
Abstract
The need to enhance food safety has led to major advancements in pesticide productions, and though many benefits have been gained, environmental contamination has also risen from these chemicals that tend to persist in the environment. Some pesticides, together with other chemicals commonly called endocrine disruptor compounds, block the receptor sites of hormones or mimic displaced hormones, leading to imbalanced hormonal levels that result in health disorders and diseases. These chemicals occur at trace levels and are not directly detected by conventional analytical methods. A dispersive liquid-liquid microextraction method was therefore developed for preconcentration of 12 analytes including hormones, endocrine disruptor compounds, and pesticides, to be analyzed by gas chromatography mass spectrometry. This was achieved by optimizing parameters such as extractor solvent type and amount, dispersive solvent type and amount, pH, and salt effect that affect extraction output. The limits of detection and quantification of the developed method were between 0.09 and 3.36 and 0.31 and 11.19 ng mL−1, respectively. The calibration plots of the analytes also showed good linearity and low percent relative standard deviations. Recovery studies were performed for tap water and wastewater samples, and the percent recoveries recorded were between 84 and 109%.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pesticides are a group of compounds used to control, prevent, or eliminate pests that affect plants and animals, or cause harm to humans (Songa and Okonkwo 2016). Pesticides are mainly used for plant protection, and they offer numerous benefits such as improved crop yield and quality, disease containment, high revenue, and control of insects that spread diseases to humans (Cooper and Dobson 2007). When classified under the target organism, pesticides include herbicides, bactericide, fungicide, termiticide, insecticide, rodenticides, amongst others (Aktar et al. 2009). These chemicals are used with the sole purpose of controlling or eliminating the target organism, but there have been reported cases of pesticides having adverse effects on human health (Neghab et al. 2014; Benedetti et al. 2013; Mostafalou and Abdollahi 2013). Pesticides reach humans through contaminated water bodies, soils, and polluted air. This has led to many legislations prohibiting the use of some pesticides (Hillocks 2012; Karabelas et al. 2009; Skevas et al. 2013; Fantke et al. 2012), especially those considered as persistent organic pollutants (POPs). The POPs show a high resistance to environmental degradation, and though most of them have been banned for many years, they can still be found in the environment (Li et al. 2016). Pesticides usually occur in low concentrations in the environment, but studies have shown that continuous intake results in bioaccumulation in organs and tissues of living organisms (Gerber et al. 2016). This risk of chronic poisoning is mostly encountered in aquatic organisms, but the same risk is encountered by other organisms that consume aquatic organisms in the food chain (Grung et al. 2015; Botaro et al. 2011; Rezg et al. 2010).
Some pesticides together with other group of chemicals affect the endocrine system of humans and other organisms. The endocrine system is a broad classification of hormone-producing glands such as the thyroid gland, adrenal glands, thymus glands, pituitary glands, and pancreas (Morgan and Tsai 2015). The normal function of the endocrine system can be disrupted by some microcontaminants having the ability to imitate hormones and block receptor cites (Mnif et al. 2011). These contaminants are commonly called endocrine disruptor compounds (EDCs) or endocrine-disrupting chemicals. These EDCs tend to hasten or block the processes of the endocrine system, and the changes associated with it are irreversible. The EDCs have been classified into three main groups namely, androgenic, thyroidal, and estrogenic (Komesli et al. 2012). This classification is based on specific hormones that are targeted by the EDCs.
EDCs and pesticides enter the environment (soil, water and air) mainly through human activities such as accidental chemical spillage, agricultural applications, and municipal sewage (Hecker and Hollert 2011; Jensen and Olesen 2014; Damalas and Eleftherohorinos 2011). About 40% of the world’s population is expected to experience water stress or scarcity by 2050 (Misra 2014; Gosain et al. 2006); hence, the treatment of municipal wastewaters for their reuse has become a viable solution to alleviate the water shortage concerns. Treated water can either be released back into the environment or instantly reused. Many plants are being used for the treatment of wastewater and those specifically used for the removal of pesticides, and EDCs include membrane bioreactors (MBRs), conventional activated sludge (CAS), and biological nutrient removing (BNR) activated sludge systems amongst others (Komesli et al. 2016). A more recent system is the multiple barrier approach which includes various processes collectively used to reduce water contamination to the lowest possible level (Health Canada 2014). A failure in one of the barriers is not expected to greatly affect the overall treatment process, and as such, the refined water would pose minimal health risk to the public. Detection of selected analytes at trace levels using a simple extraction/preconcentration method is very crucial to develop wastewater treatment strategies to get safe water.
Contaminants in water usually occur at trace and ultra trace levels which are not easily detected by direct analytical methods. There is therefore the need for various preconcentration techniques to be able to lower the limits of instrumental detection. An appropriate analytical method when developed is used to test for contaminants in wastewater before (influent) and after (effluent) the treatment process. Chromatographic methods have been the preferred techniques for the characterization and identification of not only pesticides and EDCs, but for a wide range of compounds including the analogues of homoserine lactone and nicotine (Bakaraki et al. 2016; Dobrowska et al. 2016). Those of low molecular weight and boiling point are preferably determined by gas chromatographic methods equipped mostly with flame ionization detector (FID), electron capture detector (ECD), and mass spectrometer (MS) (Zhao et al. 2016; Hassan and Sarkouhi 2016; Anjos and Andrade 2014). Instrumentations such as liquid chromatography tandem mass spectrometry (LC-MS/MS), high-performance liquid chromatography-electron spray ionization-tandem mass spectrometry (HPLC-ESI-MS/MS), and high-performance liquid chromatography inductively coupled plasma-mass spectrometry (HPLC-ICP-MS) have been used for the more polar compounds without the need for derivatization (Sönmez et al. 2012; Gui et al. 2016; Grimalt and Dehouck 2016). The introduction of dispersive liquid-liquid microextraction (DLLME) (Rezaee et al. 2006) has led to the detection of pesticides and EDCs at trace levels in comparison to those reported for solid-phase extraction/microextraction (Muz et al. 2012; Cai et al. 2013). Other advantages of the DLLME method include the usage of very low amounts of organic solvents, high recoveries, and a very short extraction period.
This study was aimed at developing a sensitive DLLME method for the trace determination of selected hormones, pesticides, and EDCs in water medium.
Materials and methods
Apparatus
The separation, identification, and quantification of analytes were achieved by an Agilent 6890 GC gas chromatograph system, fitted with an HP-5MS capillary column (30 m × 250 μm; 0.25 μm) and a mass selective detector. All injections were done in the splitless mode with a standard/sample volume of 1.0 μL and helium as carrier gas at a flow rate of 1.8 mL min−1. The injector port and transfer line temperatures were 250 and 280 °C, respectively. A ramp temperature program consisting of an initial 70 °C was increased to 180 °C (60 °C min−1) and then to 210 (4.0 °C min−1). The final ramp was to 280 °C at a rate of 40 °C min−1 and held for 2.0 min. Peaks from the total ion chromatogram were integrated according to the prominent ion fragment (m/z) of each analyte.
Chemicals
The reagents used throughout the study were of analytical grade, obtained from Merck (Germany). Acetonitrile was used to prepare all stock and working standard solutions. Methanol, chloroform, dichloromethane, ethanol, acetone, carbon tetrachloride, 1,2-dichloroethane, potassium nitrate, barium chloride, potassium chloride, sodium chloride, and potassium chloride were used in the optimization process of the DLLME. Bisphenol A (99.8%), 4-n-octylphenol (99.5%), diazinon (99%), cis-chlordane (99%), Aldrin (99%), estrone (99.5%), 17-β-estradiol (95.4%), heptachlor (99%), 4-n-nonylphenol (99.9%), and endosulfan α-β (99%) standards were all obtained from Dr. Ehrenstorfer (Augsburg, Germany). Presented in Table 1 are the uses, classification, and target hormones of each chemical.
DLLME protocol
An 8.0 mL aqueous sample/standard solution was placed in a 15-mL centrifuge tube containing 0.50 g of potassium iodide. The dispersive/extractor mixture was prepared in a separate tube by adding 200 μL chloroform to 2.0 mL methanol, and it was taken up and injected into the aqueous solution with a syringe. The resulting cloudy solution was shaken in the up-and-down motion for 30 s and centrifuged at 3461g for 2.0 min. About 50-μL volume was taken from the settled chloroform phase at the bottom of the tube and placed in microvolume insert vials for auto injections.
Samples
Municipal wastewater from a submerged-type membrane bioreactor (MBR) was obtained from an Advanced Biological Wastewater Treatment Plant and filtered with a 125-mm Sartorius filter paper and a 0.45-μm RC syringe filter. Synthetic wastewater prepared according to Bakaraki et al. (2016) was taken from a small-scale continuous-type MBR with an effective capacity of 1750 mL and filtered similarly to the municipal wastewater. The municipal wastewater obtained in bulk was stored at 4.0 °C, but the synthetic wastewater was taken freshly from the reactor for analysis. Tap water samples were taken from Bayrampaşa, Beylikduzu, Beykoz, and Maltepe districts of Istanbul.
Results and discussion
The factors affecting the extraction process were optimized to obtain high extraction outputs and recoveries for all analytes. The optimization was done by varying one parameter at a time, while keeping the other parameters constant. The reproducibility of each optimization step was determined by calculating the percent relative standard deviation (%RSD) of triplicate extractions performed for one of the varying parameters.
Extraction solvent type
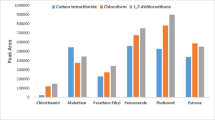
An extractor solvent should have a high affinity to dissolve the analyte(s) of interest and should be immiscible with water. A common problem encountered with extraction procedures is the volatilization of the extractor solvent after extraction, leading to a reduced volume and a more preconcentrated analyte solution. This poses challenges in determining the exact preconcentration factor of the method. Solvents with greater densities than water were therefore used to alleviate this problem, and it also provided an ease in taking up the settled organic phase from the narrow conical bottom of the centrifuge tube. The solvents tested for their extraction outputs included chloroform (ρ = 1.56), dichloromethane (ρ = 1.33), 1,2-dichloroethane (ρ = 1.25), and carbon tetrachloride (ρ = 1.59), having polarity indexes of 4.1, 3.1, 3.5, and 1.6, respectively. The polarity index of a solvent represents the degree of its interaction with various test polar solutes. With the exception of 1,2-dichloroethane, 200 μL of each solvent was mixed with 2.0 mL methanol and injected into 8.0 mL aqueous solution spiked with the mixed standards to 50 ng mL−1. 1,2-Dichloroethane was omitted because it is immiscible with methanol, but its extraction output was determined in the disperser solvent-type optimization step. Peak area results obtained from integrated chromatograms indicated that extraction efficiencies for the analytes of interest in case of chloroform were the highest if compared with other solvents used. Results can be seen in Fig. 1. The high polarity index of chloroform having higher density (ρ = 1.56) than water could have resulted in a more effective mass transfer of analytes from the aqueous solution.

Optimization of extraction solvent type: dichloromethane (1), chloroform (2), and carbon tetrachloride (3)
Extraction solvent amount
The optimum chloroform amount was thus determined by repeating the extraction process using 100-, 200-, 300-, and 400-μL volumes. The highest output was recorded for 100 μL, and the relatively lower outputs of the higher volumes can be attributed to the dilution of analytes. The final volume of chloroform that settled at the bottom of the tube was observed to have been reduced to almost half the initial volume added. The conical bottom of the 15-mL centrifuge tube used was not narrow enough to ensure an easy collection of the 100 μL settled chloroform phase. This therefore resulted in inconsistent extraction outputs for replicates of the 100 μL extractor volume. The 200-μL volume also gave a high extraction output, and the settled chloroform phase was clearly distinguished from the aqueous phase at the bottom of the tube, thereby facilitating easy analyte solution pickup for analysis. The 200-μL chloroform volume was therefore selected for further optimization studies.
Dispersive solvent type and amount
A dispersive solvent is selected based on its miscibility with both water and extractor solvent. The use of the dispersive solvent increases the surface area of contact between extractor solvent and analyte solution, thereby enhancing extraction. The dispersive solvents tested for their extraction outputs were methanol, ethanol, acetone, and acetonitrile. The extraction output of 1,2-dichloroethane/ethanol mixture was compared to the optimum result in this optimization step since it could not be performed in the solvent-type selection step. Methanol gave the highest extraction output compared to the other dispersive solvents, and it was also higher than the 1,2-dichloroethane/ethanol mixture’s extraction output. The optimum amount of methanol was therefore examined using 0.50, 1.0, 2.0, and 3.0 mL. The results obtained from integrated peak area of all analytes showed no significant increments in extraction outputs for the different volumes tested. However, 2.0 mL was selected for further optimizations due to its slightly higher output and being a more practical volume for the 15-mL centrifuge tube used.
Salt type and amount
The efficiency of an extraction depends on the ability of analytes to move from the aqueous phase into the organic phase. In order to decrease the solubility of analytes in an aqueous solution and increase their mobility into the organic phase, different salts are usually added. The salt effect was thus examined using KNO3, BaCl2, KCl, NaCl, and KCl. The 12 analytes exhibited different extraction outputs for the different types of salts used. Potassium iodide (KI) gave the highest extraction outputs for 9 out of the 12 analytes and KNO3 for the other 3. The percentage difference between the extraction outputs of KI and KNO3 and the other salts was between 9 and 67%. KI was thus selected and its optimum amount was determined using 0.50, 1.0, 2.0, and 3.0 g. A steady decrease in extraction output was observed with increasing salt amount. This can be attributed to the excess salt amount inhibiting the interaction between the extractant and the analytes. The 0.50-g KI amount was therefore selected as optimum value.
Effect of pH
The pH of an aqueous solution determines whether an analyte exists in an ionic or molecular state, which in turn affects the movement of the analyte into the extracting organic phase. Analytes in the ionic state favor the formation of complexes prior to extraction, but direct extraction of analytes into the organic solvent requires the analyte to be in the molecular form. The effect of pH on all 12 analytes was therefore examined in the range 5–9. The lowest extraction outputs were recorded at pH 5 for all analytes, indicating a significant ionization of analytes in the acidic medium. A very sharp increase in extraction output was recorded at pH 6, after which a steady decrease continued to pH 9. pH 6 was therefore selected as optimum pH value for further optimization.
Effect of mixing and mixing period
One of the most important advantages of the DLLME method is how rapid the extraction of analytes is accomplished. Even without mixing, substantive extraction outputs are recorded due to the large surface area formed between the aqueous solution and extractor solvent, by reason of the dispersive solvent. However, mixing further enhances extraction by causing more movement of the extractor solvent through the aqueous solution to increase the pickup of analytes of interest. The effect of mixing was therefore examined by subjecting the extraction mixture to 5.0 min of mechanical shaking and ultrasonication, 15 s of vigorous hand shaking, and a fourth where the mixture was allowed to stand without any form of mixing after the extractor/dispersive mixture had been injected. The mixing due to mechanical shaking gave the higher extraction outputs. The optimum period of mechanical shaking was tested between 30 s and 10 min. The extraction outputs increased steadily up to 5 min after which it begun to decrease. The 5.0-min mechanical shaking was once again compared to 30-s hand shaking, and a maximum percentage difference of 7.1% was recorded for the analyte bisphenol A. For this reason, 30-s hand shaking was the preferred choice of mixing to promote a rapid extraction process.
Analytical figures of merit
The analytical performance of the GC-MS system on the 12 analytes was determined with standards between 0.50 and 1.0 μg mL−1 using the optimized DLLME method presented in Table 2. Calibration standards were prepared in water by diluting appropriate aliquots of acetonitrile-based mixed standards. The miscibility of acetonitrile and water was ideal for preparing the water-based standards for the extraction process. However, preparing the aqueous standards from different volumes of acetonitrile-based standards would result in different analyte solubilities and consequently affect extraction outputs. All extraction solutions were therefore prepared by taking an equivalent amount from different acetonitrile-based standards. The limits of detection (LOD) and quantification (LOQ) were found to be between 0.09 and 3.4 and 0.30 and 11.2 ng mL−1, respectively. The LODs, LOQs, RSDs, and correlation coefficients (R 2) of all analytes are presented in Table 3. The total ion chromatogram of a 10 μg mL−1 mixed standard solution of all analytes is seen in Fig. 2. Tap water samples obtained from Bayrampaşa, Beylikduzu, Beykoz, and Maltepe districts of Istanbul were analyzed with the optimum method, but none of the analytes was detected.

Total ion chromatogram of 10 μg mL−1 mix standard with identified peaks
Recovery tests
An analytical method is developed for its applicability to real samples. The complicated matrix of most samples, however, imposes challenges on the amount of analyte that can be extracted from samples for analysis. It is therefore important to perform recovery studies to determine the effectiveness of the method for the intended sample matrix. The recovery test for this method was performed on tap water and wastewater by first running a blank extraction to ensure that the analytes of interest were not present, after which triplicate spike samples were extracted and analyzed. The spiking was done so as to obtain a homogenized mixture that would match real water samples contaminated with the analytes.
The percentage recovery of tap water spiked to final concentrations of 5.0, 10, 50, and 100 ng mL−1 was determined. This spiking range was specifically selected because of the linearity exhibited for all analytes and to determine the effectiveness of the recovery at different concentrations. Presented in Table 4 are the percent recoveries obtained for the different spiked concentrations and their deviations from each other, expressed as %RSD. There was no specific increasing or decreasing trend in the recovery results obtained for the analytes at different concentrations, and this can therefore be attributed to basic standard deviations in the extraction processes.
The precision of the recovery tests was also determined based on both repeatability and reproducibility. The repeatability of the recovery process was obtained from triplicate extractions performed on a 50 ng mL−1 spiked tap water sample, performed by one analyst. The %RSDs obtained were between 0.8 and 7.6%. In addition, four different analysts performed triplicate extractions for 50 ng mL−1 spiked tap water samples for the reproducibility test. As shown in Table 5, the average results from the triplicate extractions of each analyst were used to determine the %RSD of each analyte. Results from both repeatability and reproducibility tests confirm that the method can be applied for tap water determinations with high precision.
Wastewater is known to have a very complex matrix that usually interferes with the extraction of analytes. For this reason, the percent recoveries of the method for both municipal and synthetic wastewater samples were determined by spiking to 50 ng mL−1. The recovery results of analytes from the spiked synthetic wastewater were close to 100%, but the recovery results from the municipal wastewater were between 84 and 102% (Table 6). The relatively lower results of the latter suggest a more complex matrix, but the recoveries are substantial for both qualitative and quantitative determinations should the analytes be present.
Conclusion remarks
Human exposure to pesticides and EDCs has become a common phenomenon due to the persistent nature of these chemicals in the environment. A sensitive analytical method was developed for the simultaneous determination of hormones, pesticides, and EDCs at trace levels. The developed method is suitable for routine laboratory analysis due to its simplicity and fastness. It is also very economical and employs low amounts of organic solvents that pose risk to human health and the environment. The optimized method was applicable over a wide concentration range and gave very low LOD and LOQ values. The low RSD values obtained for both repeatability and reproducibility tests indicate the high precision of the method. High percent recoveries were obtained for all analytes validating the method for its purpose of real sample application. This implies that occurrence of analytes in tap water and wastewater will not be hindered by the matrix but can be readily determined and quantified.
References
Aktar, M. W., Sengupta, D., & Chowdhury, A. (2009). Impact of pesticides use in agriculture: their benefits and hazards. Interdisciplinary Toxicology, 2, 1–12.
Anjos, J. P., & Andrade, J. B. (2014). Determination of nineteen pesticides residues (organophosphates, organochlorine, pyrethroids, carbamate, thiocarbamate and strobilurin) in coconut water by SDME/GC–MS. Microchemical Journal, 112, 119–126.
Bakaraki, N., Chormey, D. S., Bakirdere, S., & Engin, G. O. (2016). Development of a sensitive liquid–liquid extraction method for the determination of N-butyryl-L-homoserine lactone produced in a submerged membrane bioreactor by gas chromatography mass spectrometry and deuterated anthracene as the internal standard. Analytical Methods, 8, 2660–2665.
Benedetti, D., Nunes, E., Sarmento, M., Porto, C., Santos, C. E. I., Dias, J. F., & Silva, J. (2013). Genetic damage in soybean workers exposed to pesticides: evaluation with the comet and buccal micronucleus cytome assays. Mutation Research, 752, 28–33.
Botaro, D., Torres, J. P. M., Malm, O., Rebelo, M. F., Henkelmann, B., & Schramm, K. W. (2011). Organochlorine pesticides residues in feed and muscle of farmed Nile tilapia from Brazilian fish farms. Food and Chemical Toxicology, 49, 2125–2130.
Cai, J., Zhu, F., Ruan, W., Lui, L., Lai, R., Zeng, F., & Ouyang, G. (2013). Determination of organochlorine pesticides in textiles using solid phase microextraction with gas chromatography-mass spectrometry. Microchemical Journal, 110, 280–284.
Cooper, J., & Dobson, H. (2007). The benefits of pesticides to mankind and the environment. Crop Protection, 26, 1337–1348.
Damalas, C. A., & Eleftherohorinos, I. G. (2011). Pesticide exposure, safety issues, and risk assessment indicators. International Journal of Environmental Research and Public Health, 8, 1402–1419.
Dobrowska, J. S., Erarpat, S., Chormey, D. S., Pyrzyńska, K., & Bakırdere, S. (2016). A novel liquid-liquid extraction for the determination of nicotine in tap water, wastewater and saliva at trace levels by GC-MS. Journal of AOAC International, 99, 806–812.
Fantke, P., Friedrich, R., & Jolliet, O. (2012). Health impact and damage cost assessment of pesticides in Europe. Environment International, 49, 9–17.
Gerber, R., Smit, N. J., Van Vuren, J. H. J., Nakayama, S. M. M., Yohannes, Y. B., Ikenaka, Y., Ishizuka, M., & Wepener, V. (2016). Bioaccumulation and human health risk assessment of DDT and other organochlorine pesticides in an apex aquatic predator from a premier conservation area. The Science of the Total Environment, 550, 522–533.
Gosain, A. K., Rao, S., & Basuray, D. (2006). Climate change impact assessment on hydrology of Indian river basins. Current Science, 90, 3.
Grimalt, S., & Dehouck, P. (2016). Review of analytical methods for the determination of pesticide residues in grapes. Journal of Chromatography A, 1433, 1–23.
Grung, M., Lin, Y., Zhang, H., Steen, A. O., Huang, J., Zhang, G., & Larssen, T. (2015). Pesticide levels and environmental risk in aquatic environments in China—a review. Environment International, 81, 87–97.
Gui, W., Tian, C., Sun, Q., Li, S., Zhang, W., Tang, J., & Zhu, G. (2016). Simultaneous determination of organotin pesticides by HPLC-ICP-MS and their sorption, desorption, and transformation in freshwater sediments. Water Research, 95, 185–194.
Hassan, J., & Sarkouhi, M. (2016). Miniaturized counter current liquid–liquid extraction for organophosphorus pesticides determination. Arabian Journal of Chemistry, 9, 38–42.
Health Canada. (2014). Guidelines for Canadian Drinking Water Quality—summary table. Water and Air Quality Bureau, Healthy Environments and Consumer Safety Branch, Health Canada, Ottawa, Ontario.
Hecker, M., & Hollert, H. (2011). Endocrine disruptor screening: regulatory perspectives and needs. Environmental Sciences Europe, 23, 15.
Hillocks, R. J. (2012). Farming with fewer pesticides: EU pesticide review and resulting challenges for UK agriculture. Crop Protection, 31, 85–93.
Jensen, P. K., & Olesen, M. H. (2014). Spray mass balance in pesticide application: a review. Crop Protection, 61, 23–31.
Karabelas, A. J., Plakas, K. V., Solomou, E. S., Drossou, V., & Sarigiannis, D. A. (2009). Impact of European legislation on marketed pesticides—a view from the standpoint of health impact assessment studies. Environment International, 35, 1096–1107.
Komesli, O. T., Bakırdere, S., Bayören, C., & Gökçay, C. F. (2012). Simultaneous determination of selected endocrine disrupter compounds in wastewater samples in ultra trace levels using HPLC-ES-MS/MS. Environmental Monitoring and Assessment, 184, 5215–5224.
Komesli, O. T., Muz, M., Ak, M. S., Bakırdere, S., & Gökçay, C. F. (2016). Comparison of EDCs removal in full and pilot scale membrane bioreactor plants: effect of flux rate on EDCs removal in short SRT. Journal of Environmental Management. doi:10.1016/j.jenvman.2016.06.004.
Li, J., Chen, C., & Li, F. (2016). Status of POPs accumulation in the Yellow River Delta: from distribution to risk assessment. Marine Pollution Bulletin, 107, 370–378.
Misra, A. K. (2014). Climate change and challenges of water and food security. International Journal of Sustainable Built Environment, 3, 153–165.
Mnif, W., Hassine, A. I. H., Bouaziz, A., Bartegi, A., Thomas, O., & Roig, B. (2011). Effect of endocrine disruptor pesticides: a review. International Journal of Environmental Research and Public Health, 8, 2265–2303.
Morgan, D., & Tsai, S. C. (2015). Sleep and the endocrine system. Critical Care Clinics, 31, 403–418.
Mostafalou, S., & Abdollahi, M. (2013). Pesticides and human chronic diseases: evidences, mechanisms, and perspectives. Toxicology and Applied Pharmacology, 268, 157–177.
Muz, M., Sönmez, M. S., Komesli, O. T., Bakırdere, S., & Gökçay, C. F. (2012). Determination of selected natural hormones and endocrine disrupting compounds in domestic wastewater treatment plants by liquid chromatography electrospray ionization tandem mass spectrometry after solid phase extraction. Analyst, 137, 884–889.
Neghab, M., Momenbella-Fard, M., Naziaghdam, R., Salahshour, N., Kazemi, M., & Alipour, H. (2014). The effects of exposure to pesticides on the fecundity status of farm workers resident in a rural region of Fars province, southern Iran. Asian Pacific Journal of Tropical Biomedicine, 4, 324–328.
Rezaee, M., Assadi, Y., Milani Hosseini, M. R., Aghaee, E., Ahmadi, F., & Berijani, S. (2006). Determination of organic compounds in water using dispersive liquid–liquid microextraction. Journal of Chromatography. A, 1116, 1–9.
Rezg, R., Mornagui, B., El-Fazaa, S., & Gharbi, N. (2010). Organophosphorus pesticides as food chain contaminants and type 2 diabetes: a review. Trends in Food Science and Technology, 21, 345–357.
Skevas, T., Oude Lansink, A. G. J. M., & Stefanou, S. E. (2013). Designing the emerging EU pesticide policy: a literature review. NJAS - Wageningen Journal of Life Sciences, 64, 95–103.
Songa, E. A., & Okonkwo, J. O. (2016). Recent approaches to improving selectivity and sensitivity of enzyme-based biosensors for organophosphorus pesticides: a review. Talanta, 155, 289–304.
Sönmez, M. S., Muz, M., Komesli, O. T., Bakırdere, S., & Gökçay, C. F. (2012). Determination of selected endocrine disrupter compounds at trace levels in sewage sludge samples. Clean-Soil, Air, Water, 40, 980–985.
Zhao, X., Zhou, Y., Kong, W., Gong, B., Chen, D., Wei, J., & Yang, M. (2016). Multi-residue analysis of 26 organochlorine pesticides in Alpinia oxyphylla by GC-ECD after solid phase extraction and acid cleanup. Journal of Chromatography B, 1017-1018, 211–220.
Acknowledgements
The authors duly acknowledge Yıldız Technical University for the financial support (Scientific Research Project, 2016-01-02-KAP04).
Author information
Authors and Affiliations
Corresponding author
Additional information
Highlights
• A dispersive liquid-liquid microextraction method was developed for the preconcentration of 12 analytes
• All of the system parameters were optimized.
• The limits of detection and quantification of the developed method were between 0.09 and 5.69 and 0.31 and 18.96 ng mL−1, respectively
• The spiking experiment at different concentrations revealed satisfactory recoveries for tap water and wastewater.
Rights and permissions
About this article

Cite this article
Chormey, D.S., Büyükpınar, Ç., Turak, F. et al. Simultaneous determination of selected hormones, endocrine disruptor compounds, and pesticides in water medium at trace levels by GC-MS after dispersive liquid-liquid microextraction. Environ Monit Assess 189, 277 (2017). https://doi.org/10.1007/s10661-017-6003-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10661-017-6003-6