS using a simple solid phase extraction system. Microchemical Journal, 116, 92–97. https://doi.org/10.1016/j.microc.2014.04.007.
Gonzalez, P. (2002). Determination of nickel by anodic adsorptive stripping voltammetry with a cation exchanger-modified carbon paste electrode. Talanta, 58(4), 679–690. https://doi.org/10.1016/s0039-9140(02)00381-8.
Haudet, S. S., Rodriguez, M. A., & Carranza, R. M. (2015). Determining the effect of the main alloying elements on localized corrosion in nickel alloys using artificial neural networks. Procedia Materials Science, 8, 21–28. https://doi.org/10.1016/j.mspro.2015.04.044.
Hol, A., Akdogan, A., Kartal, A. A., Divrikli, U., & Elci, L. (2014). Dispersive liquid–liquid microextraction of nickel prior to its determination by microsample injection system-flame atomic absorption spectrometry. Analytical Letters, 47(13), 2195–2208. https://doi.org/10.1080/00032719.2014.900777.
IUPAC. (1998). Compendium of analytical nomenclature. Qxford: Blackwell Science.
Jalbani, N., & Soylak, M. (2015a). Ligandless ultrasonic-assisted and ionic liquid-based dispersive liquid-liquid microextraction of copper, nickel and lead in different food samples. Food Chemistry, 167, 433–437. https://doi.org/10.1016/j.foodchem.2014.07.015.
Jalbani, N., & Soylak, M. (2015b). Separation-preconcentration of nickel and lead in food samples by a combination of solid-liquid-solid dispersive extraction using SiO2 nanoparticles, ionic liquid-based dispersive liquid-liquid micro-extraction. Talanta, 131, 361–365. https://doi.org/10.1016/j.talanta.2014.07.099.
Kocot, K., Pytlakowska, K., Zawisza, B., & Sitko, R. (2016). How to detect metal species preconcentrated by microextraction techniques? TrAC Trends in Analytical Chemistry, 82, 412–424. https://doi.org/10.1016/j.trac.2016.07.003.
Mettakoonpitak, J., Miller-Lionberg, D., Reilly, T., Volckens, J., & Henry, C. S. (2017). Low-cost reusable sensor for cobalt and nickel detection in aerosols using adsorptive cathodic square-wave stripping voltammetry. Journal of Electroanalytical Chemistry, 805, 75–82. https://doi.org/10.1016/j.jelechem.2017.10.026.
Özzeybek, G., Erarpat, S., Chormey, D. S., Fırat, M., Büyükpınar, Ç., Turak, F., & Bakırdere, S. (2017). Sensitive determination of copper in water samples using dispersive liquid-liquid microextraction-slotted quartz tube-flame atomic absorption spectrometry. Microchemical Journal, 132, 406–410. https://doi.org/10.1016/j.microc.2017.02.031.
Peeters, K., Zuliani, T., Zigon, D., Milacic, R., & Scancar, J. (2017). Nickel speciation in cocoa infusions using monolithic chromatography - post-column ID-ICP-MS and Q-TOF-MS. Food Chemistry, 230, 327–335. https://doi.org/10.1016/j.foodchem.2017.03.050.
Ranjbar, L., Yamini, Y., Saleh, A., Seidi, S., & Faraji, M. (2012). Ionic liquid based dispersive liquid-liquid microextraction combined with ICP-OES for the determination of trace quantities of cobalt, copper, manganese, nickel and zinc in environmental water samples. Microchimica Acta, 177(1–2), 119–127. https://doi.org/10.1007/s00604-011-0757-2.
Reclo, M., Yilmaz, E., Soylak, M., Andruch, V., & Bazel, Y. (2017). Ligandless switchable solvent based liquid phase microextraction of nickel from food and cigarette samples prior to its micro-sampling flame atomic absorption spectrometric determination. Journal of Molecular Liquids, 237, 236–241. https://doi.org/10.1016/j.molliq.2017.04.066.
Rezaee, M., Assadi, Y., Milani Hosseini, M. R., Aghaee, E., Ahmadi, F., & Berijani, S. (2006). Determination of organic compounds in water using dispersive liquid-liquid microextraction. Journal of Chromatography. A, 1116(1–2), 1–9. https://doi.org/10.1016/j.chroma.2006.03.007.
Silva, S. G., Oliveira, P. V., Nóbrega, J. A., & Rocha, F. R. P. (2009). Cloud point extraction to avoid interferences by structured background on nickel determination in plant materials by FAAS. Analytical Methods, 1(1), 68. https://doi.org/10.1039/b9ay00010k.
Uslu, H., Büyükpınar, Ç., Unutkan, T., Serbest, H., San, N., Turak, F., et al. (2018). A novel analytical method for sensitive determination of lead: Hydrogen assisted T-shape slotted quartz tube-atom trap-flame atomic absorption spectrometry. Microchemical Journal, 137, 155–159. https://doi.org/10.1016/j.microc.2017.10.015.
Viñas, P., Campillo, N., & Andruch, V. (2015). Recent achievements in solidified floating organic drop microextraction. TrAC Trends in Analytical Chemistry, 68, 48–77. https://doi.org/10.1016/j.trac.2015.02.005.
Wang, S., Meng, S., & Guo, Y. (2013). Cloud point extraction for the determination of trace amounts of cobalt in water and food samples by flame atomic absorption spectrometry. Journal of Spectroscopy, 2013, 1–7. https://doi.org/10.1155/2013/735702.
Weldeabzgi, A., Reddy, D. N., & Mekonnen, K. N. (2017). Spectrophotometric determination of nickel (II) in soil and standard alloy samples using 5-methyl-2-acetylfuran-4-methyl-3-thiosemicarbazone (5-MAFMT). Communications in Soil Science and Plant Analysis. https://doi.org/10.1080/00103624.2016.1269797.
Yang, L., Ding, J., Maxwell, P., McCooeye, M., Windust, A., Ouerdane, L., Bakirdere, S., Willie, S., & Mester, Z.́. (2011). Determination of arsenobetaine in fish tissue by species specific isotope dilution LC-LTQ-orbitrap-MS and standard addition LC-ICPMS. Analytical Chemistry, 83(9), 3371–3378. https://doi.org/10.1021/ac103258m.
Zambelli, B., Uversky, V. N., & Ciurli, S. (2016). Nickel impact on human health: an intrinsic disorder perspective. Biochimica et Biophysica Acta, 1864(12), 1714–1731. https://doi.org/10.1016/j.bbapap.2016.09.008.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest statement
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 18 kb)
Rights and permissions
About this article

Cite this article
Özzeybek, G., Alacakoç, B., Kocabaş, M.Y. et al. Trace determination of nickel in water samples by slotted quartz tube-flame atomic absorption spectrometry after dispersive assisted simultaneous complexation and extraction strategy. Environ Monit Assess 190, 498 (2018). https://doi.org/10.1007/s10661-018-6884-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10661-018-6884-z