
Abstract
Autoimmune hepatitis is associated with interactive cell populations of the innate and adaptive immune systems, and these populations are amenable to therapeutic manipulation. The goals of this review are to describe the key cell populations implicated in autoimmune hepatitis and to identify investigational opportunities to develop cell-directed therapies for this disease. Studies cited in PubMed from 1972 to 2014 for autoimmune hepatitis, innate and adaptive immune systems, and therapeutic interventions were examined. Dendritic cells can promote immune tolerance to self-antigens, present neo-antigens that enhance the immune response, and expand the regulatory T cell population. Natural killer cells can secrete pro-inflammatory and anti-inflammatory cytokines and modulate the activity of dendritic cells and antigen-specific T lymphocytes. T helper 2 lymphocytes can inhibit the cytotoxic activities of T helper 1 lymphocytes and limit the expansion of T helper 17 lymphocytes. T helper 17 lymphocytes can promote inflammatory activity, and they can also up-regulate genes that protect against oxidative stress and hepatocyte apoptosis. Natural killer T cells can expand the regulatory T cell population; gamma delta lymphocytes can secrete interleukin-10, stimulate hepatic regeneration, and induce the apoptosis of hepatic stellate cells; and antigen-specific regulatory T cells can dampen immune cell proliferation and function. Pharmacological agents, neutralizing antibodies, and especially the adoptive transfer of antigen-specific regulatory T cells that have been freshly generated ex vivo are evolving as management strategies. The cells within the innate and adaptive immune systems are key contributors to the occurrence of autoimmune hepatitis, and they are attractive therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autoimmune hepatitis is a chronic inflammatory liver disease characterized by increased serum aminotransferase levels, autoantibodies, elevated serum immunoglobulin G (IgG) concentration, lymphoplasmocytic infiltration of the portal tract, and interface hepatitis [1]. Apoptosis is the principal mechanism of hepatocyte loss, and it can extend the inflammatory and immune responses by generating apoptotic bodies that act as neo-antigens [2]. The inflammatory response to tissue injury is a complex sequence of humoral and cellular immune reactions that reflect involvement of the innate and adaptive immune systems. The humoral response involves the sensitization of CD4+ lymphocytes to self-antigens or foreign antigens that resemble self-antigens and their differentiation into antibody-producing plasma cells [3]. The cellular response involves the generation of liver-infiltrating cytotoxic CD8+ lymphocytes that are the principal perpetrators of the liver injury [3]. Stress signals released from damaged hepatocytes and chemokines that attract cells of the innate immune system initiate the process [4, 5].
The principal approach to the management of autoimmune hepatitis has been to disrupt the activated pathogenic pathways by medications with broad immunosuppressive actions [1]. Clarification and characterization of the principal cellular effectors of the disease will enhance opportunities to target these populations and alter their actions. Tools by which to manipulate the effector cell populations have emerged, and the possibility of disrupting cytokine pathways, blocking T cell antigen receptors (TCRs), strengthening favorable functions, and expanding the number and function of counter-regulatory, protective cell populations projects a new therapeutic horizon.
The goals of this review are to characterize the key components of the innate and adaptive immune systems that have been implicated in the pathogenesis of autoimmune hepatitis and to suggest investigational opportunities to develop cell-directed therapies for this disease.
Cellular Mediators in Autoimmune Hepatitis
The principal cells of the innate immune response in autoimmune liver disease are the dendritic cells and the natural killer (NK) cells [6]. The adaptive immune response emerges within the stressed microenvironment, and its highly specialized cells are able to target cells expressing “non-self” antigens and develop an immunological memory manifested as the production of autoantibodies or the development of antigen-specific TCRs. The principal components of this response are the T helper type 1 (Th1), Th2, and Th17 lymphocytes [7–9]. Natural killer T (NKT) cells, gamma delta (γδ) T cells, and regulatory T lymphocytes are also participants in the adaptive immune response [3].
Key Cellular Components of the Innate Immune System
The innate immune system constitutes the first line of defense against pathogens, and its cellular components include NK cells, mast cells, eosinophils, basophils, macrophages, neutrophils, and dendritic cells. Each cell population exhibits properties associated with both the innate and adaptive immune systems, and strict categorization by the nature of their immune response is an over simplification.
Dendritic Cells
Dendritic cells are highly specialized for uptake, processing, and presentation of foreign and self-antigens [10], and they participate in the development of immune tolerance (Table 1) [11]. Dendritic cells can respond to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) that are detected by pattern recognition receptors (PRRs) [12]. The principal PRRs on the dendritic cells are Toll-like receptors (TLRs), and they signal cell distress. Dendritic cells can recognize cell distress and migrate to areas of tissue injury (innate response), and they can present antigens to naïve lymphocytes and initiate immunological reactivity (adaptive response) [6].
The conventional dendritic cells are migratory or resident-based in lymphoid tissue, and they capture, identify, and present foreign antigens to naïve T lymphocytes (Table 1) [6]. Their major functions occur during the steady state in the absence of inflammatory signals, and they promote antigenic tolerance [6, 13]. The plasmacytoid dendritic cells respond to inflammatory signals and secrete mainly type 1 interferons (IFNs), which induce antiviral responses and promote adaptive immunity [14–17]. Plasmacytoid dendritic cells promote a Th2 immune response, and they can support the generation and function of regulatory T cells [18]. Monocyte-derived dendritic cells express high levels of the class II molecules of the major histocompatibility complex (MHC), process foreign antigens, migrate to draining lymph nodes, and sensitize naïve T lymphocytes to specific antigens (Table 1) [6].
Studies in experimental animal and human models of diverse immune-mediated diseases, including systemic lupus erythematosus, rheumatoid arthritis, type 1 diabetes, autoimmune thyroid disease, and multiple sclerosis (experimental autoimmune encephalomyelitis), have supported the role of dendritic cells in the induction of autoimmunity [6, 15, 19]. Autoimmune liver disease has been produced in animal models by the vaccination of C57BL/6 mice with dendritic cells loaded with well-differentiated hepatocellular carcinoma cells [20], and fatal autoimmune hepatitis has been induced in a mouse model from dendritic cells that induced the differentiation of naïve T lymphocytes to Th1 cells and cytotoxic T cells in response to interleukin (IL)-18 [21].
Natural Killer Cells
Natural killer (NK) cells comprise up to 15 % of the peripheral blood lymphocytes and 30–50 % of the resident hepatic lymphocytes (Table 1) [22–24]. Their number and availability attest to their importance as components of the innate immune response and protectors against invading pathogens [24]. NK cells also interact with antigen-producing cells (dendritic cells and T lymphocytes), and they can exhibit features associated with an adaptive immune response [25]. The severity of diverse tissue-specific, immune-mediated diseases, including autoimmune hepatitis [26], primary biliary cirrhosis (PBC) [27], systemic sclerosis [28], rheumatoid arthritis [29], and type 1 diabetes mellitus [30], are influenced by the activity of NK cells.
NK cells have two principal sets of receptors that modulate their activity in humans, and they are the killer immunoglobulin (Ig)-like receptors (KIRs) and the C2D natural killer group 2 member D (NKG2D) receptors (Table 1) [24]. NK cells with stimulatory KIRs can lyse distressed cells that cannot be identified as self [31], and NK cells with inhibitory KIRs can suppress cytolytic activity [32]. Self is defined mainly by the ability to express class I MHC molecules on the cell surface, and NK cells with stimulatory KIRs do not target cells expressing these molecules [33]. Combinations of KIRs can define haplotypes that reflect an inherited balance between inhibitory and stimulatory receptors, and occurrences of systemic sclerosis, type 1 diabetes mellitus, and rheumatoid vasculitis have been associated with certain KIR genotypes [33].
NKG2D receptors are expressed on all NK cells, and they direct NK cells to ligands expressed on distressed cells [34]. The soluble forms of the MHC class I-related chains A and B (MIC A and MIC B) are membrane glycoproteins that are induced by cell stress, and these glycoproteins can activate NK cells by ligating with their NKG2D receptors and inducing cytolysis [35]. Serum levels of MIC A and MIC B have been low or absent in patients with autoimmune hepatitis, PBC, and PSC [36], and the role of the NGK2D ligands in the pathogenesis of these autoimmune liver diseases is unclear. The observation that a strong activator of hepatic NK cells (polyinosinic:polycytidylic acid) can produce an NK cell-dependent hepatitis that mimics autoimmune hepatitis advances the candidacy of this population as a critical participant in the occurrence of this disease [37].
Key Cellular Components of the Adaptive Immune System
The adaptive immune system consists of T and B lymphocytes that have been differentiated into highly specialized subsets that can recognize, neutralize, and remember specific antigens [38]. The effector cell populations that can clonally expand and produce antibodies or infiltrate tissue (Th1, Th2, and Th17 lymphocytes) and the cell populations that can modulate the immune response (NKT cells, γδ cells, and regulatory T cells) are the key subsets implicated in tissue-specific autoimmune diseases and relevant to autoimmune hepatitis [38].
Th1 Lymphocytes
Th1 lymphocytes are sensitized to foreign antigens that have been presented in the antigen-binding groove of class II MHC molecules by APCs (Table 2) [39]. The activated Th1 lymphocytes then differentiate along a type 1 cytokine pathway, mediated mainly by IL-2, interferon-gamma (INF-γ), and tumor necrosis factor-alpha (TNF-α), into tissue-infiltrating cytotoxic CD8+ lymphocytes. The cytotoxic CD8+ lymphocytes target and destroy distressed cells, mainly by inducing their apoptosis [3]. The apoptotic bodies may subsequently be phagocytized and presented as neo-antigens by APCs (dendritic cells) [2]. In autoimmune hepatitis, the foreign antigens may resemble self-antigens (molecular mimicry), and repeated or intense exposures to these antigens may override self-tolerance and induce autoreactivity [3].
Th17 cells secrete the pro-inflammatory cytokines, IL-6, IL-1, and TNF-α, and they recruit Th1 cells to sites of tissue damage. The Th1 lymphocytes can in turn produce IFN-γ, and this cytokine can inhibit the further expansion of the Th17 cells in a counter-regulatory loop [7]. The pro-inflammatory cytokines, IL-2, IFN-γ, and TNF-α, secreted by the Th1 lymphocytes strengthen the adaptive immune response by boosting the expression of class I MHC molecules on APCs, increasing the expression of class II MHC molecules on hepatocytes, and promoting the proliferation of cytotoxic CD8+ lymphocytes [40]. Current management strategies for autoimmune hepatitis are directed at inhibiting the activation, differentiation, and proliferation of the Th1 lymphocytes by pharmacological agents [1].
Th2 Lymphocytes
Th2 lymphocytes are sensitized by foreign antigens presented by the class II MHC molecules expressed on APCs. In contrast to Th1 lymphocytes, these cells differentiate into B lymphocytes that can expand into clones of plasma cells (Table 2) [3]. The plasma cells in turn can produce immunoglobulin, generate antibodies, and support an antibody-dependent cell-mediated cytotoxicity [3]. Th2 lymphocytes are induced by IL-4 [8]. They also secrete IL-4, and they can expand in an autocrine fashion.
Th2 lymphocytes secrete IL-5, IL-9, IL-10, IL-13, and IL-25 in addition to IL-4 [8], and they express messenger RNA for peroxisome proliferator-activated receptor-gamma (PPAR-γ), which is a nuclear receptor that can inhibit the expression of pro-inflammatory genes [41]. IL-4 inhibits the proliferation of Th17 lymphocytes [7]; IL-5 induces the expansion of antigen-specific regulatory T cells [42]; and IL-10 dampens the inflammatory response [8, 41]. The composite effect of cytokine production by the Th2 lymphocytes is to reduce inflammation by inhibiting Th1 cells, Th17 cells, monocytes, and macrophages and by expanding the regulatory T cell population [43].
Th17 Lymphocytes
T helper 17 (Th17) lymphocytes are a subset of CD4+ lymphocytes that are generated from naïve lymphocytes by the cytokines, IL-6, and TGF-β (Table 2) [9, 44]. Th17 cells may constitute the first wave of effector cells within the liver in response to inflammatory injury. Their number is increased in the peripheral blood and liver tissue of patients with autoimmune hepatitis, and they have been associated with the severity of inflammatory activity and advanced fibrosis [45]. Th17 cells may also have a protective effect on the liver. Th17 cells secrete IL-22, and IL-22 may have anti-inflammatory effects that counterbalance the pro-inflammatory actions of IL-17 [46, 47]. In a murine model of acute alcoholic hepatitis, treatment with recombinant IL-22 reduced hepatic steatosis and up-regulated genes with anti-oxidant and anti-apoptotic effects [47].
Th17 cells have a positive feedback loop that sustains the inflammatory and immune-mediated responses within the liver (Table 2) [8, 44]. The production of IL-17 by the Th17 cells can induce the hepatic production of IL-6 [45]. Increased production of IL-6 can in turn promote the proliferation of Th17 cells by synergizing with TGF-β [45, 48]. These interactions can also intensify inflammatory activity by reciprocally suppressing regulatory T cell function [48]. The dual cytotoxic and protective actions of the Th17 cells and the dependence of these actions on the cytokine milieu afford opportunities for molecular manipulations that may have therapeutic relevance in immune-mediated diseases such as autoimmune hepatitis.
Natural Killer T Cells
NKT cells reside within the liver, and they have surface markers and functions similar to T lymphocytes and NK cells (Table 2) [49]. NKT cells respond rapidly to hepatic injury (innate immune response), and they can modulate the innate and adaptive immune responses by secreting pro-inflammatory and anti-inflammatory cytokines [50]. NKT cells can also behave as specialized, antigen-reactive T lymphocytes (adaptive immune response) [51]. The NKT cells that rapidly respond to sites of tissue damage have a semi-invariant T cell antigen receptor chain, and they have been classified as invariant NKT (iNKT) cells [49, 51]. The predominant action of the iNKT cells (stimulatory or inhibitory) is modulated in part by the nature of the sensitizing glycolipid antigens released at the sight of tissue injury [52]. iNKT cells recognize CD1d molecules [53], which are a family of class I MHC molecules that present endogenous and exogenous lipid antigens to CD1-restricted cells [52, 54].
The rapid migration of NKT cells to the injured site can extend the liver injury or promote a reparative response (Table 2) [51]. NKT cells are constitutively cytotoxic in that they can induce the apoptosis of distressed cells by releasing granzymes and perforin [54], and they can promote the inflammatory response by producing IFN-γ, IL-4, and IL-21 [55]. These stimulatory actions can in turn be modulated by the ability of NKT cells to promote the differentiation of regulatory T cells [56]. Animal models that are deficient in NKT cells do not develop experimental immune-mediated liver disease [57], and the NKT cells may influence the occurrence and severity of autoimmune hepatitis [58].
Gamma Delta T Lymphocytes
Gamma delta (γδ) T cells constitute 15–25 % of the T lymphocytes within the liver [59], and they are characterized by a TCR composed of one gamma (γ) chain and one delta (δ) chain (Table 2) [59]. The composition of the TCR distinguishes this population from the majority of T lymphocytes whose TCRs consist of one alpha (α) and one beta (β) chain. Gamma delta T cells can exhibit a range of functions that include innate and adaptive immune responses [60]. TCRs can recognize DAMPs and respond rapidly to cellular distress signals and non-peptide ligands without antigen priming [60], and they can phagocytize invading pathogens [61]. Subsets may also develop antigen recognition, specificity, and memory that are linked to the class Ib MHC molecules [62]. They can kill target cells by receptor-mediated apoptosis or the release of cytolytic granules [59], and they may protect against immune-mediated tissue damage and collagen deposition by down-regulating CD8+ cytotoxic T cells [63] and inducing the apoptosis of hepatic stellate cells [64].
Gamma delta T cells have been described in the peripheral blood and liver of patients with autoimmune liver disease, but the contribution of these cells to the disease process is unclear (Table 2). The percentage and absolute numbers of γδ T cells have been higher in the peripheral blood of children with autoimmune hepatitis or PSC compared to healthy subjects [65], and the accumulation of γδ T cells has been greater in the portal tracts of liver tissue from patients with autoimmune hepatitis, PBC, or PSC than from control specimens [66]. The increased hepatic accumulation of γδ T cells has lacked specificity for autoimmune liver disease, but it has supported speculation that the γδ T cells are effectors of liver damage [67]. Countering these pathogenic attributes have been protective actions that can reduce the inflammatory response by IL-10 production [68], impair hepatic fibrosis by inducing Fas-mediated apoptosis of hepatic stellate cells [64], and promote hepatic regeneration by secreting IL-17 and IL-22 [69]. The phenotypic and functional plasticity of γδ T cells strengthens their candidacy as important contributors to the pathogenesis of autoimmune liver disease and therapeutic targets in autoimmune hepatitis.
Regulatory T Cells
Regulatory T cells are a small but critical subset of CD4+ T lymphocytes that are characterized by a constellation of distinctive surface markers and by immunosuppressive actions that dampen the immune response (Table 2) [4, 70]. Two main subtypes of the regulatory T cells have been described, and they are the natural regulatory T cells and the induced regulatory T cells [71]. Natural regulatory T cells develop in the thymus, and they constitute 10 % of the CD4+ lymphocytes in the peripheral blood [72]. The induced regulatory T cells develop in an inflammatory milieu after antigen exposure and stimulation by TGF-β [73]. The peripherally induced regulatory T cells exert more powerful immunosuppressive effects than the natural, thymic-derived, regulatory T cells [74]. Furthermore, the induced regulatory T cells maintain phenotypic and functional stability in inflammatory conditions, whereas natural regulatory T cells can lose suppressive function when exposed to pro-inflammatory cytokines. Natural regulatory T cells can convert to Th1, Th2, and Th17 effector cells [75].
The principal function of the regulatory T cells is to suppress immune-mediated responses. Regulatory T cells expressing PPAR-γ inhibit the production of IFN-γ by CD4+ lymphocytes [41], suppress the inflammatory response in murine models [76], and inhibit the proliferation and chemotaxis of hepatic stellate cells [77]. Regulatory T cells also strengthen the type 2 anti-inflammatory cytokine pathway by producing IL-10 [78], and they limit the proliferation of Th17 cells by suppressing the production of IL-17 [79, 80]. Neonatal thymectomy in a mouse model depletes CD4+CD25+ regulatory T cells, accelerates antibody responses, and increases the frequency of pathogenic self-reactive T cells [81, 82]. In BALB/c mice with disruption of the programmed cell death-1 (PD-1 −/−) signaling pathway, neonatal thymectomy is associated with loss of natural Foxp3+ regulatory T cells and the spontaneous development of fatal autoimmune hepatitis [83–85]. These findings underscore the importance of natural, thymic-derived, regulatory T cells in maintaining immunological homeostasis and preventing an unregulated autoreactive response.
Two molecular signaling pathways influence the function of the regulatory T cells. CD39 is an ectonucleotidase that is expressed on regulatory T cells, and it catalyzes the conversion of adenosine triphosphate (ATP) and adenosine diphosphate (ADP) to adenosine monophosphate (AMP) [86]. AMP in turn is converted to the immune regulatory nucleoside, adenosine, by CD73 which is derived mainly from contiguous damaged tissue [87]. Adenosine in turn suppresses the proliferation and cytokine secretion of Th1 and Th2 lymphocytes [88].
The galectin-9-TIM-3 pathway is another mechanism by which the regulatory T cells can maintain immunological homeostasis. Galectin 9 is a beta galactosidase-binding protein expressed on regulatory T cells, and it promotes immune tolerance by binding to the T cell immunoglobulin and mucin domain-3 receptor (TIM-3) which is expressed on Th1 cells and dendritic cells [89, 90]. The ligation of galectin 9 with TIM-3 induces the apoptosis of Th1 and dendritic cells and down-regulates the immune response [90]. Deficiencies in the expression of galectin 9 on regulatory T cells and TIM-3 on the Th1 cells have been associated with autoimmune hepatitis, and correction of these deficiencies is another treatment opportunity in autoimmune hepatitis [89].
Key Chemokine Pathways Affecting Effector Cell Migration
The diverse cellular components of the innate and adaptive immune systems are directed to sites of liver injury mainly by the release of chemokines from the injured hepatic tissue [4, 5]. Chemokines function as ligands (L) that attract immune and inflammatory cells with complementary ligand receptors (R) [91]. The chemokines can be produced by injured resident cells within the liver (hepatocytes, endothelial cells, hepatic stellate cells, and dendritic cells) and by first responder cells (neutrophils and monocytes) [92]. The cellular responders can in turn generate a positive feedback loop by producing cytokines, stimulating chemokine production, and attracting effector cells [93]. The effects of the chemokines and their ligands can vary during the course of the disease, and different liver diseases may have different cytokine profiles [5, 94]. Chemokines typically promote pro-inflammatory and pro-fibrotic responses, but they can also have anti-inflammatory and anti-fibrotic actions [5].
The principal chemokine ligands implicated in immune-mediated liver diseases are CXCL9, CXCL10, CXCL11, CCL20, CXCL12 (stromal cell-derived factor-1 [SDF-1]) and CX3CL1 [fractalkine]) [4]. Serum levels of eotaxin-1 (CCL11), eotaxin-3 (CCL26), and CCL11 (macrophage-derived chemokine [MDC]) are increased in autoimmune hepatitis, primary biliary cirrhosis (PBC), and primary sclerosing cholangitis (PSC) [94]. CX3CL1 (fractalkine), which is expressed by hepatocytes, hepatic stellate cells, and biliary endothelial cells, has pro-inflammatory and anti-fibrotic effects [95, 96]. Its cognate receptor, CX3CR1, is expressed on monocytes, Kupffer cells, natural killer cells, T lymphocytes, and smooth muscle cells [97, 98]. The CX3CR1/CX3CL1 axis may protect hepatocytes from apoptosis [97, 99], limit the accumulation of monocyte-derived macrophages in the liver [97], inhibit Kupffer cell production of TGF-β [98], impair the activation of hepatic stellate cells [98], and reduce the deposition of collagen [90]. The net effects of the cell mediators in autoimmune hepatitis are influenced by the chemokine milieu associated with the hepatic injury, and therapeutic manipulations of this milieu by pharmacological agents or neutralizing antibodies are possible management strategies [4, 5].
Feasible Cell-Based Interventions in Autoimmune Hepatitis
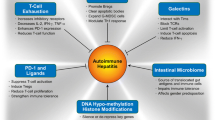
The principal cell populations that have been implicated in the pathogenesis of autoimmune hepatitis have complex, frequently opposite actions that can stimulate or inhibit the immune responses (Fig. 1). Each population and their molecular signals are feasible therapeutic targets. Intrinsic, possibly inherited, defects in cell function may be irreparable, and the restoration of immunological homeostasis may depend mainly on strengthening the salutary actions of a single population or on generating a composite beneficial effect from several populations. The challenge of immune cell manipulation is to preserve or strengthen the protective actions while restricting or isolating the deleterious effects. The ideal candidates for therapeutic manipulation in autoimmune hepatitis are well-characterized, antigen-activated, cell populations that have a broad impact on the disease and that can be accurately targeted. Regulatory T cells, cytotoxic Th1 lymphocytes, antibody-producing Th2 lymphocytes, NKT cells, and dendritic cells have these attributes.

Interactive inhibitory and stimulatory actions of the innate and adaptive immune systems in autoimmune hepatitis. Dendritic cells (DC) and natural killer (NK) cells are the principal components of the innate immune response, and they secrete interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), and interleukin (IL)-2. These cytokines have pro-inflammatory actions on the T helper type 1 (Th1) and T helper type 17 (Th17) lymphocytes. NK cells are characterized by the expression of killer immunoglobulin-like receptors (KIR) and C2D natural killer group 2 member D (NKG2D) receptors. Th1, Th type 2 (Th2), and Th17 lymphocytes; natural killer T (NKT) cells; and gamma delta lymphocytes are the principal components of the adaptive immune response. Each cell population has inhibitory (−) and stimulatory (+) actions mediated by the secretion of pro-inflammatory (IFN-γ, TNF-α, IL-2, IL-4, IL-17, IL-21) and anti-inflammatory (IL-22, IL-10) cytokines depending on the cell type. The cells of the adaptive immune response can modulate the severity of tissue injury by stimulating expansion of regulatory T cells (Treg) and inducing the apoptosis of hepatocytes and hepatic stellate cells (HSC)
Therapy Directed at Regulatory T Cells
Autoimmune hepatitis has been associated with quantitative and qualitative deficiencies in the regulatory T cell population [100], albeit these observations have been challenged [101]. CD39-positive regulatory T cells are decreased in number, hydrolyze ATP and ADP to AMP less actively, and fail to suppress IL-17 production by CD4+ lymphocytes [80]. They also have less lineage stability compared to the regulatory T cells of healthy subjects after an inflammatory challenge. The increased production of the pro-inflammatory cytokines, IFN-γ and IL-17, by the CD39-positive regulatory T cells suggests that these cells have undergone a phenotypic conversion from an immunosuppressive to a pro-inflammatory state [80]. These deficiencies and transformations within the regulatory T cell population are key aspects of autoimmune hepatitis that can be targeted by pharmacological [102, 103] and cellular interventions (Fig. 2) [104, 105].

Current and prospective cell-directed interventions in autoimmune hepatitis. Pharmacological therapies (blue) are current treatments for autoimmune hepatitis, and they have non-selective effects on dendritic cells (DC) and regulatory T cells (Tregs). Investigational interventions (black) include antibodies to cell surface markers (anti-CD3, CD20, CD80, CD83 and CD86), adoptive transfer of key cell populations, moderation of activity with engineered antigens (glycolipids), and agents that strengthen immunosuppressive actions (retinoic acid, vitamin D3). The investigational therapies have been directed mainly against the T helper type 1 (Th1) lymphocytes, Th2 lymphocytes, Tregs, natural killer T (NKT) cells, and DC
Pharmacological Agents
Corticosteroids can reconstitute the regulatory T cell population, and the regulatory T cells can in turn suppress the proliferation of CD8+ lymphocytes and induce the production of the anti-inflammatory cytokine, IL-4 (Table 3) [102]. Mycophenolate mofetil can expand the regulatory T cell population [106–108], possibly by directly inhibiting the expression of co-stimulatory molecules (CD40, CD80, CD86) by dendritic cells, impairing the presentation of antigens, and reducing the production of the pro-inflammatory cytokine, IL-12 [6, 109]. Rapamycin can also increase the population of functional antigen-specific regulatory T cells, [103, 110] and in murine models treated with various immunosuppressive agents, rapamycin has been superior to mycophenolate mofetil as an expander of this population [110].
Other less conventional agents have had similar success in restoring the function of regulatory T cells (Table 3). Retinoic acid has been able to abrogate inflammation-induced deficits in regulatory T cell function and limit the increase of Th1 and Th17 transcription factors during inflammatory activity [103]. The activated form of vitamin D3 (1, 25 dihydroxyvitamin D3) can induce the immunosuppressive actions of the regulatory T cells and promote tolerance to allografts of pancreatic tissue in murine models of transplantation (Fig. 2) [106]. Vitamin D3 inhibits antigen-presentation by dendritic cells, limits the production of IL-12, increases the secretion of IL-10, and supports the immunosuppressive actions of invariant NKT cells [106, 111].
Low serum levels of 25-hydroxyvitamin D have been found in 43 % of patients with autoimmune hepatitis, and low 25-hydroxyvitamin D levels have been associated with advanced hepatic fibrosis and severe inflammation [112]. Furthermore, polymorphisms of the vitamin D receptor gene have been described with autoimmune hepatitis [113, 114], and low serum levels of 25-hydroxyvitamin D have been identified in diverse, non-liver-related, immune-mediated diseases, including multiple sclerosis, rheumatoid arthritis, inflammatory bowel disease, and systemic lupus erythematosus [115]. The disease-specific modulatory dysfunctions of the immune system associated with vitamin D deficiency are unclear, but disturbances in the regulatory T cell population must be considered.
Current pharmacological agents may also be rendered more effective by better understanding the mechanisms for their failure and identifying strategies for re-enforcing their actions. Studies in thymectomized neonatal mice have indicated that spontaneous fatal autoimmune hepatitis is associated with splenic follicular helper T cells which can migrate into the liver via the CCR6–CCL20 axis [84, 85]. These mice have depletion of Foxp3+ regulatory T cells and disruption of the PD-1 signaling pathway, and they are resistant to treatment with dexamethasone. Splenectomy has prevented the development of autoimmune hepatitis in this experimental model, and it has suppressed liver inflammation and improved survival after administration of dexamethasone [85]. These findings identify the spleen as the induction site for experimental autoimmune hepatitis in animals deficient in regulatory T cells, and they suggest that failures of current drug regimens might be overcome by ancillary measures that not only replenish the regulatory T cell population but also target reservoirs of autoreactive cells.
Adoptive Transfer
Aberrations in the number and function of the regulatory T cells in autoimmune hepatitis can also be corrected by the adoptive transfer of regulatory T cells that have been expanded or newly generated [104, 105, 116], and this intervention has improved histological indices of disease activity and restored peripheral tolerance to liver antigens in a murine model of experimental autoimmune hepatitis (Table 3) [105]. Newly generated regulatory T cells proliferate more actively and resist apoptosis more fully than expanded populations [116]. By inhibiting IL-17 production in culture, freshly generated regulatory T cells can maintain a stable phenotype, lose pro-inflammatory properties, and improve their immunosuppressive actions [117].
Antigen-specific regulatory T cells also have greater immunosuppressive effects than non-antigen-specific polyclonal regulatory T cells [118, 119]. Antigen specificity facilitates migration of the regulatory T cells to the appropriate tissue site, and it promotes their activation in the target organ without affecting their function [120]. Regulatory T cells can be engineered to have organ specificity without knowledge of the particular antigen that actually triggers the disease [120, 121]. They can be directed to the appropriate target by the retroviral transfer of the preferred TCR [122] or the incorporation of a chimeric antigen receptor (CAR) [123].
The prospect of restoring immune tolerance favors the adoptive transfer of antigen-specific regulatory T cells over pharmacological agents with indiscriminate immunosuppressive actions in the management of autoimmune hepatitis [104]. The challenge is to universalize a treatment which is highly individualized, labor-intensive, expensive, and restricted to specialized centers [104, 124].
Therapy Directed at Th1 Lymphocytes
Non-mitogenic monoclonal antibodies to CD3 dampen tissue damage by the cytotoxic CD8+ cells through direct and indirect actions (Table 3) [125]. The antibodies target the TCR of mature T lymphocytes, and they induce degradation of deoxyribonucleic acid and promote cell death by apoptosis (direct action) (Fig. 2) [126]. They also augment the function of regulatory T cells by promoting the secretion of TGF-β by macrophages and immature dendritic cells (indirect action) [126, 127]. Macrophages and immature dendritic cells phagocytize the apoptotic bodies of the dying cytotoxic T lymphocytes, and they release TGF-β which in turn induces the activity of regulatory T cells and enhances immunosuppression. Treatment has been effective in animal models of diabetes [128] and experimental autoimmune encephalitis [126] and in patients with autoimmune diabetes [129].
Therapy Directed at Th2 Lymphocytes
Rituximab is a chimeric mouse–human monoclonal antibody that binds to CD20 on the B cell surface, induces apoptosis, and achieves rapid and sustained depletion of B cells [130]. Isolated case reports have indicated the success of rituximab in the treatment of autoimmune hepatitis and idiopathic thrombocytopenic purpura [131], autoimmune hepatitis and cryoglobulinemic glomerulonephritis [132], autoimmune hepatitis and previous B cell lymphoma [133], and autoimmune hepatitis and Evans syndrome (autoimmune hemolytic anemia and idiopathic thrombocytopenic purpura) (Table 3) [134]. Furthermore, a single-center experience involving six patients with refractory autoimmune hepatitis has demonstrated significant reductions in serum aspartate aminotransferase levels at 24 weeks, histological improvement in the 4 patients who underwent liver tissue examination, and no serious side effects after two infusions of rituximab (1,000 mg) 2 weeks apart (Fig. 2) [135].
Therapy Directed at NKT Cells
Injections of the marine sponge-derived glycosphingolipid, α-galactosylceramide, into a murine model of systemic lupus erythematosus can confer long-term protection by activating NKT cells that inhibit the proliferation of IL-10 producing B cells (Table 3) [136]. Treatments with α-galactosylceramide [137] or a synthetic analog of this same glycolipid [138] have protected mice from autoimmune diabetes and from collagen-induced arthritis (Fig. 2). Modifications in the length and structure of the acyl chain on the synthetic α-galactosylceramide molecule can alter the NKT cell response, and this molecular plasticity suggests that the structure of the glycolipid antigen can be designed to suit the disease and the individual patient [137]. Actions of the NKT cells can also be modified by blocking the production of certain cytokines and dampening their effect on the maturation and proliferation of effector T cell populations [125]. Neutralizing antibodies against pro-inflammatory (IL-21) and anti-inflammatory (IL-4, IL-10) cytokines can skew the function of T lymphocytes along a favorable pathway (Table 3) [139].
Therapy Directed at Dendritic Cells
The challenge in developing therapies directed at dendritic cells is to neutralize the target early in the disease without critically disrupting immunological homeostasis or allowing epitope spread [6]. Treatments that have been evaluated in animal models include pharmacological agents, neutralizing antibodies, tolerance induction, gene therapy, vaccination, adoptive transfer, and metabolic disruption [6, 140]. The multitude of options reflects the preliminary nature of the investigations and the lack of a clear preference (Fig. 2).
Corticosteroids impair dendritic cell function, and they are the principal medications used in the treatment of autoimmune hepatitis (Table 3) [141]. The calcineurin inhibitors down-regulate dendritic cell production of the pro-inflammatory cytokines, IL-2 and IL-12, and they inhibit the activation of memory CD8+ T lymphocytes and the differentiation of antigen-sensitized T lymphocytes [142]. Rapamycin impairs the activation of dendritic cells, prevents their maturation, and preserves tolerance to self-antigens [143]. These medications are currently used in the management of autoimmune hepatitis, and they have non-selective actions and variable results.
Molecular interventions may improve the precision of cell targeting, neutralize a critical pathogenic pathway, and achieve a durable result (Table 3) [6]. Antibodies to the surface markers of dendritic cells (CD80, CD83, and CD86) can prevent their maturation by inhibiting co-stimulatory interactions with CD28 receptors on autoreactive T lymphocytes [144], and antibodies to the delta-like ligand 4 (anti-Dll4) can block a signaling (Notch) pathway important in the development of dendritic cells [145]. The chronic stimulation of the Toll-like receptor 2 (TLR2) has prevented diabetes in a murine model by enhancing the induction of antigen tolerance by dendritic cells [146], and dendritic cells that have been generated ex vivo from harvested monocytes, transfected with genes encoding IL-10, and adoptively transferred to mice have reduced graft rejection [147]. Additional efforts of some success have included the down-regulation of dendritic cells ex vivo by oligonucleotides targeting the surface molecules, CD40, CD80, and CD86, prior to their injection into syngeneic diabetic mice [148], and the administration of unaltered autologous dendritic cells by adoptive transfer to patients with type 1 diabetes (Phase I clinical trial) [139].
Other interventions in early stages of evaluation are dendritic cell vaccines based on the linkage of a dominant autoantigen with an immune stimulatory adjuvant to inhibit dendritic cell maturation [149] and the administration of enzymes (indoleamine 2, 3 diogenase) or metabolites (3-hydroxyanthranilic acid) that inhibit the maturation of dendritic cells, their production of pro-inflammatory cytokines (IL-6, IL-12 and TNF-α), and their activation of Th1 lymphocytes (Table 3) [150].
Overview
Cells of the innate and adaptive immune systems are the principal effectors of tissue-specific immune-mediated diseases, and this highly interactive and cross-regulatory network of cell populations is amenable to therapeutic manipulation (Fig. 1). The interventions that have progressed furthest in the treatment of autoimmune hepatitis have focused on the regulatory T cell population. The adoptive transfer of regulatory T cells, especially antigen-specific cells that have been freshly generated and highly purified, has emerged as an intervention that may induce a durable, antigen-specific, immune tolerance, and it has improved the histological features of experimental autoimmune hepatitis in mice (Fig. 2) [105].
Cell-directed interventions affecting the number and actions of the Th1 and Th2 lymphocytes, NKT cells, and dendritic cells are ongoing in other immune-mediated diseases, and they may also have relevance in autoimmune hepatitis (Fig. 2). Non-mitogenic monoclonal antibodies to CD3 can target the cytotoxic CD8+ T cells that are the principal effectors of autoimmune hepatitis [129], and antibodies to CD20 have already shown promise in this disease [135]. Each intervention is poised for formal study. The contributions of the NKT cells and dendritic cells to the occurrence and severity of autoimmune hepatitis are uncertain, but their key roles in the genesis of other tissue-specific autoimmune diseases and the emergence of methods to alter them justify a continuing interest in these populations as future therapeutic targets [6, 137].
The goals of inducing a durable tolerance of pathogenic antigens and restoring immune homeostasis by correcting or counteracting deficiencies or excesses in the cellular perpetrators of the disease constitute a natural progression in the management of immune-mediated disorders. The advances that are occurring in the understanding and management of these diseases must continue to excite and energize investigational efforts in autoimmune hepatitis.
References
Manns MP, Czaja AJ, Gorham JD, et al. Diagnosis and management of autoimmune hepatitis. Hepatology. 2010;51:2193–2213.
Czaja AJ. Targeting apoptosis in autoimmune hepatitis. Dig Dis Sci. 2014;59:2890–2904.
Czaja AJ. Autoimmune hepatitis. Part A: pathogenesis. Expert Rev Gastroenterol Hepatol. 2007;1:113–128.
Czaja AJ. Review article: chemokines as orchestrators of autoimmune hepatitis and potential therapeutic targets. Aliment Pharmacol Ther. 2014;40:261–279.
Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. 2014;147:577–594, e571.
Mbongue J, Nicholas D, Firek A, Langridge W. The role of dendritic cells in tissue-specific autoimmunity. J Immunol Res. 2014;2014:857143.
Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132.
Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 2008;31:252–256.
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517.
Coquerelle C, Moser M. DC subsets in positive and negative regulation of immunity. Immunol Rev. 2010;234:317–334.
Shklovskaya E, Fazekas de St Groth B. Balancing tolerance and immunity: the role of dendritic cell and T cell subsets. Methods Mol Biol. 2007;380:25–46.
Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175.
Hubert FX, Kinkel SA, Davey GM, et al. Aire regulates the transfer of antigen from mTECs to dendritic cells for induction of thymic tolerance. Blood. 2011;118:2462–2472.
McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol. 2005;79:17–27.
Lombardi VC, Khaiboullina SF. Plasmacytoid dendritic cells of the gut: relevance to immunity and pathology. Clin Immunol. 2014;153:165–177.
Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–1226.
Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306.
Ito T, Yang M, Wang YH, et al. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J Exp Med. 2007;204:105–115.
Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol. 2013;13:566–577.
Tamaki S, Homma S, Enomoto Y, et al. Autoimmune hepatic inflammation by vaccination of mice with dendritic cells loaded with well-differentiated hepatocellular carcinoma cells and administration of interleukin-12. Clin Immunol. 2005;117:280–293.
Ikeda A, Aoki N, Kido M, et al. Progression of autoimmune hepatitis is mediated by IL-18-producing dendritic cells and hepatic CXCL9 expression in mice. Hepatology. 2014;60:224–236.
Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86:513–528.
Shi FD, Ljunggren HG, La Cava A, Van Kaer L. Organ-specific features of natural killer cells. Nat Rev Immunol. 2011;11:658–671.
Hudspeth K, Pontarini E, Tentorio P, et al. The role of natural killer cells in autoimmune liver disease: a comprehensive review. J Autoimmun. 2013;46:55–65.
Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49.
Kaneda K, Kurioka N, Seki S, Wake K, Yamamoto S. Pit cell-hepatocyte contact in autoimmune hepatitis. Hepatology. 1984;4:955–958.
Shimoda S, Harada K, Niiro H, et al. Interaction between Toll-like receptors and natural killer cells in the destruction of bile ducts in primary biliary cirrhosis. Hepatology. 2011;53:1270–1281.
Momot T, Koch S, Hunzelmann N, et al. Association of killer cell immunoglobulin-like receptors with scleroderma. Arthritis Rheum. 2004;50:1561–1565.
Yen JH, Moore BE, Nakajima T, et al. Major histocompatibility complex class I-recognizing receptors are disease risk genes in rheumatoid arthritis. J Exp Med. 2001;193:1159–1167.
van der Slik AR, Alizadeh BZ, Koeleman BP, Roep BO, Giphart MJ. Modelling KIR-HLA genotype disparities in type 1 diabetes. Tissue Antigens. 2007;69:101–105.
Moretta A, Sivori S, Vitale M, et al. Existence of both inhibitory (p58) and activatory (p50) receptors for HLA-C molecules in human natural killer cells. J Exp Med. 1995;182:875–884.
Uhrberg M, Valiante NM, Shum BP, et al. Human diversity in killer cell inhibitory receptor genes. Immunity. 1997;7:753–763.
Boyton RJ, Altmann DM. Natural killer cells, killer immunoglobulin-like receptors and human leucocyte antigen class I in disease. Clin Exp Immunol. 2007;149:1–8.
Gonzalez S, Groh V, Spies T. Immunobiology of human NKG2D and its ligands. Curr Top Microbiol Immunol. 2006;298:121–138.
Kahraman A, Fingas CD, Syn WK, Gerken G, Canbay A. Role of stress-induced NKG2D ligands in liver diseases. Liver international : official journal of the International Association for the Study of the liver.. 2012;32:370–382.
Holdenrieder S, Eichhorn P, Beuers U, et al. Soluble NKG2D ligands in hepatic autoimmune diseases and in benign diseases involved in marker metabolism. Anticancer Res. 2007;27:2041–2045.
Chen Y, Wei H, Gao B, et al. Activation and function of hepatic NK cells in hepatitis B infection: an underinvestigated innate immune response. J Viral Hepat. 2005;12:38–45.
Trivedi PJ, Adams DH. Mucosal immunity in liver autoimmunity: a comprehensive review. J Autoimmun. 2013;46:97–111.
Czaja AJ. Genetic factors affecting the occurrence, clinical phenotype, and outcome of autoimmune hepatitis. Clin Gastroenterol Hepatol. 2008;6:379–388.
Lobo-Yeo A, Senaldi G, Portmann B, et al. Class I and class II major histocompatibility complex antigen expression on hepatocytes: a study in children with liver disease. Hepatology. 1990;12:224–232.
da Rocha Junior LF, Dantas AT, Duarte AL, et al. PPARgamma agonists in adaptive immunity: what do immune disorders and their models have to tell us? PPAR Res. 2013;2013:519724.
Tran GT, Hodgkinson SJ, Carter NM, et al. IL-5 promotes induction of antigen-specific CD4+ CD25+ T regulatory cells that suppress autoimmunity. Blood. 2012;119:4441–4450.
Del Prete G, De Carli M, Almerigogna F, et al. Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. J Immunol. 1993;150:353–360.
Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057.
Zhao L, Tang Y, You Z, et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS ONE. 2011;6:e18909.
Oo YH, Banz V, Kavanagh D, et al. CXCR3-dependent recruitment and CCR6-mediated positioning of Th-17 cells in the inflamed liver. J Hepatol. 2012;57:1044–1051.
Ki SH, Park O, Zheng M, et al. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291–1300.
Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238.
Wu L, Van Kaer L. Natural killer T cells in health and disease. Front Biosci (Schol Ed). 2011;3:236–251.
Berzins SP, Smyth MJ, Baxter AG. Presumed guilty: natural killer T cell defects and human disease. Nat Rev Immunol. 2011;11:131–142.
Subleski JJ, Jiang Q, Weiss JM, Wiltrout RH. The split personality of NKT cells in malignancy, autoimmune and allergic disorders. Immunotherapy. 2011;3:1167–1184.
Van Kaer L, Parekh VV, Wu L. Invariant NK T cells: potential for immunotherapeutic targeting with glycolipid antigens. Immunotherapy. 2011;3:59–75.
Zeissig S, Blumberg RS. Primary immunodeficiency associated with defects in CD1 and CD1-restricted T cells. Ann N Y Acad Sci. 2012;1250:14–24.
Novak J, Lehuen A. Mechanism of regulation of autoimmunity by iNKT cells. Cytokine. 2011;53:263–270.
Wehr A, Baeck C, Heymann F, et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J Immunol. 2013;190:5226–5236.
Nowak M, Stein-Streilein J. Invariant NKT cells and tolerance. Int Rev Immunol. 2007;26:95–119.
Dennert G, Aswad F. The role of NKT cells in animal models of autoimmune hepatitis. Crit Rev Immunol. 2006;26:453–473.
Mattner J. Natural killer T (NKT) cells in autoimmune hepatitis. Curr Opin Immunol. 2013;25:697–703.
Hammerich L, Tacke F. Role of gamma-delta T cells in liver inflammation and fibrosis. World J Gastrointest Pathophysiol. 2014;5:107–113.
Holtmeier W, Kabelitz D. Gammadelta T cells link innate and adaptive immune responses. Chem Immunol Allergy. 2005;86:151–183.
Wu Y, Wu W, Wong WM, et al. Human gamma delta T cells: a lymphoid lineage cell capable of professional phagocytosis. J Immunol. 2009;183:5622–5629.
Morita CT, Mariuzza RA, Brenner MB. Antigen recognition by human gamma delta T cells: pattern recognition by the adaptive immune system. Springer Semin Immunopathol. 2000;22:191–217.
Rhodes KA, Andrew EM, Newton DJ, Tramonti D, Carding SR. A subset of IL-10-producing gammadelta T cells protect the liver from Listeria-elicited, CD8(+) T cell-mediated injury. Eur J Immunol. 2008;38:2274–2283.
Hammerich L, Bangen JM, Govaere O, et al. Chemokine receptor CCR6-dependent accumulation of gammadelta T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology. 2014;59:630–642.
Wen L, Peakman M, Mieli-Vergani G, Vergani D. Elevation of activated gamma delta T cell receptor bearing T lymphocytes in patients with autoimmune chronic liver disease. Clin Exp Immunol. 1992;89:78–82.
Martins EB, Graham AK, Chapman RW, Fleming KA. Elevation of gamma delta T lymphocytes in peripheral blood and livers of patients with primary sclerosing cholangitis and other autoimmune liver diseases. Hepatology. 1996;23:988–993.
Kasper HU, Ligum D, Cucus J, et al. Liver distribution of gammadelta-T-cells in patients with chronic hepatitis of different etiology. APMIS.. 2009;117:779–785.
Bonneville M, O’Brien RL, Born WK. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol. 2010;10:467–478.
Rao R, Graffeo CS, Gulati R, et al. Interleukin 17-producing gammadeltaT cells promote hepatic regeneration in mice. Gastroenterology. 2014;147:473–484, e472.
Longhi MS, Hussain MJ, Mitry RR, et al. Functional study of CD4+ CD25+ regulatory T cells in health and autoimmune hepatitis. J Immunol.. 2006;176:4484–4491.
Singer BD, King LS, D’Alessio FR. Regulatory T cells as immunotherapy. Front Immunol. 2014;5:46.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061.
Karim M, Kingsley CI, Bushell AR, Sawitzki BS, Wood KJ. Alloantigen-induced CD25+ CD4+ regulatory T cells can develop in vivo from CD25-CD4+ precursors in a thymus-independent process. J Immunol. 2004;172:923–928.
Lan Q, Fan H, Quesniaux V, et al. Induced Foxp3(+) regulatory T cells: a potential new weapon to treat autoimmune and inflammatory diseases? J Mol Cell Biol. 2012;4:22–28.
Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770.
Hontecillas R, Bassaganya-Riera J. Peroxisome proliferator-activated receptor gamma is required for regulatory CD4+ T cell-mediated protection against colitis. J Immunol. 2007;178:2940–2949.
Marra F, Efsen E, Romanelli RG, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119:466–478.
Sebastiani S, Allavena P, Albanesi C, et al. Chemokine receptor expression and function in CD4+ T lymphocytes with regulatory activity. J Immunol. 2001;166:996–1002.
Fletcher JM, Lonergan R, Costelloe L, et al. CD39+ Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol. 2009;183:7602–7610.
Grant CR, Liberal R, Holder BS, et al. Dysfunctional CD39(POS) regulatory T cells and aberrant control of T-helper type 17 cells in autoimmune hepatitis. Hepatology. 2014;59:1007–1015.
Sakaguchi S, Sakaguchi N. Thymus and autoimmunity: capacity of the normal thymus to produce pathogenic self-reactive T cells and conditions required for their induction of autoimmune disease. J Exp Med. 1990;172:537–545.
Bagavant H, Thompson C, Ohno K, Setiady Y, Tung KS. Differential effect of neonatal thymectomy on systemic and organ-specific autoimmune disease. Int Immunol. 2002;14:1397–1406.
Kido M, Watanabe N, Okazaki T, et al. Fatal autoimmune hepatitis induced by concurrent loss of naturally arising regulatory T cells and PD-1-mediated signaling. Gastroenterology. 2008;135:1333–1343.
Aoki N, Kido M, Iwamoto S, et al. Dysregulated generation of follicular helper T cells in the spleen triggers fatal autoimmune hepatitis in mice. Gastroenterology. 2011;140:1322–1333, e1321–e1325.
Maruoka R, Aoki N, Kido M, et al. Splenectomy prolongs the effects of corticosteroids in mouse models of autoimmune hepatitis. Gastroenterology. 2013;145:209–220, e209.
Borsellino G, Kleinewietfeld M, Di Mitri D, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232.
Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265.
Kobie JJ, Shah PR, Yang L, et al. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol. 2006;177:6780–6786.
Liberal R, Grant CR, Holder BS, et al. The impaired immune regulation of autoimmune hepatitis is linked to a defective galectin-9/tim-3 pathway. Hepatology. 2012;56:677–686.
Rodriguez-Manzanet R, DeKruyff R, Kuchroo VK, Umetsu DT. The costimulatory role of TIM molecules. Immunol Rev. 2009;229:259–270.
Wasmuth HE, Tacke F, Trautwein C. Chemokines in liver inflammation and fibrosis. Sem Liver Dis. 2010;30:215–225.
Lee EY, Lee ZH, Song YW. CXCL10 and autoimmune diseases. Autoimmun Rev. 2009;8:379–383.
Antonelli A, Ferrari SM, Giuggioli D, et al. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun Rev. 2014;13:272–280.
Landi A, Weismuller TJ, Lankisch TO, et al. Differential serum levels of eosinophilic eotaxins in primary sclerosing cholangitis, primary biliary cirrhosis, and autoimmune hepatitis. J Interferon Cytokine Res. 2014;34:204–214.
Bourd-Boittin K, Basset L, Bonnier D, et al. CX3CL1/fractalkine shedding by human hepatic stellate cells: contribution to chronic inflammation in the liver. J Cell Mol Med. 2009;13:1526–1535.
Shimoda S, Harada K, Niiro H, et al. CX3CL1 (fractalkine): a signpost for biliary inflammation in primary biliary cirrhosis. Hepatology. 2010;51:567–575.
Karlmark KR, Zimmermann HW, Roderburg C, et al. The fractalkine receptor CX(3)CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. 2010;52:1769–1782.
Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology. 2010;52:1390–1400.
White GE, Greaves DR. Fractalkine: a survivor’s guide: chemokines as antiapoptotic mediators. Arterioscler Thromb Vasc Biol. 2012;32:589–594.
Longhi MS, Ma Y, Bogdanos DP, et al. Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol. 2004;41:31–37.
Peiseler M, Sebode M, Franke B, et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J Hepatol. 2012;57:125–132.
Longhi MS, Ma Y, Mitry RR, et al. Effect of CD4+ CD25+ regulatory T-cells on CD8 T-cell function in patients with autoimmune hepatitis. J Autoimmun. 2005;25:63–71.
Holder BS, Grant CR, Liberal R, et al. Retinoic acid stabilizes antigen-specific regulatory T-cell function in autoimmune hepatitis type 2. J Autoimmun. 2014;53:26–32.
Longhi MS, Hussain MJ, Kwok WW, et al. Autoantigen-specific regulatory T cells, a potential tool for immune-tolerance reconstitution in type-2 autoimmune hepatitis. Hepatology. 2011;53:536–547.
Lapierre P, Beland K, Yang R, Alvarez F. Adoptive transfer of ex vivo expanded regulatory T cells in an autoimmune hepatitis murine model restores peripheral tolerance. Hepatology. 2013;57:217–227.
Gregori S, Casorati M, Amuchastegui S, et al. Regulatory T cells induced by 1 alpha,25-dihydroxyvitamin D3 and mycophenolate mofetil treatment mediate transplantation tolerance. J Immunol. 2001;167:1945–1953.
Lim DG, Joe IY, Park YH, et al. Effect of immunosuppressants on the expansion and function of naturally occurring regulatory T cells. Transpl Immunol. 2007;18:94–100.
Miroux C, Morales O, Ouaguia L, et al. Corticosteroids do not reverse the inhibitory effect of cyclosporine on regulatory T-cell activity in contrast to mycophenolate mofetil. Transpl Proc. 2012;44:2834–2839.
Mehling A, Grabbe S, Voskort M, et al. Mycophenolate mofetil impairs the maturation and function of murine dendritic cells. J Immunol. 2000;165:2374–2381.
Wu T, Zhang L, Xu K, et al. Immunosuppressive drugs on inducing Ag-specific CD4(+)CD25(+)Foxp3(+) Treg cells during immune response in vivo. Transpl Immunol. 2012;27:30–38.
Cantorna MT, Zhao J, Yang L. Vitamin D, invariant natural killer T-cells and experimental autoimmune disease. Proc Nutr Soc. 2012;71:62–66.
Efe C, Kav T, Aydin C, et al. Low serum vitamin D levels are associated with severe histological features and poor response to therapy in patients with autoimmune hepatitis. Dig Dis Sci. 2014;59:3035–3042.
Vogel A, Strassburg CP, Manns MP. Genetic association of vitamin D receptor polymorphisms with primary biliary cirrhosis and autoimmune hepatitis. Hepatology. 2002;35:126–131.
Fan L, Tu X, Zhu Y, et al. Genetic association of vitamin D receptor polymorphisms with autoimmune hepatitis and primary biliary cirrhosis in the Chinese. J Gastroenterol Hepatol. 2005;20:249–255.
Smyk DS, Orfanidou T, Invernizzi P, Bogdanos DP, Lenzi M. Vitamin D in autoimmune liver disease. Clin Res Hepatol Gastroenterol. 2013;37:535–545.
Longhi MS, Meda F, Wang P, et al. Expansion and de novo generation of potentially therapeutic regulatory T cells in patients with autoimmune hepatitis. Hepatology. 2008;47:581–591.
Longhi MS, Liberal R, Holder B, et al. Inhibition of interleukin-17 promotes differentiation of CD25(-) cells into stable T regulatory cells in patients with autoimmune hepatitis. Gastroenterology. 2012;142:1526–1535.
Penaranda C, Bluestone JA. Is antigen specificity of autoreactive T cells the key to islet entry? Immunity. 2009;31:534–536.
Sagoo P, Ali N, Garg G, et al. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med. 2011;3:83ra42.
Wright GP, Notley CA, Xue SA, et al. Adoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proc Natl Acad Sci USA. 2009;106:19078–19083.
Hombach AA, Kofler D, Rappl G, Abken H. Redirecting human CD4+ CD25+ regulatory T cells from the peripheral blood with pre-defined target specificity. Gene Ther. 2009;16:1088–1096.
Jethwa H, Adami AA, Maher J. Use of gene-modified regulatory T-cells to control autoimmune and alloimmune pathology: is now the right time? Clin Immunol. 2014;150:51–63.
Maher J. Immunotherapy of malignant disease using chimeric antigen receptor engrafted T cells. ISRN Oncol.. 2012;2012:278093.
Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308.
Czaja AJ. Nonstandard drugs and feasible new interventions for autoimmune hepatitis. Part-II. Inflamm Allergy Drug Targets. 2012;11:351–363.
Ochi H, Abraham M, Ishikawa H, et al. New immunosuppressive approaches: oral administration of CD3-specific antibody to treat autoimmunity. J Neurol Sci. 2008;274:9–12.
Perruche S, Zhang P, Liu Y, et al. CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nat Med. 2008;14:528–535.
Ishikawa H, Ochi H, Chen ML, et al. Inhibition of autoimmune diabetes by oral administration of anti-CD3 monoclonal antibody. Diabetes. 2007;56:2103–2109.
Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698.
Weiner GJ. Rituximab: mechanism of action. Semin Hematol. 2010;47:115–123.
Santos ES, Arosemena LR, Raez LE, O’Brien C, Regev A. Successful treatment of autoimmune hepatitis and idiopathic thrombocytopenic purpura with the monoclonal antibody, rituximab: case report and review of literature. Liver Int. 2006;26:625–629.
Evans JT, Shepard MM, Oates JC, Self SE, Reuben A. Rituximab-responsive cryoglobulinemic glomerulonephritis in a patient with autoimmune hepatitis. J Clin Gastroenterol. 2008;42:862–863.
Barth E, Clawson J. A Case of Autoimmune Hepatitis Treated with Rituximab. Case Rep Gastroenterol. 2010;4:502–509.
Carey EJ, Somaratne K, Rakela J. Successful rituximab therapy in refractory autoimmune hepatitis and Evans syndrome. Rev Med Chil. 2011;139:1484–1487.
Burak KW, Swain MG, Santodomino-Garzon T, et al. Rituximab for the treatment of patients with autoimmune hepatitis who are refractory or intolerant to standard therapy. Can J Gastroenterol. 2013;27:273–280.
Yang JQ, Kim PJ, Singh RR. Brief treatment with iNKT cell ligand alpha-galactosylceramide confers a long-term protection against lupus. J Clin Immunol. 2012;32:106–113.
Blumenfeld HJ, Tohn R, Haeryfar SM, et al. Structure-guided design of an invariant natural killer T cell agonist for optimum protection from type 1 diabetes in non-obese diabetic mice. Clin Exp Immunol. 2011;166:121–133.
Yoshiga Y, Goto D, Segawa S, et al. Activation of natural killer T cells by alpha-carba-GalCer (RCAI-56), a novel synthetic glycolipid ligand, suppresses murine collagen-induced arthritis. Clin Exp Immunol. 2011;164:236–247.
Maurer MF, Garrigues U, Jaspers SR, et al. Generation and characterization of human anti-human IL-21 neutralizing monoclonal antibodies. MAbs.. 2012;4:69–83.
Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. 2011;34:2026–2032.
Moser M, De Smedt T, Sornasse T, et al. Glucocorticoids down-regulate dendritic cell function in vitro and in vivo. Eur J Immunol. 1995;25:2818–2824.
Sauma D, Fierro A, Mora JR, et al. Cyclosporine preconditions dendritic cells during differentiation and reduces IL-2 and IL-12 production following activation: a potential tolerogenic effect. Transplant Proc. 2003;35:2515–2517.
Fischer R, Turnquist HR, Taner T, Thomson AW. Use of rapamycin in the induction of tolerogenic dendritic cells. Handb Exp Pharmacol. 2009;188:215–232.
Lim TS, Goh JK, Mortellaro A, et al. CD80 and CD86 differentially regulate mechanical interactions of T-cells with antigen-presenting dendritic cells and B-cells. PLoS ONE. 2012;7:e45185.
Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776.
Kim DH, Lee JC, Lee MK, Kim KW, Lee MS. Treatment of autoimmune diabetes in NOD mice by Toll-like receptor 2 tolerance in conjunction with dipeptidyl peptidase 4 inhibition. Diabetologia. 2012;55:3308–3317.
Coates PT, Krishnan R, Kireta S, Johnston J, Russ GR. Human myeloid dendritic cells transduced with an adenoviral interleukin-10 gene construct inhibit human skin graft rejection in humanized NOD-scid chimeric mice. Gene Ther. 2001;8:1224–1233.
Machen J, Harnaha J, Lakomy R, et al. Antisense oligonucleotides down-regulating costimulation confer diabetes-preventive properties to nonobese diabetic mouse dendritic cells. J Immunol. 2004;173:4331–4341.
Odumosu O, Nicholas D, Payne K, Langridge W. Cholera toxin B subunit linked to glutamic acid decarboxylase suppresses dendritic cell maturation and function. Vaccine. 2011;29:8451–8458.
Lee WS, Lee SM, Kim MK, et al. The tryptophan metabolite 3-hydroxyanthranilic acid suppresses T cell responses by inhibiting dendritic cell activation. Int Immunopharmacol. 2013;17:721–726.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Montano-Loza, A.J., Czaja, A.J. Cell Mediators of Autoimmune Hepatitis and Their Therapeutic Implications. Dig Dis Sci 60, 1528–1542 (2015). https://doi.org/10.1007/s10620-014-3473-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-014-3473-z