
Abstract
Autoimmune hepatitis is a consequence of perturbations in homeostatic mechanisms that maintain self-tolerance but are incompletely understood. The goals of this review are to describe key pathogenic pathways that have been under-evaluated or unassessed in autoimmune hepatitis, describe insights that may shape future therapies, and encourage investigational efforts. The T cell immunoglobulin mucin proteins constitute a family that modulates immune tolerance by limiting the survival of immune effector cells, clearing apoptotic bodies, and expanding the population of granulocytic myeloid-derived suppressor cells. Galectins influence immune cell migration, activation, proliferation, and survival, and T cell exhaustion can be induced and exploited as a possible management strategy. The programmed cell death-1 protein and its ligands comprise an antigen-independent inhibitory axis that can limit the performance of activated T cells by altering their metabolism, and epigenetic changes can silence pro-inflammatory genes or de-repress anti-inflammatory genes that affect disease severity. Changes in the intestinal microbiota and permeability of the intestinal mucosal barrier can be causative or consequential events that affect the occurrence and phenotype of immune-mediated disease, and they may help explain the female propensity for autoimmune hepatitis. Perturbations within these homeostatic mechanisms have been implicated in experimental models and limited clinical experiences, and they have been favorably manipulated by monoclonal antibodies, recombinant molecules, pharmacological agents or dietary supplements. In conclusion, pathogenic mechanisms that have been implicated in other systemic immune-mediated and liver diseases but under-evaluated or unassessed in autoimmune hepatitis warrant consideration and rigorous evaluation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple disturbances in immune regulatory pathways have been described in autoimmune hepatitis, but full understanding and integration of these pathogenic pathways into a cohesive knowledge base are incomplete [1, 2]. Studies have implicated molecular mimicries as a basis for epitope spread and loss of self-tolerance [3,4,5,6]. Dysregulated cytokine pathways may favor the differentiation and proliferation of liver-infiltrating cytotoxic CD8+ T lymphocytes [7,8,9,10,11,12,13,14,15], and over-expression of certain chemokines may orchestrate the migration of inflammatory and immune cells to the injured site [16]. Regulatory T cells (Tregs) may lack the number and function to suppress immune-mediated tissue damage [17,18,19,20,21], and disturbances in counter-regulatory molecules, such as microribonucleic acids (miRNAs) [22, 23], programmed cell death-1 (PD-1) protein [23,24,25,26,27,28], soluble CD163 [23, 29, 30], macrophage migration inhibitory factor (MIF) [23, 31, 32], and B cell-activating factor (BAFF) [23, 33, 34], may contribute to the homeostatic imbalance.
Genetic predispositions inside and outside the major histocompatibility complex (MHC) have been described [35,36,37,38,39,40,41]. Pathogenic processes that influence the survival of hepatocytes and activated lymphocytes (apoptosis) are being scrutinized [42,43,44]. Oxidative and nitrosative stresses that may damage cellular membranes, disrupt mitochondrial function, and promote hepatocyte loss are being defined [45,46,47,48,49], and efforts to monitor and prevent or reverse hepatic fibrosis are being developed [50,51,52,53]. Despite these investigational activities, there are several fronts that are being explored in diverse immune-mediated diseases that have been under-evaluated or not considered in autoimmune hepatitis.
The molecular signaling pathway involving the T cell immunoglobulin mucin proteins (Tims) and galectins can modulate T lymphocyte activity and the function of Tregs [54,55,56,57]. T cell exhaustion can result in a dysfunctional state characterized by sustained expression of inhibitory receptors, reduced T cell function, and altered outcome of immune-mediated diseases [58,59,60,61,62,63,64,65]. Dysregulation of antigen-independent inhibitory signals may favor autoimmunity [66,67,68], and alterations in the methylation of deoxyribonucleic acid (DNA) and modifications of the histones within nucleosomes may affect the transcriptional activity of genes that promote or suppress inflammatory activity (epigenetic changes) [69,70,71]. Furthermore, bacterial products and activated T lymphocytes may translocate from the intestinal microbiome to the peripheral lymph nodes and shape the immune response [72,73,74]. These elements have been implicated in the occurrence and severity of systemic immune-mediated and diverse liver-specific diseases, and they warrant rigorous evaluation in autoimmune hepatitis. This knowledge might in turn indicate pivotal pathogenic disturbances that could be targeted for therapeutic intervention.
The goals of this review are to describe key pathogenic pathways that have been under-evaluated or not examined in autoimmune hepatitis, indicate the potential role of these pathways in influencing disease occurrence and outcome, describe the insights in pathogenesis that may shape future therapies, and encourage the extension of investigational efforts into these areas.
Methods
English abstracts were identified in PubMed using the search words “pathogenesis of autoimmunity” and “pathogenesis of autoimmune hepatitis.” Abstracts judged pertinent to the review were identified; key aspects were recorded; and full-length articles were selected from relevant abstracts. A secondary bibliography was developed from the references cited in the selected full-length articles, and additional PubMed searches were performed to expand the concepts developed in these articles. The discovery process was repeated, and a tertiary bibliography was developed after reviewing selected articles from the secondary bibliography. The number of abstracts cited by PubMed from March 2015 to November 2017 and reviewed for pertinence to this review during the primary search was 850. The number of abstracts reviewed during the secondary and tertiary reviews exceeded 100. The abstracts judged most pertinent to the topic exceeded 300, and the number of full-length articles reviewed exceeded 100.
Results
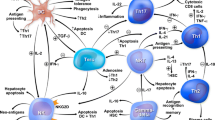
Six pathogenic pathways implicated in diverse systemic and liver-specific diseases have had little or no consideration in autoimmune hepatitis (Fig. 1). These pathways can modulate the adaptive immune response [54,55,56,57], alter the responsiveness of T lymphocytes after chronic stimulation [59, 61], weaken antigen-independent co-inhibitory signals that check immune activity [66, 67], influence the transcriptional activity of pivotal regulatory genes [69,70,71], and promote autoimmunity after translocation of gut-derived bacterial antigens and activated immune cells to the systemic circulation [72]. Perturbations within these pathways have possible pathogenic implications in autoimmune hepatitis, and their role in this disease warrants scrutiny.

Under-evaluated or unassessed pathogenic factors in autoimmune hepatitis. The family of T cell immunoglobulin mucin proteins (Tims) promotes the secretion of interleukin (IL)-10 by regulatory B cells (Bregs), clears apoptotic bodies, expands the population of granulocytic myeloid-derived suppressor cells (G-MDSCs), and modulates the response of T helper 1 (Th1) lymphocytes. These combined actions could reduce the severity of autoimmune hepatitis (blunted arrow). The family of galectins can interact with the Tims family and block T cell antigen receptors (TCRs), limit T cell activation, induce T cell apoptosis, and reduce the production of interferon gamma (IFN-γ). These combined actions could also have an inhibitory effect on disease activity (blunted arrow). In contrast, changes in the composition of the intestinal microbiome (dysbiosis) and increased permeability of the intestinal mucosal barrier could enhance disease activity (sharp arrow) by facilitating translocation of gut-derived microbial antigens and activated immune cells into the systemic circulation, impairing immune tolerance, and affecting gender predisposition for the disease. Epigenetic changes manifested by hypomethylation of deoxyribonucleic acid (DNA) and histone modifications could also enhance disease activity by altering the transcriptional activity of key genes. Vitamin D status is one factor that may affect the gene response (sharp arrow). The programmed cell death-1 protein (PD-1) and its ligands and T cell exhaustion might help counter the immune stimulatory effects on disease severity (blunted arrows) by reducing T cell activation, inducing the formation of regulatory T cells (Tregs), and limiting the production of pro-inflammatory cytokines, including IL-2, IFN-γ, and tumor necrosis factor alpha (TNF-α). Perturbations in these interactive regulatory mechanisms could influence the persistence and severity of autoimmune hepatitis, and their investigation could suggest corrective interventions
Pathway Involving T Cell Immunoglobulin Mucin Proteins
The T cell immunoglobulin mucin proteins (Tims) constitute a family of three glycoproteins expressed on the surface of T helper-1 (Th1) lymphocytes, Th2 lymphocytes, and dendritic cells [54, 75, 76] (Table 1). Tim-2 shares 85% nucleotide sequence homology with Tim-1 [77], and Tim-1, Tim-3, and Tim-4 are the family members recognized in humans [78]. All Tim proteins have common structural motifs that characterize the family, and an immunoglobulin variable (IgV) domain constitutes a cleft that can serve as a receptor for ligand binding [79,80,81]. Each Tim family member influences the immune response, and together they modulate peripheral immune tolerance. Their pathogenic roles in autoimmune hepatitis are speculative.
Tim-1
Tim-1 polarizes the differentiation of activated CD4+ lymphocytes, and it is preferentially expressed on Th2 lymphocytes [82] (Table 1). The secretion of interleukin (IL)-10 by B cells is promoted by Tim-1 [83], and regulatory B cells (Bregs) expressing Tim-1 have broad anti-inflammatory actions. Bregs reduce the secretion of IL-17 and interferon gamma (IFN-γ), and they favor the generation of Tregs [84,85,86,87,88]. Phosphatidylserine is present on apoptotic cells, and the IgV domain of Tim-1 serves as a receptor for phosphatidylserine. Bregs expressing Tim-1 can bind apoptotic bodies, and this ligation increases the production of IL-10 [88] which in turn can suppress immune reactivity [89, 90].
Tim-1 is also present on Th1 cells, and it can help regulate the activity of this population (Table 1). Blockade of Tim-1 expression with a Tim-1-specific antibody has inhibited Tim-1-dependent Th1 responses while preserving Th2 responses in a murine model of cardiac transplantation [91]. The net result has been prolonged survival of the fully mismatched cardiac allografts [91]. These findings suggest that Tim-1 may have positive or negative inhibitory effects on different T cell populations.
The avidity with which Tim-1 is engaged on the surface of the T cell is a major determinant of T cell behavior (Table 1). Anti-Tim-1 antibodies with a high infinity for Tim-1 can increase the population of antigen-specific T cells, the secretion of IFN-γ and IL-17, and the severity of experimental autoimmune encephalitis [78]. In contrast, low affinity antibodies can prevent these pro-inflammatory responses and promote a Th2 response [78]. Clarification of the immunomodulatory effects of Tim-1 in autoimmune hepatitis might suggest opportunities for targeted intervention.
Tim-3
Tim-3 is expressed on activated Th1 lymphocytes, Tregs, natural killer (NK) cells, and antigen-presenting dendritic cells [92] (Table 1). The ligation of Tim-3 with galectin-9 modulates the Th1 immune response directly by triggering the death of Th1 lymphocytes that secrete IFN-γ [55, 93, 94]. IFN-γ stimulates the secretion of galectin-9 by Th1 lymphocytes, and the galectin-9 in turn marks the Th1 lymphocytes for destruction [93, 94].
Tim-3 can also dampen the Th1 response indirectly by expanding the population of granulocytic myeloid-derived suppressor cells (G-MDSCs) that are produced in high levels in cancer, inflammation, and infection [94,95,96,97,98] (Table 1). These cells constitute a population of neutrophils that are close to naïve neutrophils [98], but they are characterized by the surface markers CD11b+Ly6G+ (lymphocyte antigen 6 complex, locus G) [94, 99].
G-MDSCs can suppress T lymphocyte activity by generating reactive oxygen species (ROS) [95], by secreting immunosuppressive molecules such as CD200 that can inhibit monocytes, granulocytes, and T lymphocytes [97, 100, 101], and by promoting the dissociation of antigenic peptides from the T cell antigen receptor (TCR) of CD8+ T lymphocytes [102]. Tim-3 on the surface of macrophages and dendritic cells can also bind apoptotic bodies, and it can reduce autoantibody production by promoting phagocytosis of the apoptotic bodies and eliminating them as potential autoantigens [103]. The role of G-MDSCs in autoimmune hepatitis is unknown.
Tim-4
Tim-4 is present on macrophages and dendritic cells [81], and it is the natural ligand for Tim-1 [104] (Table 1). Tim-4 can also bind to phosphatidylserine on the surface of apoptotic cells in conjunction with Tim-1, and it can promote the phagocytosis and clearance of these potential antigens [81]. The amount of Tim-4 influences the degree of its ligation with Tim-1, and this interaction in turn affects T lymphocyte proliferation. High doses of Tim-4 promote T lymphocyte proliferation, whereas low doses are inhibitory [78, 104]. In this fashion, Tim-4 can modulate the anti-inflammatory effects of Tim-1. Interventions that manipulate Tim-4 levels might be useful in managing immune-mediated diseases, and this possibility is another reason to consider evaluation of the Tim family members in autoimmune hepatitis.
Pathway Involving Galectins
Galectins are a family of guanosine diphosphates that are secreted directly into the extracellular environment without passing through the endoplasmic reticulum and Golgi apparatus [105, 106]. Galectins can bind β-galactosides in glycoproteins, and they can influence molecular interactions at the cell surface of immune cells [107] (Table 1). Alterations in the glycosylation of cell surface molecules can affect immune cell migration, activation of Toll-like receptors (TLRs), and differentiation of naïve T lymphocytes into Th1, Th2, and Th17 populations [107,108,109,110]. Macrophages, dendritic cells, B and T lymphocytes, Kupffer cells, and endothelial cells express galectins, and these molecules are abundant in the liver [106, 111].
Galectin-3 has sequence homology with the anti-apoptotic B cell leukemia/lymphoma-2 molecule (Bcl-2), and it can promote T cell proliferation or hepatocyte survival by preventing apoptosis [112]. Galectin-3 can also form a lattice with glycoprotein which restricts the presentation of antigen by TCRs. Galectin-3 may thereby increase the threshold for T lymphocyte activation and limit the autoreactive response [113].
Galectin-9 is the ligand of Tim-3 [79], and studies are beginning to clarify its role in liver disease [19, 106, 114, 115] (Table 1). The interaction between Tim-3 and galectin-9 increases the flux of calcium into Th1 lymphocytes and selectively induces their aggregation and death. Th1 immune responses are inhibited, and the production of IFN-γ is reduced [55, 116]. Galectin-9 can also modulate signaling pathways that influence cytokine profiles and induce apoptosis independently of Tim-3 [107, 117].
In experimental rheumatoid arthritis, galectin-9 has decreased the production of pro-inflammatory cytokines, IL-12, IL-17, and IFN-γ, in synovial fluid. It has also increased the in vitro differentiation of naïve T lymphocytes into Tregs while suppressing their differentiation into pro-inflammatory Th17 cells [118]. In a murine model of chronic eye infection by herpes simplex virus (HSV), the administration of galectin-9 has reduced the severity of keratitis by increasing apoptosis of effector T cells, reducing the production of pro-inflammatory cytokines, and expanding the population of Tregs [119].
Paradoxically, galectin-9 in the absence of Tim-3 has increased in a dose-dependent fashion the secretion of the pro-inflammatory cytokines IFN-γ and tumor necrosis factor alpha (TNF-α) by cultured T helper lymphocytes [117]. These observations underscore the variable and inverse actions of certain immune modulators depending on their amount and microenvironment. They also indicate the difficulty in developing molecular interventions that are precisely targeted and dosed to minimize unsuspected and unwanted consequences [120].
Tim-3 and Galectin-9 in Non-autoimmune Liver Diseases
The Tim-3/galectin-9 pathway is being investigated in diverse non-autoimmune liver diseases [106]. Plasma levels of galectin-9 are upregulated in chronic hepatitis C, and recombinant galectin-9 can expand Tregs and promote the apoptosis of virus-specific CD8+ cytotoxic T lymphocytes [121]. The reduced immune reactivity may in turn facilitate the maintenance of chronic infection with the hepatitis C virus (HCV) [106, 122].
Galectin-9 and Tim-3 are upregulated in nonalcoholic fatty liver disease (NAFLD), and the apoptosis of natural killer T (NKT) cells is promoted [106, 123]. The production of IL-15 is also stimulated by Tim-3 and galectin-9, and IL-15 induces proliferation of NKT cells. The counter-regulatory actions of NKT cell depletion and stimulation may modulate inflammatory activity in NAFLD [123].
Galectin-9 levels are also increased in the plasma of patients with acute alcoholic hepatitis compared to patients with alcoholic cirrhosis and healthy individuals [106, 124]. High levels of galectin-9 and Tim-3 are expressed by T lymphocytes from these patients, and they may weaken the innate immune response that protects against septic complications [124].
Tim-3 and Galectin-9 in Autoimmune Hepatitis
Concanavalin A induces a severe immune-mediated liver injury that is dependent on activated T lymphocytes, and recombinant galectin-9 has ameliorated this injury in a murine model [114, 115]. Circulating levels of pro-inflammatory cytokines (TNF-α, IFN-γ, and IL-6) are reduced; serum levels of alanine (ALT) and aspartate (AST) aminotransferase improve; concanavalin A-specific CD4+ T cells undergo apoptosis; and the number of Tregs is expanded [114]. Furthermore, survival has been prolonged, and histological features improved in treated mice compared to control mice [115].
In patients with autoimmune hepatitis, the expression of galectin-9 on Tregs is reduced, and the presentation of Tim-3 on the surface of naïve CD4+CD25− T cells is downregulated [19]. These changes may diminish the function of the Tregs and render T lymphocytes less able to be regulated. The net effect may be worsening of disease activity as suggested by the inverse correlation between Tim-3 downregulation and serum AST levels [19].
Further insights into the regulatory deficiencies and homeostatic imbalances of the Tims and galectins involved in autoimmune hepatitis may identify factors that can be manipulated to attenuate disease activity.
Pathway Influencing T Cell Exhaustion
CD4+ and CD8+ T lymphocytes are the principal cell populations that influence the occurrence and severity of autoimmune hepatitis [125], and multiple molecular and cellular interactions influence the activation, differentiation, proliferation, and survival of these cells [2, 16]. Evolving management strategies in autoimmune hepatitis have been designed to disrupt or modulate these activation pathways [126], but none has been formulated to therapeutically induce T cell exhaustion.
T cell exhaustion has been well described in experimental models and patients with persistent antigen stimulation associated with chronic infection (animal models) [58, 127, 128] and malignancy (animal models and patients) [60, 65, 129] (Table 1). It has been recognized as a deleterious consequence of persistent antigen exposure that weakens immune-mediated defense mechanisms, and reversal of T cell exhaustion has been a therapeutic goal in these instances [62, 65]. Intentional induction of T cell exhaustion as a management strategy for autoimmune disease has only recently be considered, and the concept has been justified by the recognized plasticity of the CD4+ and CD8+ T cell populations and the multiplicity of factors that modulate their exhaustion [61, 63, 64].
T cell exhaustion in autoimmune disease may have the beneficial effect of attenuating disease activity and preventing relapse [61, 63, 64]. Studies have already demonstrated that T cell exhaustion can be reversed [130, 131] and induced [61]. This plasticity of the CD4+ and CD8+ T cells suggests that manipulations are possible to adjust effector T cell activity by direct intervention [64]. The mechanisms that induce or prevent T cell exhaustion in autoimmune hepatitis have not been evaluated, but their clarification might direct future management strategies.
Characteristics of T Cell Exhaustion
T cell exhaustion connotes a dysfunctional state in which immune responsiveness becomes progressively diminished during persistent antigen exposure and stimulation of TCRs [59, 61,62,63, 129, 132, 133]. The exhausted phenotype is characterized by the expression of multiple inhibitory receptors on the cell surface, progressive loss of cytokine production (IL-2, IFN-γ, and TNF-α), and altered metabolic and transcriptional activity [62, 65, 134] (Table 1).
The expression of the programmed cell death-1 (PD-1) protein is a key manifestation of T cell exhaustion [135,136,137], but 12 other inhibitory receptors have been described to date. These co-inhibitory surface molecules are differentially expressed and associated with the exhausted state, and they include cytotoxic T lymphocyte antigen-4 (CTLA-4), lymphocyte-activated gene-3 (LAG-3), NK cell receptor 2B4 (CD244), Tim-3, B and T lymphocyte activator (BTLA), NK cell receptor BY55 (CD160), and CD200 [58, 65, 134, 138, 139]. The co-expression of multiple inhibitory receptors is the requisite feature of T cell exhaustion, and the number of these inhibitory receptors reflects the severity of the exhaustion [62, 138].
Both CD4+ and CD8+ T cells can become progressively exhausted, but their exhausted phenotypes may differ [134, 139]. The expression of inhibitory molecules and transcriptional factors are similar between the two T cell populations, but CD4+ T cells exhibit more pronounced expression of CTLA-4, BTLA-4, and CD200 [134, 139]. Exhausted CD8+ T cells have reduced expression of the interleukin-7 receptor (IL-7R), and this reduction correlates with the duration of antigen exposure. The reduction of IL-7R is also associated with decreased expression of the anti-apoptotic Bcl-2 and a shortened survival of the exhausted CD8+ T cells [140].
The expression of IL-7R is a feature of memory T cells whose proliferation after clearance of antigen and ability to reactivate with restimulation are maintained by IL-7 and IL-15 [61, 62]. The level of IL-7R expression is a distinguishing feature between exhausted CD8+ T cells and memory T cells, and the cell surface display of IL-7RlowPD-1high has been proposed as the phenotype of exhausted CD8+ T cells [61].
Severity of T Cell Exhaustion
T cells do not exhibit uniform responses during chronic antigen exposure, and subsets expressing different co-stimulatory molecules may have different proliferative potentials and propensities for exhaustion [65, 141]. Furthermore, exhausted T cells do not have the same severity of exhaustion or ability to recover. Exhausted T cells that have highly expressed T-bet (T-box transcription factor) and intermediately expressed PD-1 (T-bethighPD-1int) are still able to secrete IFN-γ and TNF-α, proliferate at a reduced level, and recover from exhaustion [65, 142]. In contrast, T cells with the highly expressed transcription factor, eomesodermin (eomes), and inhibitory factor, PD-1, are in a severe state of exhaustion with limited reversibility, and they are defined by the surface markers, eomeshighPD-1high [62, 143, 144]. Variations among T cell subsets in their propensity to become exhausted and in the severity and reversibility of the exhaustion that develops can influence the effectiveness of interventions designed to induce or reverse the exhausted state.
Modulators of T Cell Exhaustion
Co-stimulatory signals can override the propensity for CD8+ T cell exhaustion, and activated CD4+ T cells are the main promoters of continuous CD8+ T cell activity [61, 62]. In the absence of the co-stimulatory effect of CD4+ T cells, CD8+ T cells develop an exhausted phenotype during persistent antigen exposure [145] (Table 1). IL-21, produced by activated CD4+ T cells, induces the proliferation of CD8+ T cells, and it thereby preserves their identity as effector cells [146,147,148,149,150,151]. CD2 is also instrumental in preventing the exhaustion of CD8+ T cells in vitro [61].
CD2 is an adhesion molecule on the surface of T cells, and it maintains their function [152]. T cells with low levels of CD2 are associated with increased viral loads and disease progression in untreated patients with human immunodeficiency virus (HIV) [153]. Similarly, patients with advanced melanoma and recurrent disease have fewer CD2-positive lymphocytes and shorter survivals than patients without recurrent disease [154]. In these instances of chronic infection and malignancy, low levels of CD2 expression may have contributed to exhaustion of the immune defenses and worsening of the disease.
Other molecules that favor T cell exhaustion are IL-10 and PD-1 [135, 155] (Table 1). The inhibitory effects of IL-10 and PD-1 on CD8+ T cells are independent and synergistic. Simultaneous blockade of both inhibitory pathways invigorates antiviral T cell responses more than blockade of any individual pathway [155]. Transforming growth factor beta (TGF-β) has been associated with increased apoptosis of virus-specific CD8+ T cells, but it has not been essential for functional exhaustion [156], as confirmed by blocking experiments [157, 158].
Therapeutic induction of T cell exhaustion is a fresh concept in the management of autoimmune diseases, and there are multiple modulators within the network that maintains T cell activity [61,62,63,64]. Investigations of T cell exhaustion in autoimmune hepatitis may clarify mechanisms that perpetuate effector cell activity and survival despite chronic antigen stimulation. These insights may help explain variations in disease severity and propensities for spontaneous exacerbation or relapse after drug withdrawal, and they may also direct interventional strategies.
Pathway Involving Programmed Cell Death-1 (PD-1) Protein and Its Ligands
PD-1 (CD279) is a 288 amino acid protein that is expressed on the surface of activated T cells, B cells, NK cells, Kupffer cells, and thymocytes [24, 67, 68]. It is an antigen-independent inhibitory receptor that suppresses T cell activation, decreases lymphocyte proliferation, and maintains immune tolerance [66, 159] (Table 1). The actions of PD-1 can also synergize with signals mediated by TGF-β [159], and this synergy can induce naïve T cells to transform into Tregs [160].
The ligands of PD-1 are programmed death ligand-1 (PD-L1) [161, 162] and programmed death ligand-2 (PD-L2) [163]. These ligands are each preferentially expressed on different cell surfaces, including hepatic sinusoidal endothelial cells, Kupffer cells, hepatocytes, and hepatic stellate cells (PD-L1) [164] and dendritic cells and activated macrophages (PD-L2) [165]. The expression of each ligand is enhanced by pro-inflammatory stimuli (lipopolysaccharide [LPS], IFN-γ, and IL-4) [166], and their ligation with PD-1 dampens the adaptive immune response [136] (Table 1). The principal inhibitory mechanism of the ligation is to change the metabolism of activated T cells from aerobic glycolysis to fatty acid oxidation [167,168,169,170]. This metabolic switch to fatty acid oxidation may also induce the formation of Tregs [170, 171].
The expression of PD-1 is the hallmark of T cell exhaustion [59, 61, 63, 135,136,137], and the PD-1/PD-L1/2 pathway has also been incriminated by preliminary studies in autoimmune hepatitis [24,25,26,27,28]. For these reasons, the PD-1 axis warrants further investigation as a potential target of therapeutic interventions designed to strengthen its immune inhibitory effects.
PD-1 Inhibitory Axis and Autoimmune Hepatitis
Serum antibodies to PD-1 (anti-PD-1) have been present more frequently in patients with autoimmune hepatitis (63%) than in normal individuals (3%) and patients with drug-induced liver injury (8%), acute viral hepatitis (13%), or primary sclerosing cholangitis (18%) [26] (Table 1). The anti-PD-1 levels have also correlated with serum concentrations of bilirubin and ALT, and the presence of anti-PD-1 has been associated with a slow response to corticosteroid treatment and relapse after drug withdrawal [26]. The pathogenic effects of circulating anti-PD-1 in autoimmune hepatitis are unknown, but one possibility is that the disease-related antibodies block the inhibitory signals generated by PD-1 and thereby promote T cell activation and proliferation.
Variant forms of the PD-1 molecule can be expressed by the PD-1 gene, and one variant is soluble because the domain encoded by exon 3 that anchors the molecule to the cell membrane has been spliced out [172]. This variant soluble form (PD-1Δex3) has an intact extracellular domain and ligates normally with PD-L1 and PD-L2. Patients with autoimmune hepatitis who have untreated active disease, disease in exacerbation, and incomplete responses to standard corticosteroid therapy have had increased plasma levels of soluble PD-1 [28] (Table 1). These patients have also had a > threefold upregulated expression of PD-1 on CD4+ T cells compared to healthy subjects.
The apparent paradoxical association of increased plasma levels of soluble PD-1 with disease activity in autoimmune hepatitis is unexplained, but the findings suggest that the variant soluble form interferes with the binding of full-length PD-1 with its ligands. In patients with rheumatoid arthritis, the levels of soluble PD-1 in synovial fluid have correlated with synovial levels of TNF-α, and the PD-1Δex3 splice variant has blocked the inhibitory effect of membrane-bound PD-1 on T cell proliferation [173].
Additional evidence of a disturbed PD-1 axis in autoimmune hepatitis has been evident in assessments of liver tissue. Messenger ribonucleic acid (mRNA) for PD-L2 has been less abundant in liver tissue from patients with autoimmune hepatitis than in liver samples from patients with primary biliary cholangitis (PBC) and chronic hepatitis C [24] (Table 1). Furthermore, IL-4, the principal inducer of PD-L2, has been absent in the liver tissue of patients with autoimmune hepatitis. These findings suggest that patients with autoimmune hepatitis have a deficiency of one of the key ligands for PD-1 in liver tissue and that the absence of PD-L2 may compromise the inhibitory effects of PD-1 on the adaptive immune response.
Implications in Autoimmune Hepatitis
The PD-1 axis has been implicated as an inhibitory mechanism by which to induce T cell exhaustion and suppress T cell activation and proliferation. These attributes may be deficient in autoimmune hepatitis as a result of antibodies to PD-1 that interfere with its inhibitory function [26], over-expression of variant soluble PD-1 molecules that block PD-1 signaling [28], or deficient expression of ligands that are essential for PD-1 activity [24]. The results of the preliminary studies disclosing these deficiencies in autoimmune hepatitis warrant further clarification to translate the correlative findings into pathogenic mechanisms. These investigations might also support future efforts to bolster the PD-1 axis [61, 64].
Pathway Involving Epigenetic Changes
Changes in chromatin structure can alter the transcriptional activity of promoter genes without altering the sequence of deoxyribonucleic acid (DNA) [69,70,71, 174, 175]. These epigenetic changes involve alterations in the methylation status of DNA and modifications of the histones that constitute the chromatin (Table 1). Such changes can silence pro-inflammatory genes or de-repress anti-inflammatory genes and alter the clinical manifestations and outcome of immune-mediated diseases. The changes can be induced by cues from the environment [69,70,71], and this interaction can strengthen hypotheses that link the occurrence and outcome of autoimmune disease with exogenous factors such as drugs, pollutants, infections, and diet [176,177,178,179].
Epigenetic changes can also occur as a natural consequence of aging. Genomic instabilities can be induced by repeated cycles of DNA replication and the cumulative effects of protracted exposure to toxic environmental elements [180,181,182] (Table 1). A stable inheritable trait may be a consequence of these epigenetic perturbations and influence disease risk in progeny [69, 174, 175]. Individual variations in phenotype, disparities in disease occurrence, and dissimilarities in outcome may also be attributable in part to epigenetic alterations [178].
Interventions designed to promote the expression of tumor suppressor genes and upregulate the expression of tumor rejection antigens are being evaluated in malignancy [183,184,185,186,187,188]. These efforts at epigenetic manipulation suggest that therapeutic interventions that modulate the transcriptional activity of pro- and anti-inflammatory genes in autoimmune disease may be possible [189,190,191]. Epigenetic changes have been described in systemic lupus erythematosus (SLE) [192, 193], rheumatoid arthritis [194], and systemic sclerosis [195], and they have also been recognized in primary biliary cholangitis (PBC) [196] and NAFLD [197]. Similar investigations are warranted in autoimmune hepatitis to clarify pathogenic mechanisms, risk factors, and management possibilities.
DNA Demethylation
DNA within chromatin is methylated at the site of a cytosine nucleotide that is separated by a phosphate from a guanine nucleotide (CpG site) [69]. These CpG sites are in a linear sequence along the DNA, and their methylation inhibits the binding of transcription factors to the gene [69, 198] (Table 1). Gene activity relates to the density of DNA methylation. Heavily methylated DNA inhibits the binding of transcription factors to the gene promoter and reduces production of gene product. In contrast, hypomethylated DNA accommodates the transcription factors and sustains or promotes gene activity. The gene promoters implicated in SLE, rheumatoid arthritis, and multiple sclerosis are hypomethylated [176, 199].
The methylation status of DNA is modulated by methyl-DNA binding domain proteins (MBDs) which adhere to the methylated CpG site [200, 201] and by the ten eleven translocation oxygenases (TET1-3) [202,203,204,205] (Table 1). Some MBDs have DNA demethylase activity which promotes the reversal of methylation and an increase in transcriptional activity, whereas other members of the same family suppress gene activity [206,207,208]. The TET family of enzymes catalyze the oxidation of 5-methylcytosine to 5-hydroxymethylcytosine, 5-formylcytosine, or 5-carboxylcytosine, and they can also reverse methylation and increase gene activity [202,203,204,205].
Countering the stimulatory actions of certain MBDs and the TET enzymes are the DNA methyltransferases that methylate DNA using S-adenosylmethionine as the principal methyl donor [176, 209, 210] (Table 1). Perturbations in this counter-regulatory interactive network can upset homeostatic balance and contribute to the development and severity of autoimmune disease. The counter-regulatory proteins affecting DNA methylation are logical targets for therapeutic manipulation [189].
Histone Modifications
Four histones arranged as an octamer constitute a nucleosome around which the DNA molecule within the chromatin is wrapped [71, 211]. These histones can be modified by processes (phosphorylation, methylation, acetylation, and ubiquitination) which alter the amino acids residing in the tail of the molecule [212,213,214]. Counter-regulatory enzymes shape the histone modifications, and their cumulative effects influence the transcriptional activity of the gene [69, 214].
Histone acetyltransferase can transfer an acetyl group to an N-terminal lysine [215], and histone deacetylase can remove the acetyl group [216, 217] (Table 1). Histone methyltransferases can methylate arginine residues [213], and the peptidylarginine deiminases (PADs) can convert histone methylarginine to citrulline [218]. The amino acid alterations induced by the interactive enzymes do not have a predictable effect on gene activity, and the suppression or promotion of transcriptional activity can depend on the nature and location of the amino acid transformations [70, 217, 219]. Strategies to modulate specific enzymes and favorably alter histone structure may have a less certain effect on gene activity than altering the methylation status of DNA [189].
Considerations in Autoimmune Hepatitis
Vitamin D deficiency (serum 25-hydroxyvitamin D3 level, < 50 nmol/L) has been found in 51–68% of patients with non-cholestatic chronic liver disease [220, 221], and the vitamin D level is < 30 μg/L in 81% of Turkish patients with autoimmune hepatitis [46]. The liver hydroxylates the natural skin-derived vitamin D3 to the metabolically inactive circulating form of 25-hydroxyvitamin D3. This circulating form can then be activated in the kidney to 1, 25-dihydroxyvitamin D3 [222, 223]. The active metabolite is a ligand for the vitamin D receptor (VDR) which can form a heterodimer with the retinoid X receptor (RXR) and regulate gene expression by binding to the vitamin D response element (VDRE) located in introns [224, 225]. The binding of VDR to VDRE attracts co-activators and enzymes that can induce histone acetylation, alter chromatin structure, and promote gene transcriptional activity [224]. Genetic polymorphisms can influence the structure of the VDR, and the association of these polymorphisms with autoimmune hepatitis suggests that individuals may have different inherited VDR binding avidity and different epigenetic effects on gene activity [226, 227].
Vitamin D deficiency has been associated mainly with cirrhosis, and it may reflect an inability of the liver to hydroxylate vitamin D3 [220, 221]. Other factors, including diet, malabsorption, and medications that inhibit the cytochrome P450 enzymes involved with vitamin D metabolism, may also contribute [220, 228]. 1, 25-Dihydroxyvitamin D3 has been ascribed diverse immune, inflammatory, and anti-fibrotic effects in chronic liver disease [229, 230], and future investigations of epigenetic changes in autoimmune hepatitis must examine the role of vitamin D deficiency in modulating gene function and promoting disease activity and progression.
Requisites for Therapeutic Intervention
Therapeutic interventions must be directed at the principal epigenetic change that contributes to the disease, and they must be designed for precision targeting and trans-generational tolerance [71]. Induced undirected epigenetic changes may disrupt homeostatic pathways and lead to unwanted consequences and an undesired inheritable trait. Regions near the binding sites for gene regulation are the preferred targets, and most epigenetic variations in the immune-mediated diseases have been in these areas [231] (Table 1). Multiple areas that are collectively arrayed in highly expressed genes are super-enhancer regions, and they are likely to modulate disease-relevant gene function [232,233,234]. The super-enhancer regions are prized targets for manipulation.
The enzymes that modify the DNA and histones within the chromatin are active at certain sites because of particular genomic sequences and sequence-specific binding factors [189] (Table 1). Different isotypes of the same enzyme family have cell and site specificity because of these genomic sequences and the binding factors that characterize the cell’s microenvironment [235, 236]. Interventions can be designed to modulate the particular isotype of the enzyme contributing to the undesired gene effect. In this fashion, the cell type and culpable genomic sequence can be precisely targeted [189].
Progress in designing epigenetic interventions for autoimmune hepatitis requires delineation of the changes that are affecting the disease-pertinent genes and recognition of the enzymatic isotypes and binding proteins shaping those changes. This knowledge will help explain disparities in disease risk between different populations and clarify pathogenic mechanisms.
Pathway Involving Dysbiosis and Translocation of Gut-Derived Antigens and Immune Cells
The human intestinal tract harbors 10–11 trillion bacteria representing 500–1500 species that can be organized in four main phyla (Actinobacteria, Firmicutes, Proteobacteria, and Bacteroidetes) [74, 237,238,239]. This vast microbial population constitutes the intestinal microbiome, and it remains relatively stable from late childhood through adulthood becoming less diverse and more variable over short intervals in the elderly [239,240,241,242]. Multiple environmental factors can influence the composition of the microbiome, including diet, antibiotics, and socioeconomic status [243], but the commensal populations are generally well tolerated because of an acquired immune tolerance [244].
Changes in the composition of the intestinal microflora (dysbiosis) have been associated with diverse autoimmune diseases (type 1 diabetes, rheumatoid arthritis, multiple sclerosis, and inflammatory bowel disease) [245,246,247,248,249], and these findings have suggested that changes in the intestinal microbiome may overcome intestinal immune tolerance and become a pathogenic factor. The intestinal microbiome has also been implicated in several liver diseases, including autoimmune hepatitis [72, 73, 250, 251] (Table 1).
An animal model of NAFLD has demonstrated the presence of microbial products in the portal circulation and the release of pro-inflammatory cytokines that sustain the disease [252,253,254]. TLRs in biliary epithelial cells (BECs) respond to lipopolysaccharide (LPS), a major component of gram-negative bacteria, by producing IFN-γ in a murine model of primary sclerosing cholangitis (PSC) [255]. BECs in PBC express TLRs that respond to gut-derived bacterial products (LPS, flagellin, and cytosine-phosphorothioate-guanine oligonucleotide), and the BECs produce the pro-inflammatory cytokines, IL-1β, IL-6, IL-8, and TNF-α [256,257,258]. Commensal intestinal microflora have had a protective effect in a model of PSC [259], and LPS-stimulated NK cells have contributed to bile duct destruction and disease progression in a model of PBC [260, 261].
The intestinal microbiome is now recognized as a reservoir of immunogenic substrates and activated immune cells that can shape systemic innate and adaptive immune responses by mechanisms of translocation [74]. Gut-derived activated immune cells have been demonstrated in peripheral lymph nodes [262], and bacterial products have been found in the systemic circulation of patients and experimental models with liver disease [72, 254, 263, 264]. In autoimmune hepatitis, a pathogenic role for the intestinal microbiome has been proposed [72, 73] and this possibility warrants further evaluation [265].
Intestinal Microbiome and Autoimmune Hepatitis
Dysbiosis has been described in an animal model and in patients with autoimmune hepatitis (Table 1). The diversity and total load of intestinal bacteria have been reduced in a murine model of autoimmune hepatitis, and the dysbiosis has been associated with disease activity, Th1 response, and reduced number of Tregs in the liver tissue [73]. Dysbiosis, manifested by a reduced number of intestinal anaerobes (Bifidobacterium and Lactobacillus), has also distinguished patients with autoimmune hepatitis from healthy subjects [72].
Plasma levels of LPS have been higher in patients with autoimmune hepatitis than in healthy individuals, and these findings have justified speculation that translocation of gut-derived microbial products and activated immune cells may be instrumental in the disease [72]. This possibility has been supported by evidence that the intestinal mucosal barrier may be disrupted in these patients [72]. Histological examination of duodenal mucosa has disclosed small, irregularly arranged, duodenal villi with inflammatory cells in the lamina propria and damaged tight junctions (Table 1). Immunohistochemical analyses of the proteins that maintain tight junctions within the duodenal mucosa have also indicated decreased expression of zona occludens-1 and occludin [72]. These findings cannot be extrapolated to the colonic mucosa, but they should encourage comparable assessments of the colonic mucosa and the integrity of its barrier function.
Intestinal Microbiome and Gender Bias
The composition of the intestinal microbiome may influence the female bias for autoimmune disease, and it is a consideration that has not been evaluated in autoimmune hepatitis (Table 1). Female non-obese diabetic (NOD) mice have a 1.3–4.4-fold greater incidence of type 1 diabetes than male NOD mice, and this difference in susceptibility is absent in germ-free animals [266]. Differences in the composition of the intestinal microflora between genders of NOD mice have been eliminated by male castration [266], and transfer of intestinal microbiota from adult male mice to immature female mice has resulted in elevated serum testosterone levels in the females and protection against type 1 diabetes [267]. Furthermore, blockade of the androgen receptor with flutamide has attenuated the changes associated with the male-specific microbiome in the female NOD mice [267]. The transfer of T cells from male mice to flutamide-treated female mice has not protected against the development of diabetes [267].
These demonstrations in genetically susceptible murine models of autoimmune disease indicate that the composition of the intestinal microbiome can vary between genders, be influenced by male hormones, and also produce testosterone that can alter disease risk in females [266,267,268]. Similar observations in experimental models and patients with autoimmune hepatitis could not only transform current pathogenic concepts of autoimmune hepatitis and its female propensity but also prove pivotal in developing new management strategies [74, 265].
Therapeutic Implications
The integration of previously under-evaluated or unassessed pathogenic mechanisms into the knowledge base of autoimmune hepatitis could suggest new management strategies (Table 2). The challenges are to identify the predominant mechanism to target in an individual patient, design interventions that are directed with precision, anticipate and minimize side effects, and institute the intervention at the appropriate time and for the optimal duration.
Pathogenic mechanisms may be variably active at different stages of the disease. Inhibitory receptors may not be uniformly expressed on activated T cells or displayed consistently [62]. The numbers and functions of CD4+, CD8+, and regulatory T cells may change during the inflammatory process. Effector cells may enter or reverse exhaustion, and apoptotic activity may fluctuate [62]. Inhibitory molecules may paradoxically have stimulatory effects in certain microenvironments [57, 78], and interventions may be efficacious only when the pathogenic mechanisms are robust [61]. Most importantly, infection and malignancy may be consequences of the disease or its prospective intervention [189]. These factors complicate the emergence of novel therapies, and they must be considered in evaluating the performance of any new management strategy.
Prospective Interventions that Modulate the Tim Proteins
Tim-1 can inhibit or stimulate T cell proliferation and the production of pro-inflammatory cytokines depending on the affinity with which Tim-1 is engaged with its Tim-4 ligand [78, 269]. Two monoclonal antibodies specific for Tim-1 have opposite effects on T cell proliferation, production of IFN-γ and IL-17, and the severity of experimental autoimmune encephalomyelitis [78] (Table 2). Both antibodies have closely related epitopes in the IgV domain of the Tim-1 molecule, but the activating antibody has an avidity for the molecule that is 17 times greater than the inhibitory antibody. These findings suggest that blocking antibodies can be designed with an avidity that promotes the desired action of the Tim-1 molecule. A blocking anti-Tim-1 monoclonal antibody has prolonged allograft survival by downregulating Th1 responses, promoting Th2 responses, and enhancing the regulatory activity of Tregs in a murine model of mismatched cardiac transplantation [91].
Prospective Interventions that Modulate Galectin Levels
Galectin-9 administered to murine models has improved the keratitis associated with HSV infection [119] and the histological features and survival of concanavalin-induced fulminant hepatitis [115], and it has reduced the pro-inflammatory cytokines associated with rheumatoid arthritis [118] (Table 2). These findings in experimental models of immune-mediated disease suggest that galectin-9 may evolve as a therapeutic intervention in autoimmune hepatitis [55, 57, 118]. Countering this prospect is an awareness that galectin-9 can act independently of Tim-3 and increase the production of the pro-inflammatory cytokines INF-γ in Th1 cells and TNF-α in Th2 cells in a dose-dependent fashion [107, 117].
Prospective Interventions that Induce T Cell Exhaustion
The known factors that promote the activity of CD8+ T cells and prevent their exhaustion (activated CD4+ T cells, IL-21, and CD2) [61, 146,147,148,149,150,151,152] and those that favor exhaustion (IL-10 and PD-l) [135, 155] are potential targets for interventions that induce T cell exhaustion. Preliminary studies have indicated that chimeric PD-L1 can skew the differentiation of non-exhausted robust CD8+ T cells with IL-7RhighPD-1low surface markers toward the exhausted phenotype of IL-7RlowPD-1high [61] (Table 2). Furthermore, alefacept, a recombinant fusion molecule consisting of lymphocyte function-associated antigen-3 (LFA-3) and immunoglobulin-1 (IgG1), has been engineered to block the CD2 molecule on the surface of T cells [270,271,272,273,274,275]. Alefacept has already been used clinically to induce histological remission in patients with psoriasis [273] and reduce insulin use and the frequency of hypoglycemic events in patients with recent onset type 1 diabetes [275]. IL-21, a product of activated human CD4+ T cells [146, 147, 276], is another potential therapeutic target as mice that are genetically deficient in IL-21 experience severe exhaustion of CD8+ T cells during chronic infection [149,150,151].
Prospective Interventions that Strengthen the PD-1 Axis
PD-1 is the hallmark of T cell exhaustion, and advances in promoting T cell exhaustion by suppression of CD4+ T cells, blockage of CD2, or inhibition of IL-21 may be the most promising methods to enhance PD-1 expression and improve immune tolerance [61, 62, 64]. The addition of PD-L1 to cell cultures has moved CD8+ T cells toward exhaustion [61] (Table 2).
Prospective Interventions that Reverse Epigenetic Changes
Investigational efforts to reverse epigenetic changes that suppress anti-inflammatory gene activity or promote the expression of pro-inflammatory gene products have centered mainly on manipulations of the MBDs [208, 277] and the bromodomain and extra-terminal (BET) family of proteins [278,279,280] (Table 2). DNA hypomethylation and increased transcriptional activity have been associated with MBD2 in patients with SLE [281], and anti-sense oligonucleotides that inhibit MBD2 have limited the growth of human cancer cells in a murine model, possibly by de-repressing tumor suppressor genes [282].
The BET family has histone acetyltransferase activity, and it can increase gene activity by acetylating lysine residues in the histone proteins [278, 280]. Molecular inhibitors of the BET proteins have reduced the activity of super-enhancer regions near gene promoters [279], and they have decreased disease-associated gene expression in patients with juvenile idiopathic arthritis [283], protected against endotoxic shock in animal models [284], and improved experimental autoimmune encephalomyelitis [279].
Powerful methyl donors, such as S-adenosylmethionine, may inhibit DNA demethylase activity and help restore immune homeostasis [285]. These supplements are available, and their evaluation in a well-designed, placebo-controlled, clinical trial would constitute an epigenetic intervention that might be applied to autoimmune hepatitis [285].
The administration of 1, 25-dihydroxyvitamin D3 for 4 weeks to a murine model of cholestatic liver disease has inhibited the activation and proliferation of hepatic stellate cells, increased mRNA levels of metalloproteinase, and reduced serum ALT levels [286]. Similarly, the administration of 1, 25-dihydroxyvitamin D3 to a murine model of toxin-induced liver injury has prevented hepatic fibrosis, inhibited the production of TGF-β, and suppressed the expression of tissue inhibitors of metalloproteinases [287]. In each study, treatment had preventive rather than remedial effects as short-term therapy failed to reduce overall hepatic fibrosis or reverse cirrhosis [286, 287]. Clarification of the role of 1, 25-dihydroxyvitamin D3 in the pathogenesis of autoimmune hepatitis could lead to the evaluation of preventive and remedial management strategies based on vitamin D supplementation.
Prospective Interventions that Manipulate the Intestinal Microbiome or Prevent Translocation
Modification of the intestinal microbiome has already been evaluated as a therapeutic intervention in multiple experimental models and small clinical trials, and it awaits study in autoimmune hepatitis (Table 2). Allergic diarrhea and colitis have improved in a murine model after intestinal recolonization with 17 strains of human-derived Clostridia selected by their ability to expand Tregs [288]. Two strains of Bifidobacterium have been used as a probiotic supplement to increase the secretion of soluble factors that favor the generation of Tregs in cultures of intestinal epithelial cells [289], and a probiotic enriched in Lactobacillaceae has promoted the generation of tolerogenic dendritic cells, reduced the expansion of Th1 and Th17 cells, and prevented diabetes in NOD mice [290].
Blockade of TLRs that recognize molecular patterns associated with intestinal microbes has attenuated the renal and pulmonary manifestations of experimental SLE [291], and the administration of gelatin tannate, a protector of the intestinal mucosal barrier, has lowered blood levels of LPS and decreased the activity of experimental acute colitis [292]. A meta-analysis of 10 randomized clinical trials in patients with rheumatoid arthritis has supported the use of tetracycline or minocycline [293], and a randomized pilot study of 35 patients with PSC has demonstrated that the administration of vancomycin or metronidazole for 12 weeks reduced serum alkaline phosphatase and bilirubin levels, improved risk scores, and ameliorated pruritus [294].
Dietary adjustments, probiotic preparations, antibiotics, intestinal recolonization, TLR blockade, and pharmacological agents that strengthen the intestinal mucosal barrier have all been mustered to modify the effects of the intestinal microbiome on systemic immune responses and the occurrence of immune-mediated disease [74]. Progress in the management of autoimmune hepatitis by altering the intestinal microbiome must begin by defining the dysbiosis, determining its gender specificity, and establishing dysbiosis as a cause rather than consequence of the disease.
Overview
Autoimmune hepatitis is a consequence of perturbations in homeostatic mechanisms that maintain self-tolerance [2]. Recognition of these regulatory deficiencies is essential in understanding the key mechanisms that induce, sustain, and advance the disease. Identification of pivotal aberrations is also essential in designing corrective strategies [126], and it may generate international collaborations like the multicenter treatment trial of antibodies to BAFF receptor that is about to launch in autoimmune hepatitis (NCT03217422).
Assessments of the Tim, galectin, and PD-1 pathways are opportunities to discover and manipulate deficiencies in the signaling networks that modulate the adaptive immune response [67, 76, 105, 106]. Assessments of the mechanisms that prevent or induce T cell exhaustion may suggest therapeutic interventions that can modify disease severity or terminate the disease [61, 64]. Assessments of epigenetic changes may identify unsuspected inheritable traits for autoimmune hepatitis, clarify the burden of risk for this disease, and generate studies that discover environmental cues for its occurrence, including acquired vitamin D deficiency [69,70,71]. Assessments of the intestinal microbiome and the permeability of the intestinal mucosal barrier may help explain loss of tolerance for self-antigens, female propensity for autoimmune hepatitis, and varied susceptibility patterns among different age groups, genders, ethnicities, and geographical regions [74, 265].
Autoimmune hepatitis should be fully integrated into the family of immune-mediated diseases, and pathogenic insights made within this family should be recognized as opportunities to expand disease-specific concepts. Shared insights from studies and experiences within the family of autoimmune diseases can contribute to a positive feedback loop that expedites investigational progress, enhances understanding, and enriches management options for each family member.
References
Liberal R, Longhi MS, Mieli-Vergani G, Vergani D. Pathogenesis of autoimmune hepatitis. Best Prac Res Clin Gastroenterol. 2011;25:653–664.
Czaja AJ. Transitioning from idiopathic to explainable autoimmune hepatitis. Dig Dis Sci. 2015;60:2881–2900.
Bogdanos DP, Choudhuri K, Vergani D. Molecular mimicry and autoimmune liver disease: virtuous intentions, malign consequences. Liver. 2001;21:225–232.
Bowen DG. Of mice and molecular mimicry: modeling autoimmune hepatitis. Hepatology. 2008;48:1013–1015.
Hintermann E, Holdener M, Bayer M, et al. Epitope spreading of the anti-CYP2D6 antibody response in patients with autoimmune hepatitis and in the CYP2D6 mouse model. J Autoimmun. 2011;37:242–253.
Ehser J, Holdener M, Christen S, et al. Molecular mimicry rather than identity breaks T-cell tolerance in the CYP2D6 mouse model for human autoimmune hepatitis. J Autoimmun. 2013;42:39–49.
Lohr H, Manns M, Kyriatsoulis A, et al. Clonal analysis of liver-infiltrating T cells in patients with LKM-1 antibody-positive autoimmune chronic active hepatitis. Clin Exp Immunol. 1991;84:297–302.
Schlaak JF, Lohr H, Gallati H, Meyer zum Buschenfelde KH, Fleischer B. Analysis of the in vitro cytokine production by liver-infiltrating T cells of patients with autoimmune hepatitis. Clin Exp Immunol. 1993;94:168–173.
Maggiore G, De Benedetti F, Massa M, Pignatti P, Martini A. Circulating levels of interleukin-6, interleukin-8, and tumor necrosis factor-alpha in children with autoimmune hepatitis. J Pediatr Gastroenterol Nutr. 1995;20:23–27.
Cookson S, Constantini PK, Clare M, et al. Frequency and nature of cytokine gene polymorphisms in type 1 autoimmune hepatitis. Hepatology. 1999;30:851–856.
Czaja AJ, Sievers C, Zein NN. Nature and behavior of serum cytokines in type 1 autoimmune hepatitis. Dig Dis Sci. 2000;45:1028–1035.
Kamijo A, Yoshizawa K, Joshita S, et al. Cytokine profiles affecting the pathogenesis of autoimmune hepatitis in Japanese patients. Hepatol Res. 2011;41:350–357.
Longhi MS, Liberal R, Holder B, et al. Inhibition of interleukin-17 promotes differentiation of CD25(-) cells into stable T regulatory cells in patients with autoimmune hepatitis. Gastroenterology. 2012;142:1526–1535.
Landi A, Weismuller TJ, Lankisch TO, et al. Differential serum levels of eosinophilic eotaxins in primary sclerosing cholangitis, primary biliary cirrhosis, and autoimmune hepatitis. J Interferon Cytokine Res. 2014;34:204–214.
Behfarjam F, Sanati MH, Nasseri Moghaddam S, et al. Role of Th1/Th2 cells and related cytokines in autoimmune hepatitis. Turk J Gastroenterol. 2017;28:110–114.
Czaja AJ. Review article: chemokines as orchestrators of autoimmune hepatitis and potential therapeutic targets. Aliment Pharmacol Ther. 2014;40:261–279.
Longhi MS, Ma Y, Bogdanos DP, et al. Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol. 2004;41:31–37.
Longhi MS, Ma Y, Mitry RR, et al. Effect of CD4 + CD25 + regulatory T-cells on CD8 T-cell function in patients with autoimmune hepatitis. J Autoimmun. 2005;25:63–71.
Liberal R, Grant CR, Holder BS, et al. The impaired immune regulation of autoimmune hepatitis is linked to a defective galectin-9/tim-3 pathway. Hepatology. 2012;56:677–686.
Longhi MS, Ma Y, Grant CR, et al. T-regs in autoimmune hepatitis-systemic lupus erythematosus/mixed connective tissue disease overlap syndrome are functionally defective and display a Th1 cytokine profile. J Autoimmun. 2013;41:146–151.
Liberal R, Grant CR, Holder BS, et al. In autoimmune hepatitis type 1 or the autoimmune hepatitis-sclerosing cholangitis variant defective regulatory T-cell responsiveness to IL-2 results in low IL-10 production and impaired suppression. Hepatology. 2015;62:863–875.
Migita K, Komori A, Kozuru H, et al. Circulating microRNA profiles in patients with type-1 autoimmune hepatitis. PLoS One. 2015;10:e0136908.
Czaja AJ. Emerging therapeutic biomarkers of autoimmune hepatitis and their impact on current and future management. Expert Rev Gastroenterol Hepatol. 2018;. https://doi.org/10.1080/17474124.2018.1453356.
Mataki N, Kikuchi K, Kawai T, et al. Expression of PD-1, PD-L1, and PD-L2 in the liver in autoimmune liver diseases. Am J Gastroenterol. 2007;102:302–312.
Oikawa T, Takahashi H, Ishikawa T, et al. Intrahepatic expression of the co-stimulatory molecules programmed death-1, and its ligands in autoimmune liver disease. Pathol Int. 2007;57:485–492.
Matsumoto K, Miyake Y, Matsushita H, et al. Anti-programmed cell death-1 antibody as a new serological marker for type 1 autoimmune hepatitis. J Gastroenterol Hepatol. 2014;29:110–115.
Miyake Y, Yamamoto K, Matsushita H, et al. Multicenter validation study of anti-programmed cell death-1 antibody as a serological marker for type 1 autoimmune hepatitis. Hepatol Res. 2014;44:1299–1307.
Aarslev K, Dige A, Greisen SR, et al. Soluble programmed death-1 levels are associated with disease activity and treatment response in patients with autoimmune hepatitis. Scand J Gastroenterol. 2017;52:93–99.
Gronbaek H, Kreutzfeldt M, Kazankov K, et al. Single-centre experience of the macrophage activation marker soluble (s)CD163—associations with disease activity and treatment response in patients with autoimmune hepatitis. Aliment Pharmacol Ther. 2016;44:1062–1070.
Moller HJ. Soluble CD163. Scand J Clin Lab Invest. 2012;72:1–13.
Assis DN, Leng L, Du X, et al. The role of macrophage migration inhibitory factor in autoimmune liver disease. Hepatology. 2014;59:580–591.
Assis DN, Takahashi H, Leng L, et al. A macrophage migration inhibitory factor polymorphism is associated with autoimmune hepatitis severity in US and Japanese patients. Dig Dis Sci. 2016;61:3506–3512.
Migita K, Abiru S, Maeda Y, et al. Elevated serum BAFF levels in patients with autoimmune hepatitis. Hum Immunol. 2007;68:586–591.
Nishikawa H, Enomoto H, Iwata Y, et al. B-cell activating factor belonging to the tumor necrosis factor family and interferon-gamma-Inducible protein-10 in autoimmune hepatitis. Medicine (Baltimore). 2016;95:e3194.
Strettell MD, Donaldson PT, Thomson LJ, et al. Allelic basis for HLA-encoded susceptibility to type 1 autoimmune hepatitis. Gastroenterology. 1997;112:2028–2035.
Czaja AJ, Strettell MD, Thomson LJ, et al. Associations between alleles of the major histocompatibility complex and type 1 autoimmune hepatitis. Hepatology. 1997;25:317–323.
Yokosawa S, Yoshizawa K, Ota M, et al. A genomewide DNA microsatellite association study of Japanese patients with autoimmune hepatitis type 1. Hepatology. 2007;45:384–390.
Mells GF, Kaser A, Karlsen TH. Novel insights into autoimmune liver diseases provided by genome-wide association studies. J Autoimmun. 2013;46:41–54.
de Boer YS, van Gerven NM, Zwiers A, et al. Genome-wide association study identifies variants associated with autoimmune hepatitis type 1. Gastroenterology. 2014;147:443–452.
van Gerven NM, de Boer YS, Zwiers A, et al. HLA-DRB1*03:01 and HLA-DRB1*04:01 modify the presentation and outcome in autoimmune hepatitis type-1. Genes Immun. 2015;16:247–252.
Umemura T, Joshita S, Hamano H, et al. Association of autoimmune hepatitis with Src homology 2 adaptor protein 3 gene polymorphisms in Japanese patients. J Hum Genet. 2017;62:963–967.
Fox CK, Furtwaengler A, Nepomuceno RR, Martinez OM, Krams SM. Apoptotic pathways in primary biliary cirrhosis and autoimmune hepatitis. Liver. 2001;21:272–279.
Bai J, Odin JA. Apoptosis and the liver: relation to autoimmunity and related conditions. Autoimmun Rev. 2003;2:36–42.
Czaja AJ. Targeting apoptosis in autoimmune hepatitis. Dig Dis Sci. 2014;59:2890–2904.
Sanz-Cameno P, Medina J, Garcia-Buey L, et al. Enhanced intrahepatic inducible nitric oxide synthase expression and nitrotyrosine accumulation in primary biliary cirrhosis and autoimmune hepatitis. J Hepatol. 2002;37:723–729.
Efe C, Kav T, Aydin C, et al. Low serum vitamin D levels are associated with severe histological features and poor response to therapy in patients with autoimmune hepatitis. Dig Dis Sci. 2014;59:3035–3042.
Beyazit Y, Kocak E, Tanoglu A, Kekilli M. Oxidative stress might play a role in low serum vitamin D associated liver fibrosis among patients with autoimmune hepatitis. Dig Dis Sci. 2015;60:1106–1108.
Beyazit Y, Efe C, Tanoglu A, et al. Nitric oxide is a potential mediator of hepatic inflammation and fibrogenesis in autoimmune hepatitis. Scand J Gastroenterol. 2015;50:204–210.
Czaja AJ. Nature and implications of oxidative and nitrosative stresses in autoimmune hepatitis. Dig Dis Sci. 2016;61:2784–2803.
Czaja AJ, Carpenter HA. Decreased fibrosis during corticosteroid therapy of autoimmune hepatitis. J Hepatol. 2004;40:646–652.
Czaja AJ. Rapidity of treatment response and outcome in type 1 autoimmune hepatitis. J Hepatol. 2009;51:161–167.
Montano-Loza AJ, Thandassery RB, Czaja AJ. Targeting hepatic fibrosis in autoimmune hepatitis. Dig Dis Sci. 2016;61:3118–3139.
Wang J, Malik N, Yin M, et al. Magnetic resonance elastography is accurate in detecting advanced fibrosis in autoimmune hepatitis. World J Gastroenterol. 2017;23:859–868.
Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The TIM gene family regulates autoimmune and allergic diseases. Trends Mol Med. 2005;11:362–369.
Zhu C, Anderson AC, Schubart A, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252.
Zhu C, Anderson AC, Kuchroo VK. TIM-3 and its regulatory role in immune responses. Curr Top Microbiol Immunol. 2011;350:1–15.
Zhang Y, Ma CJ, Wang JM, et al. Tim-3 regulates pro- and anti-inflammatory cytokine expression in human CD14 + monocytes. J Leukoc Biol. 2012;91:189–196.
Wherry EJ, Ha SJ, Kaech SM, et al. Molecular signature of CD8 + T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499.
Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014;35:51–60.
McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523:612–616.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–499.
McKinney EF, Smith KG. T-cell exhaustion: understanding the interface of chronic viral and autoinflammatory diseases. Immunol Cell Biol. 2016;94:935–942.
McKinney EF, Smith KG. T cell exhaustion and immune-mediated disease-the potential for therapeutic exhaustion. Curr Opin Immunol. 2016;43:74–80.
Catakovic K, Klieser E, Neureiter D, Geisberger R. T cell exhaustion: from pathophysiological basics to tumor immunotherapy. Cell Commun Signal. 2017;15:1.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242.
Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. 2016;7:550.
Zamani MR, Aslani S, Salmaninejad A, Javan MR, Rezaei N. PD-1/PD-L and autoimmunity: a growing relationship. Cell Immunol. 2016;310:27–41.
Mann DA. Epigenetics in liver disease. Hepatology. 2014;60:1418–1425.
Jeffries MA, Sawalha AH. Autoimmune disease in the epigenetic era: how has epigenetics changed our understanding of disease and how can we expect the field to evolve? Expert Rev Clin Immunol. 2015;11:45–58.
Czaja AJ. Epigenetic changes and their implications in autoimmune hepatitis. Eur J Clin Invest. 2018;48:e12899.
Lin R, Zhou L, Zhang J, Wang B. Abnormal intestinal permeability and microbiota in patients with autoimmune hepatitis. Int J Clin Exp Pathol. 2015;8:5153–5160.
Yuksel M, Wang Y, Tai N, et al. A novel “humanized mouse” model for autoimmune hepatitis and the association of gut microbiota with liver inflammation. Hepatology. 2015;62:1536–1550.
Czaja AJ. Factoring the intestinal microbiome into the pathogenesis of autoimmune hepatitis. World J Gastroenterol. 2016;22:9257–9278.
Kuchroo VK, Umetsu DT, DeKruyff RH, Freeman GJ. The TIM gene family: emerging roles in immunity and disease. Nat Rev Immunol. 2003;3:454–462.
Kuchroo VK, Meyers JH, Umetsu DT, DeKruyff RH. TIM family of genes in immunity and tolerance. Adv Immunol. 2006;91:227–249.
Chakravarti S, Sabatos CA, Xiao S, et al. Tim-2 regulates T helper type 2 responses and autoimmunity. J Exp Med. 2005;202:437–444.
Xiao S, Najafian N, Reddy J, et al. Differential engagement of Tim-1 during activation can positively or negatively costimulate T cell expansion and effector function. J Exp Med. 2007;204:1691–1702.
Cao E, Zang X, Ramagopal UA, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity. 2007;26:311–321.
Santiago C, Ballesteros A, Tami C, et al. Structures of T Cell immunoglobulin mucin receptors 1 and 2 reveal mechanisms for regulation of immune responses by the TIM receptor family. Immunity. 2007;26:299–310.
Kobayashi N, Karisola P, Pena-Cruz V, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940.
Umetsu SE, Lee WL, McIntire JJ, et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat Immunol. 2005;6:447–454.
Xiao S, Brooks CR, Zhu C, et al. Defect in regulatory B-cell function and development of systemic autoimmunity in T-cell Ig mucin 1 (Tim-1) mucin domain-mutant mice. Proc Natl Acad Sci U S A. 2012;109:12105–12110.
DiLillo DJ, Matsushita T, Tedder TF. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann N Y Acad Sci. 2010;1183:38–57.
Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–241.
Bouaziz JD, Le Buanec H, Saussine A, Bensussan A, Bagot M. IL-10 producing regulatory B cells in mice and humans: state of the art. Curr Mol Med. 2012;12:519–527.
Yang M, Rui K, Wang S, Lu L. Regulatory B cells in autoimmune diseases. Cell Mol Immunol. 2013;10:122–132.
Xiao S, Brooks CR, Sobel RA, Kuchroo VK. Tim-1 is essential for induction and maintenance of IL-10 in regulatory B cells and their regulation of tissue inflammation. J Immunol. 2015;194:1602–1608.
Gray M, Miles K, Salter D, Gray D, Savill J. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc Natl Acad Sci U S A. 2007;104:14080–14085.
Miles K, Heaney J, Sibinska Z, et al. A tolerogenic role for Toll-like receptor 9 is revealed by B-cell interaction with DNA complexes expressed on apoptotic cells. Proc Natl Acad Sci U S A. 2012;109:887–892.
Ueno T, Habicht A, Clarkson MR, et al. The emerging role of T cell Ig mucin 1 in alloimmune responses in an experimental mouse transplant model. J Clin Invest. 2008;118:742–751.
Monney L, Sabatos CA, Gaglia JL, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541.
Imaizumi T, Kumagai M, Sasaki N, et al. Interferon-gamma stimulates the expression of galectin-9 in cultured human endothelial cells. J Leukoc Biol. 2002;72:486–491.
Dardalhon V, Anderson AC, Karman J, et al. Tim-3/galectin-9 pathway: regulation of Th1 immunity through promotion of CD11b + Ly-6G + myeloid cells. J Immunol. 2010;185:1383–1392.
Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802.
Movahedi K, Guilliams M, Van den Bossche J, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–4244.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174.
Fridlender ZG, Sun J, Mishalian I, et al. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS One. 2012;7:e31524.
Deniset JF, Kubes P. Recent advances in understanding neutrophils. F1000Res. 2016;5:2912.
Wright GJ, Cherwinski H, Foster-Cuevas M, et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol. 2003;171:3034–3046.
Fischer MA, Davies ML, Reider IE, et al. CD11b(+), Ly6G(+) cells produce type I interferon and exhibit tissue protective properties following peripheral virus infection. PLoS Pathog. 2011;7:e1002374.
Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010;184:3106–3116.
Nakayama M, Akiba H, Takeda K, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113:3821–3830.
Meyers JH, Chakravarti S, Schlesinger D, et al. TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat Immunol. 2005;6:455–464.
John S, Mishra R. Galectin-9: From cell biology to complex disease dynamics. J Biosci. 2016;41:507–534.
Golden-Mason L, Rosen HR. Galectin-9: Diverse roles in hepatic immune homeostasis and inflammation. Hepatology. 2017;66:271–279.
Vaitaitis GM, Wagner DH Jr. Galectin-9 controls CD40 signaling through a Tim-3 independent mechanism and redirects the cytokine profile of pathogenic T cells in autoimmunity. PLoS One. 2012;7:e38708.
Morgan R, Gao G, Pawling J, et al. N-acetylglucosaminyltransferase V (Mgat5)-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J Immunol. 2004;173:7200–7208.
Grigorian A, Lee SU, Tian W, et al. Control of T Cell-mediated autoimmunity by metabolite flux to N-glycan biosynthesis. J Biol Chem. 2007;282:20027–20035.
Marth JD, Grewal PK. Mammalian glycosylation in immunity. Nat Rev Immunol. 2008;8:874–887.
Bacigalupo ML, Manzi M, Rabinovich GA, Troncoso MF. Hierarchical and selective roles of galectins in hepatocarcinogenesis, liver fibrosis and inflammation of hepatocellular carcinoma. World J Gastroenterol. 2013;19:8831–8849.
Yang RY, Hsu DK, Liu FT. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc Natl Acad Sci U S A. 1996;93:6737–6742.
Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5N-glycosylation. Nature. 2001;409:733–739.
Lv K, Zhang Y, Zhang M, Zhong M, Suo Q. Galectin-9 ameliorates Con A-induced hepatitis by inducing CD4(+)CD25(low/int) effector T-Cell apoptosis and increasing regulatory T cell number. PLoS One. 2012;7:e48379.
Tadokoro T, Morishita A, Sakamoto T, et al. Galectin9 ameliorates fulminant liver injury. Mol Med Rep. 2017;16:36–42.
Kashio Y, Nakamura K, Abedin MJ, et al. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J Immunol. 2003;170:3631–3636.
Su EW, Bi S, Kane LP. Galectin-9 regulates T helper cell function independently of Tim-3. Glycobiology. 2011;21:1258–1265.
Seki M, Oomizu S, Sakata KM, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol. 2008;127:78–88.
Sehrawat S, Suryawanshi A, Hirashima M, Rouse BT. Role of Tim-3/galectin-9 inhibitory interaction in viral-induced immunopathology: shifting the balance toward regulators. J Immunol. 2009;182:3191–3201.
Wiersma VR, de Bruyn M, Helfrich W, Bremer E. Therapeutic potential of Galectin-9 in human disease. Med Res Rev. 2013;33:E102–E126.
Mengshol JA, Golden-Mason L, Arikawa T, et al. A crucial role for Kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis C infection. PLoS One. 2010;5:e9504.
Rosen HR. Emerging concepts in immunity to hepatitis C virus infection. J Clin Invest. 2013;123:4121–4130.
Tang ZH, Liang S, Potter J, et al. Tim-3/galectin-9 regulate the homeostasis of hepatic NKT cells in a murine model of nonalcoholic fatty liver disease. J Immunol. 2013;190:1788–1796.
Markwick LJ, Riva A, Ryan JM, et al. Blockade of PD1 and TIM3 restores innate and adaptive immunity in patients with acute alcoholic hepatitis. Gastroenterology. 2015;148:590–602.
Montano-Loza AJ, Czaja AJ. Cell mediators of autoimmune hepatitis and their therapeutic implications. Dig Dis Sci. 2014;60:1528–1542.
Czaja AJ. Evolving paradigm for treatment of autoimmune hepatitis. Expert Rev Clin Immunol. 2017;13:781–798.
Zajac AJ, Blattman JN, Murali-Krishna K, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213.
Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927.
Davoodzadeh Gholami M, Kardar GA, Saeedi Y, et al. Exhaustion of T lymphocytes in the tumor microenvironment: significance and effective mechanisms. Cell Immunol 2017;322:1–14.
Brooks DG, McGavern DB, Oldstone MB. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J Clin Invest. 2006;116:1675–1685.
Lee J, Ahn E, Kissick HT, Ahmed R. Reinvigorating exhausted T cells by blockade of the PD-1 pathway. For Immunopathol Dis Therap. 2015;6:7–17.
Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2009;106:8623–8628.
Kahan SM, Wherry EJ, Zajac AJ. T cell exhaustion during persistent viral infections. Virology. 2015;479–480:180–193.
Bellon M, Nicot C. Telomere dynamics in immune senescence and exhaustion triggered by chronic viral infection. Viruses. 2017;9:289.
Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687.
Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–245.
Wei F, Zhong S, Ma Z, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci U S A. 2013;110:E2480–E2489.
Blackburn SD, Shin H, Haining WN, et al. Coregulation of CD8 + T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37.
Crawford A, Angelosanto JM, Kao C, et al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity. 2014;40:289–302.
Lang KS, Recher M, Navarini AA, et al. Inverse correlation between IL-7 receptor expression and CD8 T cell exhaustion during persistent antigen stimulation. Eur J Immunol. 2005;35:738–745.
Im SJ, Hashimoto M, Gerner MY, et al. Defining CD8 + T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421.
Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 2008;105:15016–15021.
Intlekofer AM, Takemoto N, Wherry EJ, et al. Effector and memory CD8 + T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–1244.
Paley MA, Kroy DC, Odorizzi PM, et al. Progenitor and terminal subsets of CD8 + T cells cooperate to contain chronic viral infection. Science. 2012;338:1220–1225.
Matloubian M, Concepcion RJ, Ahmed R. CD4 + T cells are required to sustain CD8 + cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68:8056–8063.
Parrish-Novak J, Dillon SR, Nelson A, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408:57–63.
Parrish-Novak J, Foster DC, Holly RD, Clegg CH. Interleukin-21 and the IL-21 receptor: novel effectors of NK and T cell responses. J Leukoc Biol. 2002;72:856–863.
Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol. 2005;175:2261–2269.
Johnson LD, Jameson SC. Immunology. A chronic need for IL-21. Science. 2009;324:1525–1526.
Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science. 2009;324:1569–1572.
Yi JS, Du M, Zajac AJ. A vital role for interleukin-21 in the control of a chronic viral infection. Science. 2009;324:1572–1576.
Wilkins AL, Yang W, Yang JJ. Structural biology of the cell adhesion protein CD2: from molecular recognition to protein folding and design. Curr Protein Pept Sci. 2003;4:367–373.
Bolduan S, Koppensteiner H, Businger R, et al. T cells with low CD2 levels express reduced restriction factors and are preferentially infected in therapy naive chronic HIV-1 patients. J Int AIDS Soc. 2017;20:21865.
Harcharik S, Bernardo S, Moskalenko M, et al. Defining the role of CD2 in disease progression and overall survival among patients with completely resected stage-II to -III cutaneous melanoma. J Am Acad Dermatol. 2014;70:1036–1044.
Brooks DG, Ha SJ, Elsaesser H, et al. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci U S A. 2008;105:20428–20433.
Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8 + T cell deletion and viral persistence in vivo. Immunity. 2009;31:145–157.
Garidou L, Heydari S, Gossa S, McGavern DB. Therapeutic blockade of transforming growth factor beta fails to promote clearance of a persistent viral infection. J Virol. 2012;86:7060–7071.
Boettler T, Cheng Y, Ehrhardt K, von Herrath M. TGF-beta blockade does not improve control of an established persistent viral infection. Viral Immunol. 2012;25:232–238.
Patsoukis N, Brown J, Petkova V, et al. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5:ra46.
Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029.
Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369.
Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034.
Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268.
Chen CL, Pan QZ, Zhao JJ, et al. PD-L1 expression as a predictive biomarker for cytokine-induced killer cell immunotherapy in patients with hepatocellular carcinoma. Oncoimmunology. 2016;5:e1176653.
Tseng SY, Otsuji M, Gorski K, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–846.
Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003;100:5336–5341.
Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol. 2004;172:4661–4665.
Chang CH, Curtis JD, Maggi LB Jr, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251.
Tkachev V, Goodell S, Opipari AW, et al. Programmed death-1 controls T cell survival by regulating oxidative metabolism. J Immunol. 2015;194:5789–5800.
Patsoukis N, Bardhan K, Chatterjee P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692.
Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4 + T cell subsets. J Immunol. 2011;186:3299–3303.
Nielsen C, Ohm-Laursen L, Barington T, Husby S, Lillevang ST. Alternative splice variants of the human PD-1 gene. Cell Immunol. 2005;235:109–116.
Wan B, Nie H, Liu A, et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J Immunol. 2006;177:8844–8850.
Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398.
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783.
Javierre BM, Hernando H, Ballestar E. Environmental triggers and epigenetic deregulation in autoimmune disease. Discov Med. 2011;12:535–545.
Meda F, Folci M, Baccarelli A, Selmi C. The epigenetics of autoimmunity. Cell Mol Immunol. 2011;8:226–236.
Heyn H, Moran S, Hernando-Herraez I, et al. DNA methylation contributes to natural human variation. Genome Res. 2013;23:1363–1372.
Canas CA, Canas F, Bonilla-Abadia F, Ospina FE, Tobon GJ. Epigenetics changes associated to environmental triggers in autoimmunity. Autoimmunity. 2016;49:1–11.
Issa JP. Aging and epigenetic drift: a vicious cycle. J Clin Invest. 2014;124:24–29.
Dozmorov MG, Coit P, Maksimowicz-McKinnon K, Sawalha AH. Age-associated DNA methylation changes in naive CD4 + T cells suggest an evolving autoimmune epigenotype in aging T cells. Epigenomics. 2017;9:429–445.
Maegawa S, Gough SM, Watanabe-Okochi N, et al. Age-related epigenetic drift in the pathogenesis of MDS and AML. Genome Res. 2014;24:580–591.
Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–1803.
Fang F, Balch C, Schilder J, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116:4043–4053.
Konkankit VV, Kim W, Koya RC, et al. Decitabine immunosensitizes human gliomas to NY-ESO-1 specific T lymphocyte targeting through the Fas/Fas ligand pathway. J Transl Med. 2011;9:192.
Scandura JM, Roboz GJ, Moh M, et al. Phase 1 study of epigenetic priming with decitabine prior to standard induction chemotherapy for patients with AML. Blood. 2011;118:1472–1480.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27.
Zeng W, Dai H, Yan M, et al. Decitabine-induced changes in human myelodysplastic syndrome cell line SKM-1 are mediated by FOXO3A activation. J Immunol Res. 2017;2017:4302320.
Szyf M. Epigenetic therapeutics in autoimmune disease. Clin Rev Allergy Immunol. 2010;39:62–77.
Radic M, Muller S. Epigenetics of autoantigens: new opportunities for therapy of autoimmune diseases. Genet Epigenet. 2013;5:63–70.
Tough DF, Prinjha RK. Immune disease-associated variants in gene enhancers point to BET epigenetic mechanisms for therapeutic intervention. Epigenomics. 2017;9:573–584.
Somers EC, Richardson BC. Environmental exposures, epigenetic changes and the risk of lupus. Lupus. 2014;23:568–576.
Relle M, Foehr B, Schwarting A. Epigenetic aspects of systemic lupus erythematosus. Rheumatol Ther. 2015;2:33–46.
Liu Y, Aryee MJ, Padyukov L, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31:142–147.
Lian X, Xiao R, Hu X, et al. DNA demethylation of CD40 l in CD4 + T cells from women with systemic sclerosis: a possible explanation for female susceptibility. Arthritis Rheum. 2012;64:2338–2345.
Lleo A, Zhang W, Zhao M, et al. DNA methylation profiling of the X chromosome reveals an aberrant demethylation on CXCR3 promoter in primary biliary cirrhosis. Clin Epigenetics. 2015;7:61.
Murphy SK, Yang H, Moylan CA, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. 2013;145:1076–1087.
Cedar H, Bergman Y. Programming of DNA methylation patterns. Annu Rev Biochem. 2012;81:97–117.
Javierre BM, Fernandez AF, Richter J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20:170–179.
Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547.
Hendrich B, Abbott C, McQueen H, et al. Genomic structure and chromosomal mapping of the murine and human Mbd1, Mbd2, Mbd3, and Mbd4 genes. Mamm Genome. 1999;10:906–912.
Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935.
Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303.
Guo JU, Su Y, Zhong C, Ming GL, Song H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle. 2011;10:2662–2668.
Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479.
Gunther K, Rust M, Leers J, et al. Differential roles for MBD2 and MBD3 at methylated CpG islands, active promoters and binding to exon sequences. Nucleic Acids Res. 2013;41:3010–3021.
Baubec T, Ivanek R, Lienert F, Schubeler D. Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell. 2013;153:480–492.
Menafra R, Stunnenberg HG. MBD2 and MBD3: elusive functions and mechanisms. Front Genet. 2014;5:428.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068.
Baubec T, Colombo DF, Wirbelauer C, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015;520:243–247.
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260.
Osley MA, Fleming AB, Kao CF. Histone ubiquitylation and the regulation of transcription. Results Probl Cell Differ. 2006;41:47–75.
Wysocka J, Allis CD, Coonrod S. Histone arginine methylation and its dynamic regulation. Front Biosci. 2006;11:344–355.
Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491–507.
Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120.
de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749.
Thiagalingam S, Cheng KH, Lee HJ, et al. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100.
Knight JS, Zhao W, Luo W, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest. 2013;123:2981–2993.
Kondo Y, Shen L, Cheng AS, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet. 2008;40:741–750.
Fisher L, Fisher A. Vitamin D and parathyroid hormone in outpatients with noncholestatic chronic liver disease. Clin Gastroenterol Hepatol. 2007;5:513–520.
Miroliaee A, Nasiri-Toosi M, Khalilzadeh O, et al. Disturbances of parathyroid hormone-vitamin D axis in non-cholestatic chronic liver disease: a cross-sectional study. Hepatol Int. 2010;4:634–640.
DeLuca HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80:1689S–1696S.
Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289:F8–F28.
Kim S, Shevde NK, Pike JW. 1,25-dihydroxyvitamin D3 stimulates cyclic vitamin D receptor/retinoid X receptor DNA-binding, co-activator recruitment, and histone acetylation in intact osteoblasts. J Bone Miner Res. 2005;20:305–317.
Ramagopalan SV, Heger A, Berlanga AJ, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 2010;20:1352–1360.
Vogel A, Strassburg CP, Manns MP. Genetic association of vitamin D receptor polymorphisms with primary biliary cirrhosis and autoimmune hepatitis. Hepatology. 2002;35:126–131.
Fan L, Tu X, Zhu Y, et al. Genetic association of vitamin D receptor polymorphisms with autoimmune hepatitis and primary biliary cirrhosis in the Chinese. J Gastroenterol Hepatol. 2005;20:249–255.
Ohyama Y, Yamasaki T. Eight cytochrome P450s catalyze vitamin D metabolism. Front Biosci. 2004;9:3007–3018.
Zhang Y, Leung DY, Richers BN, et al. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J Immunol. 2012;188:2127–2135.
Luong KV, Nguyen LT. The role of vitamin D in autoimmune hepatitis. J Clin Med Res. 2013;5:407–415.
Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343.
Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319.
Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947.
Parker SC, Stitzel ML, Taylor DL, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013;110:17921–17926.
Di Croce L, Raker VA, Corsaro M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002;295:1079–1082.
Vire E, Brenner C, Deplus R, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874.
Zoetendal EG, Vaughan EE, de Vos WM. A microbial world within us. Mol Microbiol. 2006;59:1639–1650.
Booijink CC, Zoetendal EG, Kleerebezem M, de Vos WM. Microbial communities in the human small intestine: coupling diversity to metagenomics. Future Microbiol. 2007;2:285–295.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230.
Mariat D, Firmesse O, Levenez F, et al. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009;9:123.
Claesson MJ, Cusack S, O’Sullivan O, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A. 2011;108:4586–4591.
Jalanka-Tuovinen J, Salonen A, Nikkila J, et al. Intestinal microbiota in healthy adults: temporal analysis reveals individual and common core and relation to intestinal symptoms. PLoS One. 2011;6:e23035.
Miller GE, Engen PA, Gillevet PM, et al. Lower neighborhood socioeconomic status associated with reduced diversity of the colonic microbiota in healthy adults. PLoS One. 2016;11:e0148952.
Power SE, O’Toole PW, Stanton C, Ross RP, Fitzgerald GF. Intestinal microbiota, diet and health. Br J Nutr. 2014;111:387–402.
Wen L, Ley RE, Volchkov PY, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113.
Mejia-Leon ME, Barca AM. Diet, microbiota and immune system in type 1 diabetes development and evolution. Nutrients. 2015;7:9171–9184.
Miyake S, Kim S, Suda W, et al. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS One. 2015;10:e0137429.
Rogier R, Koenders MI, Abdollahi-Roodsaz S. Toll-like receptor mediated modulation of T cell response by commensal intestinal microbiota as a trigger for autoimmune arthritis. J Immunol Res. 2015;2015:527696.
Sokol H, Seksik P, Rigottier-Gois L, et al. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:106–111.
Miyake Y, Yamamoto K. Role of gut microbiota in liver diseases. Hepatol Res. 2013;43:139–146.
Henao-Mejia J, Elinav E, Thaiss CA, Licona-Limon P, Flavell RA. Role of the intestinal microbiome in liver disease. J Autoimmun. 2013;46:66–73.
Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–334.
Csak T, Ganz M, Pespisa J, et al. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–144.
Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185.
Mueller T, Beutler C, Pico AH, et al. Enhanced innate immune responsiveness and intolerance to intestinal endotoxins in human biliary epithelial cells contributes to chronic cholangitis. Liver Int. 2011;31:1574–1588.
Wang AP, Migita K, Ito M, et al. Hepatic expression of toll-like receptor 4 in primary biliary cirrhosis. J Autoimmun. 2005;25:85–91.
Kikuchi K, Lian ZX, Yang GX, et al. Bacterial CpG induces hyper-IgM production in CD27(+) memory B cells in primary biliary cirrhosis. Gastroenterology. 2005;128:304–312.
Honda Y, Yamagiwa S, Matsuda Y, et al. Altered expression of TLR homolog RP105 on monocytes hypersensitive to LPS in patients with primary biliary cirrhosis. J Hepatol. 2007;47:404–411.
Tabibian JH, O’Hara SP, Trussoni CE, et al. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology. 2016;63:185–196.
Shimoda S, Harada K, Niiro H, et al. Interaction between Toll-like receptors and natural killer cells in the destruction of bile ducts in primary biliary cirrhosis. Hepatology. 2011;53:1270–1281.
Shimoda S, Hisamoto S, Harada K, et al. Natural killer cells regulate T cell immune responses in primary biliary cirrhosis. Hepatology. 2015;62:1817–1827.
Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc Natl Acad Sci U S A. 2005;102:17729–17733.
Chan CC, Hwang SJ, Lee FY, et al. Prognostic value of plasma endotoxin levels in patients with cirrhosis. Scand J Gastroenterol. 1997;32:942–946.
Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology. 2005;41:422–433.
Czaja AJ. Review article: next-generation transformative advances in the pathogenesis and management of autoimmune hepatitis. Aliment Pharmacol Ther. 2017;46:920–937.
Yurkovetskiy L, Burrows M, Khan AA, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity. 2013;39:400–412.
Markle JG, Frank DN, Mortin-Toth S, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013;339:1084–1088.
Markle JG, Frank DN, Adeli K, von Bergen M, Danska JS. Microbiome manipulation modifies sex-specific risk for autoimmunity. Gut Microbes. 2014;5:485–493.
de Souza AJ, Oriss TB, O’Malley KJ, Ray A, Kane LP. T cell Ig and mucin 1 (TIM-1) is expressed on in vivo-activated T cells and provides a costimulatory signal for T cell activation. Proc Natl Acad Sci U S A. 2005;102:17113–17118.
Miller GT, Hochman PS, Meier W, et al. Specific interaction of lymphocyte function-associated antigen 3 with CD2 can inhibit T cell responses. J Exp Med. 1993;178:211–222.
Majeau GR, Meier W, Jimmo B, Kioussis D, Hochman PS. Mechanism of lymphocyte function-associated molecule 3-Ig fusion proteins inhibition of T cell responses. Structure/function analysis in vitro and in human CD2 transgenic mice. J Immunol. 1994;152:2753–2767.
da Silva AJ, Brickelmaier M, Majeau GR, et al. Alefacept, an immunomodulatory recombinant LFA-3/IgG1 fusion protein, induces CD16 signaling and CD2/CD16-dependent apoptosis of CD2(+) cells. J Immunol. 2002;168:4462–4471.
Chamian F, Lowes MA, Lin SL, et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci U S A. 2005;102:2075–2080.
Weaver TA, Charafeddine AH, Agarwal A, et al. Alefacept promotes co-stimulation blockade based allograft survival in nonhuman primates. Nat Med. 2009;15:746–749.
Rigby MR, Harris KM, Pinckney A, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest. 2015;125:3285–3296.
Kuchen S, Robbins R, Sims GP, et al. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4 + T cell-B cell collaboration. J Immunol. 2007;179:5886–5896.
Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304.
Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002;513:124–128.
Bandukwala HS, Gagnon J, Togher S, et al. Selective inhibition of CD4 + T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc Natl Acad Sci U S A. 2012;109:14532–14537.
Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–736.
Balada E, Ordi-Ros J, Serrano-Acedo S, Martinez-Lostao L, Vilardell-Tarres M. Transcript overexpression of the MBD2 and MBD4 genes in CD4 + T cells from systemic lupus erythematosus patients. J Leukoc Biol. 2007;81:1609–1616.
Campbell PM, Bovenzi V, Szyf M. Methylated DNA-binding protein 2 antisense inhibitors suppress tumourigenesis of human cancer cell lines in vitro and in vivo. Carcinogenesis. 2004;25:499–507.
Peeters JG, Vervoort SJ, Tan SC, et al. Inhibition of super-enhancer activity in autoinflammatory site-derived T cells reduces disease-associated gene expression. Cell Rep. 2015;12:1986–1996.
Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123.
Detich N, Hamm S, Just G, Knox JD, Szyf M. The methyl donor S-adenosylmethionine inhibits active demethylation of DNA: a candidate novel mechanism for the pharmacological effects of S-adenosylmethionine. J Biol Chem. 2003;278:20812–20820.
Reiter FP, Hohenester S, Nagel JM, et al. 1,25-(OH)(2)-vitamin D(3) prevents activation of hepatic stellate cells in vitro and ameliorates inflammatory liver damage but not fibrosis in the Abcb4(-/-) model. Biochem Biophys Res Commun. 2015;459:227–233.
Abramovitch S, Sharvit E, Weisman Y, et al. Vitamin D inhibits development of liver fibrosis in an animal model but cannot ameliorate established cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2015;308:G112–G120.
Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236.
Lopez P, Gonzalez-Rodriguez I, Sanchez B, et al. Interaction of Bifidobacterium bifidum LMG13195 with HT29 cells influences regulatory-T-cell-associated chemokine receptor expression. Appl Environ Microbiol. 2012;78:2850–2857.
Dolpady J, Sorini C, Di Pietro C, et al. Oral probiotic VSL#3 prevents autoimmune diabetes by modulating microbiota and promoting indoleamine 2,3-dioxygenase-enriched tolerogenic intestinal environment. J Diabetes Res. 2016;2016:7569431.
Pawar RD, Ramanjaneyulu A, Kulkarni OP, et al. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol. 2007;18:1721–1731.
Scaldaferri F, Lopetuso LR, Petito V, et al. Gelatin tannate ameliorates acute colitis in mice by reinforcing mucus layer and modulating gut microbiota composition: Emerging role for ‘gut barrier protectors’ in IBD? United Eur Gastroenterol J. 2014;2:113–122.
Stone M, Fortin PR, Pacheco-Tena C, Inman RD. Should tetracycline treatment be used more extensively for rheumatoid arthritis? Metaanalysis demonstrates clinical benefit with reduction in disease activity. J Rheumatol. 2003;30:2112–2122.
Tabibian JH, Weeding E, Jorgensen RA, et al. Randomised clinical trial: vancomycin or metronidazole in patients with primary sclerosing cholangitis—a pilot study. Aliment Pharmacol Ther. 2013;37:604–612.
Author information
Authors and Affiliations
Contributions
AJC researched, designed, and wrote this article. The tables are original, constructed by AJC, fully referenced, and developed solely for this review. The figure was drawn by AJC, and it has never been published previously. The review article is original, current, comprehensive, and previously unpublished.
Corresponding author
Ethics declarations
Conflict of interest
Author has no conflict of interests to declare.
Rights and permissions
About this article

Cite this article
Czaja, A.J. Under-Evaluated or Unassessed Pathogenic Pathways in Autoimmune Hepatitis and Implications for Future Management. Dig Dis Sci 63, 1706–1725 (2018). https://doi.org/10.1007/s10620-018-5072-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-018-5072-x