
Abstract
Several lines of evidence suggest the involvement of disturbance in epigenetic processes in autoimmune disease. Most noteworthy is the global DNA hypomethylation seen in lupus. Epigenetic states in difference from genetic lesions are potentially reversible and hence candidates for pharmacological intervention. Potential targets for drug development are histone modification and DNA methylating and demethylating enzymes. The most advanced set of drugs in clinical development are histone deacetylase (HDAC) inhibitors. However, the prevalence of DNA hypomethylation in lupus suggests that we should shift our attention from HDAC inhibitors to DNA demethylation inhibitors. MBD2 was recently proposed to be involved in demethylation in T cells in lupus and is, therefore, a candidate target. Although this field is at its infancy, it carries great promise.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Changes in the normal programming of gene expression lie at the basis of autoimmunity. Gene function is obviously disrupted by DNA sequence changes; however, it is becoming clear that differences in epigenomic programming could also alter gene function in a stable and long-term fashion and play an important role in autoimmune disease. An excellent example is the established role of demethylation of regulatory regions of genes such as lymphocyte function-associated antigen 1 (LFA-1; CD11a/CD18) [1] IL4 and IL6 [2] in T cells from lupus patients.
DNA methylation plays an important role in the differentiation of T cells as well as in maintaining the gene expression profile of mature T cells. For example, the normal programmed maturation of CD4+ cells into either Th1/T2 cells involves DNA demethylation and epigenetic activation of a subset of cytokines [3]. Abnormal demethylation could, on the other hand, lead to aberrant activation of several genes in T cells leading to autoreactivity in vitro and autoimmunity in vivo [4]. Two lines of evidence support a causal role for DNA demethylation in lupus. First, DNA demethylating drugs could induce lupus-like symptoms in patients. Second, T cell DNA from lupus patients is hypomethylated relative to control cells. There are other data supporting DNA demethylation involvement in atopy and autoimmunity. The interferon gamma (IFN-gamma) promoter was demethylated in CD8+ T cells from atopic children in the age range in which hyperproduction of IFN-gamma was recently been identified as a common feature of the atopic phenotype [5]. Changes in DNA methylation could occur not only in T cells but also in the target tissue. For example, in multiple sclerosis (MS) DNA methylation of the peptidyl arginine deiminase 2 (PAD2) promoter is decreased to one third of the level of that in DNA from normal white matter [6]. This enzyme catalyzes the citrullination of myelin basic protein and is increased in MS brain white matter resulting in loss of myelin stability. In this example, the demethylation of PAD2 was associated with increased DNA demethylase activity in white brain matter [6].
There is evidence also for the involvement of chromatin and its remodeling and modification in autoimmune disease. Abnormal patterns of histone modifications occur in CD4+ T cells from lupus patients that include H3 and H4 histone hypoacetylation and H3K9 histone hypomethylation [7].
The basic hypothesis driving epigenetic pharmacology is that, since epigenetic programming is reversible in difference from sequence differences, drugs that target one or more of the enzymes of the epigenetic machinery would restore a “normal” gene expression program. Perhaps the best illustration of the potential of this approach is in cancer therapy where DNA methylation inhibitors and histone deacetylase (HDAC) inhibitors were shown to activate tumor suppressor genes and block tumor growth in animals as well as in several clinical trials [8]. It is clear, however, that global interference with epigenetic programming could result in both therapeutic as well as adverse changes in gene expression. A good example are DNA demethylation drugs which activate tumor suppressor genes and thus slow down tumor growth but might at the same time demethylate and activate prometastatic genes and promote metastasis [9]. One of the cardinal challenges in the field is how to achieve specificity and avoid toxicity. It is critical, therefore, to delineate the detailed involvement of epigenetic enzymes, the upstream signals which act upon them, and the factors that target them to specific genes. More importantly, it is important to understand how these processes are disrupted in specific diseases such as autoimmune disease. This review will discuss these questions.
The epigenome consists of the chromatin including an assortment of histone modifications as well as a covalent modification of cytosines residing at the dinucleotide sequence CG in DNA by methylation [10]. The pattern of histone and DNA modifications varies from gene to gene and the same gene could have diverse patterns of chromatin and DNA modifications in different tissues. The epigenome determines the accessibility of the transcription machinery to the genome and thus programs gene expression. We, therefore, distinguish between open and closed configuration of chromatin [11–15]. Large chromatin domains, which are tightly silenced, are termed heterochromatin; these regions are located around centromeres and telomeres. Regions of chromatin which are permissive for gene expression are termed euchromatin. However, silent genes are found in euchromatic regions and are marked by similar marks to those which are involved in heterochromatin silencing [16]. Recently, a new level of epigenetic regulation by small noncoding RNAs termed microRNA has been discovered [17]. A large number of loci in the human genome encode noncoding RNAs, which are processed to short RNAs and target-specific genes for silencing. microRNAs regulate gene expression at different levels: silencing of chromatin, degradation of mRNA, and blocking translation. microRNAs were found to play an important role in cancer [18] and could potentially play an important role in autoimmunity as well [19–21]. Other classes of small RNAs such as piRNA were recently discovered in the rat, which are thought to participate in silencing of gene expression [22]. Additional forms of noncoding RNA play a role in programming gene expression such as the Air RNA regulating IgfIIR gene expression in a manner which is dependent on the parental origin of the allele [23] and Xist RNA which is involved in the inactivation of the X chromosome [24]. Our understanding of how noncoding RNAs are integrated into long-term programming of gene expression is still preliminary. microRNA processing complexes such as dicer and silencers physically interact with chromatin modification machineries [25, 26]. microRNA expression is itself regulated by epigenetic factors such as DNA methylation and chromatin structure [27] and thus could mediate the impact of epigenetic reprogramming in response to environmental exposure on a panel of other genes. Since microRNAs also act by changing chromatin structure, they should be considered as well under the headings of chromatin and DNA methylation [28–30]. The DNA methylation and chromatin modification machineries are thus candidates for pharmacological manipulation as a potential approach to autoimmune disease. This review will discuss possible targets for epigenetic drugs and their potential utility in autoimmune disease.
Chromatin and chromatin-modifying enzymes
Abnormal chromatin modification was reported in lupus as well as MS and, since chromatin modification plays an important role in programming the expression of cytokines during T cell maturation, it stands to reason that chromatin-modifying enzymes are targets for therapeutic agents in therapy of autoimmune disease and allergy [31, 32].
The DNA is wrapped around a protein-based structure termed chromatin. The basic building block of chromatin is the nucleosome, which is formed of an octamer of histone proteins. There are five basic forms of histone proteins termed H1, H2A, H2B, H3, and H4 [33], as well as other minor variants, which are involved in specific functions such as DNA repair or gene activation [34]. The octamer structure of the nucleosome is composed of a H3–H4 tetramer flanked on either side with a H2A–H2B dimer [33]. The N-terminal tails of these histones are extensively modified by methylation with different numbers of methyl residues: monomethyl, dimethyl, and trimethyl per lysine [35], phosphorylation, acetylation [36], sumoylation, [37] and ubiquitination [38]. The state of modification of these tails plays an important role in defining the accessibility of the DNA to the transcription machinery and the transcription state of genes. Recent genome-wide mapping of 39 known histone modifications has illustrated that a large number of combinatorial patterns of different histone modification are associated with promoters and enhancers. Some combinatorial patterns distinguish highly active promoters [39]. Different histone variants, which replace the standard isoforms, also play a regulatory role and mark active genes in some instances [40]. Histone variants are incorporated into specific regions of the genome throughout the cell cycle in contrast to the canonical histones, which are deposited during DNA replication [41].
An important principle of epigenetic mechanism is reversibility. All histone modifications reflect a balance of modifying and demodifying enzymes. Thus, the state of modification of chromatin could be tilted in either direction by blocking the different modification enzymes with specific antagonists. Another important principle to note is that histone-modifying enzymes are generally not strictly sequence-specific; they are targeted to specific positions in the genome by sequence-specific binding factors. The state of modification at specific loci is defined through recruitment of chromatin-modifying and chromatin-demodifying enzymes by sequence-specific factors. Many repressors and repressor complexes recruit HDACs to genes, thus causing their inactivation [42]. For example, in response to TGF beta receptor signaling, SMAD2 moves into the nucleus and recruits TGIF and HDACs to form a repressor complex at SMAD targets in the genome [43]. The requirement for targeting has important pharmacological implications since blocking a general histone-modifying enzyme would not have a general effect on gene expression and would affect only certain classes of genes. The gene-specific effects would be defined by the milieu of targeting proteins in a given cell. For example, inhibition of HDAC would result in hyperacetylation exclusively in genes that are targeted by histone acetyltransferases (HAT).
Histone acetylation as a therapeutic target
The most investigated histone modification is histone acetylation. Histone acetylation is catalyzed by HAT, which transfer an acetyl group from the cofactor acetyl CoA onto the ε-amino position on lysine and is reversed by HDAC [44]. H3 and H4 histones are acetylated at different positions, especially in the N-terminus tail; H3 at K9 residue as well as other residues (4, 14, 18, 23, 27) and H4 tails at a number of residues (K-5, 8, 12, 16, 20) [44, 45]. A long list of studies has consistently demonstrated that histone acetylation is a hallmark of active genes [46, 47]. The ubiquitous association of histone acetylation with active genes was recently confirmed in whole-genome analyses [48–50]. Thus, this histone modification is an important potential pharmacological target in inflammatory and autoimmune diseases. It is, therefore, not surprising that the first class of pharmacological inhibitor that were tested were HDAC inhibitors [31, 51–55]. The balance between HATs and HDACs defines the state of acetylation of the given loci they associate with. Thus, inhibitors of HDACs will trigger histone acetylation around genes that have HATs in close proximity and HAT inhibitors would trigger histone deacetylation of genes that are associated with HDACs. Since the targeting of HDACs and HATs to genes is delineated by DNA binding factors, the outcome of pharmacological intervention with these enzymes would be defined by the milieu of these factors in the cells. Thus, the history of the transcription programming in a cell would define the specificity of the response to histone acetylation modulators.
Two phylogenic superfamilies of HAT are known to date: the GCN5 N-acetyltransferases-related family GNAT and the MYST family (for a review, see [56]). PCAF is a mammalian transcriptional coactivator, which was discovered based on its homology to GCN5-HAT. PCAF acts as a transcriptional coactivator in many processes and is known to interact with the CREB binding protein (CBP) and its homolog p300. p300 and CBP are ubiquitously expressed transcription coactivators. CBP is an example of targeting of HATs to specific sequences in response to firing of a signaling pathway. CBP associates with CREB a sequence-specific factor that recognizes cAMP responsive elements in the genome. The MYST superfamily includes the human acetyltransferases MOZ and Tip60 [57]. Tip60 acetylates H4 histone tails and is involved in transcriptional activation of genes encoding proteins responsible for DNA damage repair [58, 59]. MOZ is involved in acute myeloid leukemia as suggested by the observation that MOZ and CBP are found fused in this leukemia. The fused protein is mistargeted, resulting in aberrant activation of gene expression [57]. HATs could also have nonhistone and thus nonepigenetic substrates. For example, Tip60 acetylates the nuclear damage response protein kinase ATM [60] and p53 [61, 62].
HATs are reasonable targets for pharmacological manipulation in autoimmunity since aberrant activation of gene expression is involved in T cell autoreactivity. The idea that aberrant gene activation is involved in autoimmunity is supported by the well-established observation that DNA methylation inhibitors such as 5-azacytidine (5-azaC) which are potent inducers of silent genes induce autoreactivity in T cells [63–65]. The close bilateral relationship between DNA methylation and histone acetylation and its implications on the use of histone acetylation modulators as therapeutic agents will be discussed below. Thus, it is conceivable that inhibitors of HAT would trigger inhibition of such aberrantly activated genes and reversal of T cells autoreactivity. Several attempts of developing HAT inhibitors were reported recently and preclinical activities were observed [66]. A list of small molecules and natural products were shown to inhibit HAT activity, such as 4-hydroxyquinolines inhibitors of p300/CBP[67], anacardic acid [68] and benzamides related to anacardic acid with alkyl chains of defined length [69], agarcinol and isogarcinol [70] and the natural product Rosa rugosa Thunb. methanol extract [71], and glycosaminoglycans inhibitors of p300 [72]. Anacardic acid, for example, inhibits the Tip60 HAT in vitro and blocks the Tip60-dependent activation of the ATM and DNA-Pkc protein kinases by DNA damage in vivo compromising the repair process and radiosensitizing the cells [73]. This field is still in its infancy but it definitely deserves attention especially in the context of autoimmune disease.
The main focus in histone acetylation has been directed toward HDAC inhibitors. Several HDACs which are divided into four distinct phylogenic classes were characterized (for a review, see Holbert [74]). Thus, it is critical to identify the specific isotypes involved in pathology to gain drug selectivity. It is believed that isotypic-specific inhibitors of HDACs will exhibit increased specificity and reduced toxicity.
Class 1 HDACs includes HDAC 1–3 and 8, class 2 includes the human HDACs 4–7, 9, and 10, class 3 includes the SIRTUINS and class 4 includes the recently identified HDAC 11. The mechanism of action of class 1 and 2 HDACs seems to involve a mode of catalysis in which the bound zinc ion mediates the nucleophilic attack of a water molecule on the acetylated lysine substrate. Class 3 HDACs, on the other hand, require the presence of the oxidized form of the cofactor nicotinamide adenine dinucleotide (NAD+) as a cofactor for the reaction [74].
A combination of gene expression profiling, functional genomics using knock down approaches [75, 76], and chromatin immunoprecipitation (ChIP) with HDAC isotypic-specific antibodies and genome-wide arrays allow the deciphering of the specific HDAC isotypes involved in specific pathological states. Class 1 and 2 HDACs were shown to be involved in malignancies in humans and are the target of the HDAC inhibitors TSA and SAHA [76–78]. HDAC7 was shown to be involved in angiogenesis and specifically cell migration [79]. Alterations in class 3 HDACs or SIRTUINS were shown to be associated with age-associated diseases, obesity, and neurodegenerative disorders [80] and are recognized as longevity factors [81]. To my knowledge, although HDAC inhibitors were tested in vitro and in vivo in autoimmunity models, we do not have a clear indication of the HDAC isotypes involved in autoimmunity and in the response of autoimmune models to HDAC inhibitors. However, since TSA and SAHA have shown antiautoimmunity activity, it stands to reason that class 1and 2 HDACs are involved. Perhaps the main targets of these HDAC inhibitors in autoimmunity are similar to the targets involved in the anticancer activity and include proapoptotic genes and cell growth regulators. It is imperative to delineate the HDAC involvement in autoimmunity and develop isotypic-specific inhibitors for any further development of rational HDAC-based antiautoimmune therapy.
Interestingly, HDAC inhibitors induce Treg cells suppressive function, thus inhibiting the transplant response. HDAC inhibitors activate the transcription factor FOXP3 which plays a cardinal role in the immunosuppressive function of Treg cells [82]. HDAC9 proved particularly important in negatively regulating FOXP3-dependent suppression [82], thus raising the attractive possibility that isotypic-specific HDAC9 inhibitors might serve as excellent agents for suppressing antitransplant response in transplantation therapy. In addition, it might be potentially important in the suppression of other autoimmune and proinflammatory conditions. Recent preclinical trials with the HDAC inhibitors SAHA in rhesus demonstrated efficacy in primates in induction of Treg function.
Not all HDAC activity is directed toward histones. Several nonhistone proteins are known to be acetylated by HATs and deacetylated by HDACs. Some HDACs are cytosolic and mitochondria. For example, the SIRTUINS are thought to have several nonhistone targets that are critical for their involvement in metabolic response and aging [83–85]. These will not be discussed here as the focus of this review is on the epigenome. However, it should be noted that the nonhistone targets of HDAC inhibitors confound the interpretation of their effects on cells and animals.
HDAC inhibitors that are presently at different stages of development fall into five structural groups. The classic HDAC inhibitors TSA and SAHA are hydroxamate based and they inhibit class 1 and 2 HDACs [86, 87]. SAHA is the first clinically approved HDAC inhibitors [88]. A second group includes hydroxamate-based HDAC inhibitors (LBH589, PXD101) that are currently in different stages of development and inhibit class 1 and 2 HDACs. The third group are aliphatic based and inhibit class 1 and 2 HDACs. This group includes sodium butyrate, one of the earliest HDAC inhibitors, as well as the mood stabilizer and antiepileptic valproic acid. The fourth group includes cyclic peptides-based HDAC inhibitors (FK228) that inhibit class 1 and 2 HDACs. The fifth class is benzamide-based HDAC inhibitors that inhibit class 1, 2, and 3 HDACs. An interesting example of this class is MGCD0103 which showed isotypic specificity against class 1 HDACs and a broad spectrum of antitumor activity [89]. HDAC inhibitors have exhibited anticancer activity in preclinical tumor models and phase 1 and 2 clinical trials (for a review, see [90]) and the first HDAC inhibitor, vorinostat (SAHA), was recently approved for clinical use in cutaneous T cell lymphoma [91]. Vorinostat was safe and effective at an oral dose of 400 mg/day with an overall response rate of 30–31% in refractory-advanced patients with cutaneous T cell lymphoma [91]. The HDAC1 isotypic-specific MGCD0103 is now being tested in phase 1 and phase 2 clinical trials in solid and hematological tumors and has shown some response [92, 93].
The rationale for using HDAC inhibitors in autoimmune disease is based on the documented effect of this approach in cancer cells [94, 95]. In cancer, one of the main impacts of HDAC inhibition is induction of apoptosis and cell cycle arrest through the activation of gene expression of tumor suppressor proteins such as p21 [87, 96]. Induction of apoptosis and cell cycle arrest might also be a mechanism through which HDAC inhibitors would block and kill activated T cells involved in autoimmunity. What is counterintuitive, however, is the effect that HDAC inhibitors have on suppressing cytokine expression since HDAC inhibition should lead to induction of gene activity [54]. Perhaps the answer to this paradox rests in the complex impact that HDAC inhibition has on gene expression circuitries. In a mouse model of MS, it was shown that HDAC inhibitors have a complex effect on gene expression profiles. While the HDAC inhibitor TSA upregulates antioxidant, antiexcitotoxicity, and proneuronal growth and differentiation mRNAs, it also inhibits caspase activation and downregulates gene targets of the proapoptotic E2F transcription factor pathway. In splenocytes, TSA reduces chemotactic, pro-Th1, and proproliferative mRNAs [31]. HDAC inhibitors were shown to be effective as well in a MRL-lpr/lpr murine model of lupus [97]. In this model, the HDAC inhibitors TSA and SAHA inhibited the expression of IL-12, IFN-gamma, IL-6, and IL-10 mRNA and increased the accumulation of acetylated histones H3 and H4 in total cellular chromatin. A 5-week treatment resulted in a significant reduction in proteinuria, glomerulonephritis, and spleen weight [97]. Although the overall immediate output of the complex gene expression alterations induced by HDAC inhibitors seems to be in the direction of reducing autoimmunity, it is unclear whether HDAC inhibition would also lead to aberrant activation of genes in T cells that might eventually promote autoimmunity in a more extended time scale. This should be of concern since DNA demethylating drugs whose impact on gene expression parallels and complements HDAC inhibitors are known to promote systemic lupus erythematosus [63].
In addition to targeting HDAC and HATs, histone acetylation could potentially be modulated by targeting cellular inhibitors of HAT activity such as the SET/TAF-Ibeta/INHAT complex [98] and NIR [99]. Blocking HAT inhibitors should result in which might serve as targets of novel inhibitors. Inhibition of inHATs could result in hyperacetylation as well.
In summary, although it stands to reason that histone acetylation plays a critical role in the gene expression reprogramming involved in autoimmunity, the identity of the main actors is still mostly unknown. Most of the effort is focused on HDAC inhibitors emulating the cancer model but the rationale is not completely clear. The focus on HDAC inhibition might not be fully warranted since this approach might cause long-term adverse effects, stimulating rather than suppressing autoimmunity. HATs and inHATs are untapped targets that warrant consideration.
Histone methylation enzymes and their inhibitors
In difference from histone acetylation, which is generally associated with active genes, different histone methylation marks are associated with either gene activity or gene silencing. Monomethyl, dimethyl, or trimethyl moieties could decorate lysine residues with different functional consequences. These serve as different biological signals. Histone methylation at lysine residues H3-K9Me, H3-K27Me, and H4-K20Me are associated with gene repression. However, it is unclear whether this rule applies in all cases. Surprisingly, a recent study suggests that H3K9Me3 is also associated with active genes rather than being exclusively a repressive marking as previously thought [100]. In general, these modifications are hallmarks of gene silencing as validated by genome-wide ChIP on chip analyses [101]. H3-K4Me, H3-K36Me, and H3-K79Me are associated, on the other hand, with gene activity and transcription elongation [102] and this is confirmed by genome-wide analyses [101]. There is a cross-talk between histone acetylation and histone methylation. H3-K4Me3 and H3K4Me2 peaks are found to occur in close proximity to the transcriptional start sites [103].
Interestingly, critical genes in stem cells are found in a bivalent state with chromatin marks of silencing by PcG such as H3-K27Me and activation such as H3-K4Me. It is believed that this bivalent state maintains these genes poised for either full silencing or activation once these pluripotent cells differentiate [104, 105].
Histone methylation is catalyzed by histone methyltransferases (HMT) and histone demethylation by histone demethylases. Like acetylation, the state of histone methylation is a balance of methylating and demethylating enzymes. Therefore, it stands to reason that one could affect the state of histone methylation by pharmacologically modifying either the methylating or demethylating enzymes and tilting the methylation equilibrium in either direction.
Arginine and lysine HMT catalyze the transfer of a methyl group from the methyl donor SAM to either arginines or lysines. Mammalian homologs of Suv3-9 that methylate lysine 9 on the tail of H3 histones were the first histone lysine methyltransferases (HKMT) to be identified [106]. SET-containing HKMT methylate Lys-4, Lys-9, Lys-27, or Lys-36 of histone H3 and lys-20 of histone H4. There are two distinct families of lysine methyltransferases SET which contain a SET domain of approximately 130 amino acids and Dot1p [107]. All arginine methyltransferases are members of the PRMT family (for a review, see [108]). PRMT1 and PRMT4/CARM1 methylate histone H3, H4, and H2B.
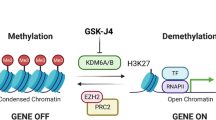
A new area that is of potential interest is the development of HMTase inhibitors. H3K9Me2 histone dimethylation is a hallmark of gene silencing and was shown to mark silenced tumor suppressor genes [109–111]. H3K27Me3 methylation, which is targeted by EZH2, is another interesting target for inhibition. EZH2 associates with DNA methyltransferases (DNMTs) in silencing of tumor suppressor genes [112]. HMTase inhibitors could be therapeutically used to activate silenced tumor suppressor genes. HMTase inhibitors are far less developed than HDAC inhibitors and we know very little on their anticancer activity in either preclinical or clinical models. H3K9 methylation is catalyzed by SUV39 and G9a HMTases. Two HMTase inhibitors were recently described: The fungal mycotoxin chaetocin, which belongs to the class of 3,6-epidithio-diketopiperazines (ETPs) specifically inhibits the Drosophila HMTAse dSU(VAR)3–9 and its human homolog. It also inhibits other HMTase belonging to the class of the SUV39 family such as G9a or DIM5 with weaker potency. Other HMTase containing the SET domain were inhibited at significantly higher IC50 (490 mM for the dE(Z) complex and 4,180 mM for PRSET7 and SET7/9) [113].
A small-molecule inhibitor of G9a histone methyltransferase identified by screening a chemical library was reported last year and was shown to block H3K9Me2 in vitro and in cell culture [114]. However, it is still unknown whether these compounds have anticancer activity or systemic toxicity. There is significant activity in this area and it is anticipated that several HMTase inhibitors including EZH2 inhibitors will be in preclinical and clinical development in the next few years.
Another interesting group of targets are histone demethylases. H3K4Me3 and H4K4Me2 are hallmarks of active promoters in plants [115] and human cells [103]. Since the state of histone methylation is a balance of methylation and demethylation reactions, inhibition of H3K4 demethylase would result in increased H3K4 histone methylation and activation of genes including tumor suppressor genes.
Histone methylation has been considered for a long time as a stable mark that coated stably suppressed heterochromatic regions. The fact that HP-1, the protein recognizing H3-K9Me was a hallmark of heterochromatin, helped in entrenching the idea that histone methylation was extremely stable as are heterochromatin regions and that it could be reversed only by histone protein turnover and not with an enzymatic process. However, the discovery of histone demethylases in recent years [116, 117], including demethylase that could demethylate H3-K9Me3 and H3-K36me36 [118], laid to rest the dogma that histone methylation is irreversible.
Two classes of histone demethylases were recently discovered: (a) FAD-dependent amine monooxygenases which demethylate histones via an oxidation reaction releasing formaldehyde as the leaving group and (b) JmjC domain demethylases which also release formaldehyde but require < ketoglutarate and Fe(II) to drive the reaction. Lysine-specific demethylase 1 (LSD1) is a FAD-dependent enzyme and demethylates monomethyl and dimethyl H3 K4 (for a review, see [74]). Since K4 methylation marks histones associated with active genes, it was anticipated that demethylation of this mark would have a repressive effect on gene expression. In agreement with this prediction, LSD1 was found to reside in the CoREST complex, which represses neuronal genes in non-neuronal cells [119]. However, LSD1 could also function in gene activation when it is associated with the androgen receptor by removing repressive H3-K9Me marks [117]. This dual action of LSD1 illustrates an important principle that extends to several chromatin and DNA modification proteins as will be discussed below. The same protein can perform conflicting roles in both gene activation and gene repression. LSD1 resides in different complexes programming either gene activation or repression during development [120]. This obviously has important therapeutic implications. Inhibition of histone demethylases might affect both gene activation and repression and thus the overall output of the effect of a histone modification drug must be considered when weighing its therapeutic value.
A representative of the second class of histone demethylases, JmjC domain-containing histone demethylase 1 (JHDM1), that specifically demethylates histone H3 at lysine 36 (H3-K36) was recently discovered [121]. H3-K36 is probably involved in promoting transcription elongation and thus maintenance of a transcriptional active state [107]. Removal of this mark would result in transcription repression. Since other methylation sites are known to exist in histones, it is anticipated that more demethylases with different specificities will be discovered in the coming years.
Delineating the specific roles of the different HMT and histone demethylases in the aberrant gene activation leading to autoreactivity of T cells in autoimmune disease is an unexplored area that would potentially play an important role in the pharmacology of autoimmune disease. The field of HMT inhibitors is still in its infancy and there is no evidence as of yet on the possible effect of HMT inhibitors in autoimmune disease but it stands to reason that isotypic HMT inhibitors would have important effects on autoimmune disease.
Interestingly, however, novel biguanide and bisguanidine polyamine analogs were shown to be inhibitors of LSD1 and to induce expression of several genes, which are aberrantly silenced in colorectal cancer [122]. An interesting twist in this story of monoamine oxidases rediscovered as histone demethylases (LSD1) is that certain nonselective monoamine oxidase antidepressants such as tranylcypromine that were clinically used for some time and believed to be acting on monoamine oxidases also appear to inhibit LSD1 demethylase [123]. It is tempting to speculate that inhibition of LSD1 is part of the mechanism of action of these antidepressants through the activation of critical genes suppressed by the H3-K4me demethylating activity of LSD1 in the brain [116] or by repressing genes activated by the H3-K9Me demethylation activity of LSD1 [117]. These agents were not yet tested in autoimmune disease. It is predicted that they would have an impact similar to HDAC inhibitors. However, proper utilization of histone demethylase inhibitors will require deciphering the specific histone demethylase isotypes that are involved in the aberrant activation of genes in autoreactive T cells. Isotypic-specific histone demethylase inhibitors will significantly add to the arsenal of drugs targeting antiautoimmune diseases.
DNA methylation and demethylation enzymes and their inhibitors
The pattern of DNA methylation, the distribution of CG dinucleotides in the genome that bear a methyl moiety is cell type-specific. This pattern of methylation is generated during gestation. Several promoters that are critical for T cell activation are methylated. The process of T cell maturation involves a programmed changed in the state of methylation of specific promoters. Methylation of regulatory regions of these genes is most probably involved in their programmed silencing during gestation and demethylation is related to programmed activation of these genes during T cell maturation. For example, the 5′ region of the IL-4 locus is hypermethylated in naive T cells and becomes specifically demethylated in Th2 cells [124]. CD4+ T cell differentiation to Th2 cells is accompanied with demethylation of Th2-specific cytokines, a process that coincides with acquisition of DNase hypersensitivity, a mark of open chromatin configuration [125]. IFN gamma is demethylated progressively during differentiation of CD4+ T cells down the Th1 pathway but not the Th2 pathway [5]. T cell DNA from lupus patients is hypomethylated [64, 126, 127]. Environmental agents associated with lupus, such as procainamide, hydralazine, and ultraviolet light, were shown to inhibit T cell DNA methylation, increase LFA-1 expression, and induce autoreactivity [63]. Moreover, treatment of activated CD4+ T cells with the DNA demethylating drug 5-azaC resulted in the induction of L-4, IL-6, and IFN-gamma. Adoptive transfer of 5-azaC-treated or procainamide-treated cells into unirradiated syngeneic recipients induced an immune complex glomerulonephritis and IgG anti-DNA and antihistone antibodies characteristic of lupus [128]. Paradoxically, however, treatment of mice with 5-azaC inhibits the lpr gene-induced lymphadenopathy and acceleration of lupus-like syndrome in MRL/MpJ-lpr/lpr mice [129]. These effects resemble the effects of HDAC inhibitors on the same mouse model [97] and most probably involve the antiproliferative activity of 5-azaC and not its DNA demethylating activity. One could predict that the changes in gene expression caused by 5-azaC-induced DNA demethylation would trigger aberrant gene activation in T cells and long-term autoimmunity. Therefore, although the antiproliferative effects of HDAC inhibitors and 5-azaC might have a short-term impact on the rate of proliferation and apoptosis of autoreactive T cells, the reprogramming of gene expression caused by these agents might outweigh the impact of the antiproliferative activity. If DNA demethylation plays a causal role in autoimmunity as suggested by these data, then blocking DNA demethylation rather than promoting DNA demethylation should be a therapeutic goal.
DNA methylating and demethylating enzymes
The DNA methylation reaction is catalyzed by DNA methyltransferases (DNMT) [130]. Methylation of DNA occurs immediately after replication by a transfer of a methyl moiety from the donor S-adenosyl-l-methionine (SAM, AdoMet) in a reaction catalyzed by DNMT. Three distinct phylogenic DNMTs were identified in mammals. DNMT1 shows preference for hemimethylated DNA in vitro, which is consistent with its role as a maintenance DNMT, whereas DNMT3a and DNMT3b methylate unmethylated and methylated DNA at an equal rate, which is consistent with a de novo DNMT role [131]. Two additional DNMT homologs were found: DNMT2 whose substrate and DNA methylation activity is unclear [132] but was shown to methylate tRNA [133, 134] and DNMT3L which is essential for the establishment of maternal genomic imprints but lacks key methyltransferase motifs and is possibly a regulator of methylation rather than an enzyme that methylates DNA [135].
Razin and Riggs proposed that the DNA methylation pattern is accurately inherited during replication since maintenance DNMT1 could only methylate hemimethylated sites. Hemimethylated sites are generated on the nascent DNA strand during DNA replication when a methylated CG dinucleotide in the template strand is replicated. DNA methylation was, therefore, proposed to be truly heritable by a semiconservative mechanism similar to DNA replication [136]. This concept has led to the basic notion that, although DNA methylation patterns are sculpted during development by demethylases and de novo methyltransferases, they are fixed thereafter and are inherited faithfully similar to the genetic sequence.
If this model is indeed true, the pharmacological options of modulating DNA methylation would be limited to dividing cells and to reducing the levels of DNA methylation by blocking DNMT1 during cell division. Indeed, this has been the dominant thinking in the field of DNA methylation inhibitors, that it should be impossible to increase DNA methylation or to block DNA methylation in nondividing cells. The most widely used pharmacological agent in DNA methylation is the nucleoside analog 5-azaC. It is believed that 5-azaC passively inhibits DNA methylation during DNA synthesis once it is incorporated into the nascent strand of DNA by trapping the DNMT enzyme [137]. However, we proposed a decade ago that the DNA methylation pattern is found in a dynamic equilibrium between methylating and demethylating enzymes [138]. This concept was supported by recent data showing rapid changes in DNA methylation in the brain [139]. Similarly, rapid replication-independent changes in DNA methylation in the IL2 gene promoter occur in T cells upon activation [140]. Several lines of evidence suggest that even the process of replicating the DNA methylation pattern during cell division is not an automatic process. It has recently been demonstrated that an additional factor is required for targeting DNMT1 to newly replicating hemimethylated DNA, the protein ubiquitin-like, containing PHD and RING finger domains 1 (UHRF1), also known as NP95 in mouse and ICBP90 in human [141]. Several lines of evidence indicate that DNMTs are targeted to specific sequences by sequence-specific factors, which recognize specific sequences of DNA. For example, the histone methyltransferase EZH2 or the oncoprotein PML-RAR target DNMTs to specific sequences in DNA [142, 143]. Thus, maintenance of the DNA methylation pattern is at least partly an active and targeted process rather than an automatic process as previously thought.
The model that the DNA methylation pattern is in a dynamic equilibrium that is maintained by DNMTs, targeting proteins and demethylases has important implications for the pharmacology of DNA methylation. Similar to histone acetylation, it should be possible to tilt the DNA methylation equilibrium in both directions even in cells that do not divide. Inhibition of DNMT should result in reduced DNA methylation while inhibition of demethylases would result in increased DNA methylation. Given that aberrant induction of genes by DNA demethylation is consistently seen in autoreactive CD4+ T cells [1, 125, 144], DNA demethylation inhibitors that prompt an increase in DNA methylation should be of special interest in autoimmune disease.
Since it is becoming clear that the DNA methylation equilibrium in somatic tissues involves both methylation and demethylation, it is critical to identify the enzymatic machinery responsible for demethylation of distinct classes of genes such as cytokines in autoreactive T cells. The identity of the enzymes required for DNA demethylation has been extremely controversial and there was reluctance in the field to accept the notion of enzymatic demethylation. Several candidates were proposed in the last decade but all proposed candidates were contested immediately after publication. Part of the problem is the difficulty encountered in the purification of active demethylation enzymes and the difficulty in developing a consistent cell-free assay of DNA demethylation. Nevertheless, several cell-free assays were published. In 1982, Gjerset and Martin used a cell-free radioactive assay that measured release of radioactive methyl groups from DNA to demonstrate 5-methylcytosine demethylase activity in erythrocytes extracts [145], demethylase activity was shown in nuclear extract from RAS-transfected p19 cells using a radioactively labeled methylated CG oligonucleotide [146], A549 human lung cancer cell line [138], prostate cancer cells [147], and normal white matter from the brain using the radioactively labeled methylated CG as a substrate [6]. The nature of these assays excluded the possibility that the demethylation was caused by a nuclease or base removal.
Since there was a reluctance to accept that DNA methylation is a reversible reaction, alternative “repair” enzymatic pathways that involve removal and replacement of the methylated cytosine with an unmethylated cytosine were proposed. One proposal has been that a G/T mismatch repair glycosylase also functions as a 5-methylcytosine DNA glycosylase, recognizes methyl cytosines, and cleaves the bond between the sugar and the 5-methylcytosine base. The abasic site is then repaired and replaced with a nonmethylated cytosine, resulting in demethylation by a repair process [148]. An additional protein with a similar activity was recently identified, the methylated DNA binding protein 4 (MBD4) [149]. A protein involved in DNA damage response, GADD45A, was proposed to trigger active DNA demethylation through a repair-mediated process [150]. However, this was contested by a later study [151]. More recently, it was proposed that deamination of the 5-methylcytosine to thymidine by DNMT3A and DNMT3B followed by mismatch repair was involved in the oscillating state of methylation of estrogen receptor target genes [152, 153]. A very recent study suggested that demethylation in zebra fish embryos involved a complex sequence of coupled enzymatic reactions; a 5-meC deaminase (AID, which converts 5-meC to thymine) and a G:T mismatch-specific thymine glycosylase (Mbd4) and repair promoted by GAD45 [154].
The only bona fide demethylation enzyme proposed to date was methylated binding protein 2 (MBD2) [155]. A mechanism for demethylation by this enzyme was recently proposed involving oxidation of the 5-methylcytosine to 5-hydroxymethylcytosine followed by release of the oxidized methyl group as formaldehyde [156]. Interestingly, recent studies provided evidence for the presence of hydroxymethylcytosine in mammalian DNA [157]. Although the 5-hydroxymethylcytosine modification is proposed to be catalyzed by a dedicated enzyme, it is possible that a fraction of the 5-hydroxymethylcytosine in the genome is an intermediate in the DNA demethylation reaction.
The role of MBD2 in demethylation was disputed [158]. The MBD2 knockout mouse DNA methylation was not different than the control; however, the assay used looked at the global state of methylation at MspI and HpaII sites in spleen and liver [159]. This assay does not measure the details of the DNA methylation pattern at single-site resolution. Thus, it is possible that numerous changes in DNA methylation patterns go undetected using this assay. A more comprehensive analysis of methylation has not been performed and needs to be done to assess the effects of MBD2 depletion on DNA methylation patterns. However, although global changes in methylation were not altered in MBD2−/−, hypermethylation of several tumor suppressor genes was observed in adenomas that arose in APC Min−/+ Mbd2−/− mice [160]. Several follow-up studies have continued to show that MBD2 could trigger DNA demethylation in vitro [156, 161, 162] and in cells [163]. Knock down of MBD2 in colorectal, lung, breast, and prostate cancer cells led to inhibition of tumor growth [164, 165], invasion, and metastasis, as well as silencing and hypermethylation of hypomethylated prometastatic genes [166, 167] supporting the involvement of MBD2 in demethylation, cancer growth, and metastasis.
Evidence suggests that MBD2 might be playing an important role in demethylation of DNA in lupus and perhaps other autoimmune disease. Higher levels of MBD2 mRNA were found in T cells from lupus patients that correlated with the extent of genomic hypomethylation [127, 168]. More recently, it was shown that demethylation of Th2 cytokines during maturation of T cells involves a noncoding region (CNS-1) that interacts with MBD2 in mature thymocytes, suggesting that this protein may regulate the demethylation of this region [3].
Inhibitors of DNA methylation and demethylation
Most of the attention in the field of DNA methylation pharmacology has been directed at developing DNA methylation inhibitors. The driving force behind this effort was the idea that tumor suppressor genes are silenced in cancer by DNA methylation of their promoters. Therefore, blocking DNMT during DNA synthesis would result in passive demethylation in dividing cancer cells and reactivation of these genes triggering suppression of tumor growth [169]. The first DNA methylation inhibitor 5-azaC (DAC) and its deoxy analog 5-deoxycytidine (5-azaCdR) [170] were recently approved by the FDA for treatment of myelodysplastic syndromes (MDS) [171].
The three most commonly used catalytic inhibitors of DNMTs are the nucleoside analogs: 5-azaC, 5-azaCdR, and zebularine. The mechanism of action of these inhibitors is somewhat unique. They are first phosphorylated to the triphosphate nucleotide form and are then incorporated into DNA during DNA synthesis. DNMT1 forms a covalent bond with the carbon at position 6 of the cytosine as well as 5-aza-cytosine ring. Under normal conditions, the enzyme transfers the methyl group from SAM to the five-carbon position of the cytosine ring. This enables the release of the enzyme from its covalent bond with cytosine. When a 5′-aza-cytosine ring replaces cytosine in the DNA, the methyl transfer does not take place and the DNMT is trapped on the DNA [137]. The replication fork progresses in the absence of DNMT, resulting in passive loss of DNA methylation in the nascent strand but not the template.
Zebularine is a nucleoside analog which unlike 5-azaC is chemically stable and is orally bioavailable. Zebularine has been originally identified as a cytidine deaminase inhibitor [172]. This compound exhibits DNA demethylation activity with reduced potency and toxicity in comparison to 5-azaC. This mechanism of action implies that 5-azaC will only be active in cells that are synthesizing DNA when the analog is incorporated into an extending nascent DNA strand. However, surprisingly, 5-azaC was reported to cause demethylation in nondividing neurons [139, 173]. Demethylation in nondividing cells in response to 5-azaC could only come about if 5-azaC would block DNMT activity without being incorporated into the DNA and if neurons express a constitutive tone of DNA demethylase activity that acts in the opposite direction to DNMT. There is some evidence indeed that 5-azaC triggers proteasomal degradation of DNMT1 [174]. It is not clear whether this proteasomal degradation of DNMT1 requires incorporation of 5-azaC into DNA. Blocking DNMT would then tilt the balance of the DNA methylation equilibrium towards DNA demethylation by resident DNA demethylases. Such a mechanism has implications on autoimmune disease since drug-driven DNA demethylation was associated with lupus. It might imply that 5-azaC could cause demethylation in resting T cells as well as activated T cells.
Since both 5-azaC and zebularine are incorporated into DNA, they might have additional toxicities that are independent of their effects on DNA methylation that result from trapping DNMT1 onto DNA and perhaps the trapping of other DNA binding proteins as well [175]. Non-nucleoside-based inhibitors of DNMT1 that inhibit DNMT catalytic activity without incorporation into DNA are, therefore, of much interest. Such a compound was described but its efficacy and potency in whole animals and humans is unclear [176].
Several clinical trials have been launched with a nucleoside analog pan DNMT inhibitor 5-azaC and its deoxy analog 2′-deoxy-5-azacytidine (DAC). Responses with tolerable adverse effects were reported in clinical trials in hematological malignancies especially in MDS [177]. However, there was no significant success reported in solid tumors [178].
It is unclear whether DNA methylation inhibition would have any positive therapeutic impact in autoimmune disease. Although the antiproliferative activity of DNMT inhibitors [179] might reduce autoimmunity by blocking activated T cells, a long line of data suggests that DNA methylation inhibitors would trigger iatrogenic lupus and it stands to reason that other autoimmune disease might be induced by hypomethylating therapy [4]. Surprisingly, however, 5-azaC had a therapeutic effect on the lpr gene-induced lymphadenopathy and acceleration of lupus-like syndrome in MRL/MpJ-lpr/lpr mice [129]. This effect might have resulted from the DNA methylation-independent activities of 5-azaC. As DNA demethylating drugs are entering clinical development, it is critical to address the issue of the potential adverse effects on the immune system of these drugs and to identify drugs that have reduced risk of inducing autoimmunity by indiscriminate demethylation.
Other commonly used drugs were shown to bring about demethylation. For example, procainamide, the widely used antiarrhythmic drug, inhibits DNMT activity and promotes hypomethylation [63, 180]. Analogs of procainamide were recently synthesized and one lead was reported to inhibit DNMT1 and to cause global hypomethylation [181]. Hydralazine, an antidiuretic, induces hypomethylation [63]. Valproic acid, a widely used antiepileptic and mood stabilizer, was shown to cause demethylation [162, 182]. These data raise the concern that many other heavily used drug affect the DNA methylation pattern and thus can promote the expression of disease-promoting genes [183]. Drug-induced autoimmunity was traced several decades ago to the ability of these agents to trigger DNA demethylation [63]. Future drug safety tests should include measures of DNA demethylation especially in the context of the potential for triggering T cell autoreactivity.
Currently available DNA methylation inhibitors inhibit all the DNMT isoforms. It is important to delineate the specific DNMTs involved in the control of genes involved in T cell autoreactivity. Isotypic-specific DNMT inhibitors might exhibit a reduced risk for induction of autoimmunity.
The strong data linking hypomethylation and lupus point to a new direction in epigenetic approach to autoimmunity. We suggest that it might be worthwhile to shift from the standard copying of current anticancer approaches, which invariably focus on DNA methylation inhibitors and HDAC inhibitors to DNA demethylation inhibitors and HAT inhibitors. We proposed a similar shift in focus in anticancer therapy as well [179, 184, 185].
The main challenge here is to characterize the demethylase that is mainly involved in T cell activation, maturation, and autoreactive T cells. MBD2 seems to be a good first target. We have previously developed antisense oligonucleotide inhibitors of MBD2 and have shown that these reduce MBD2 levels and tumorigenesis of a human tumor xenoplant in nude mice [186]. We also demonstrated that MBD2 antisense treatment of breast cancer cells and prostate cancer cells resulted in increased methylation of prometastatic genes and inhibition of cell invasiveness and metastasis [167, 187]. It will be interesting to test whether MBD2 inhibitors would reverse the demethylation of cytokines and other genes in CD4+ T cells from lupus patients. If this is indeed the case, MBD2 inhibition might be a feasible approach to treat the epigenetic defects in several autoimmune diseases. It is also critical to develop small molecule inhibitors of MBD2 and other demethylases and demethylase-associated proteins as possible tools in the arsenal against autoimmune disease.
Another candidate inhibitor of DNA demethylation is the methyl donor of the DNA methylation reaction: S-adenosylmethionine (SAM), which is available in North America as a dietary supplement. Interestingly, SAM was also shown to inhibit MBD2/demethylase activity in vitro and to reverse gene demethylation in living cells [188]. Treatment of metastatic breast and prostate cancer cells with SAM resulted in silencing of demethylated genes and inhibition of invasiveness and metastasis in vivo [167, 187]. Metastatic breast cancer cells exhibit widespread hypomethylation that is alike the general hypomethylation seen in T cells in lupus patients [1, 189]. SAM or SAM analogs might be candidate agents for treating autoimmune disease. Although SAM is highly unstable under physiological conditions, it was administered either orally or intravenously for treating migraines and depression with some documented clinical effects [190–192]. Dietary supplementation with SAM has shown some efficacy in clinical studies in osteoarthritis [193]. Developing SAM analogs that exhibit higher stability and potency in inhibiting demethylation might be of value in the treatment of lupus and perhaps other autoimmune diseases.
Bilateral relationship between chromatin structure and DNA methylation and its implications for pharmacology of autoimmune disease
The two components of the epigenome, DNA methylation and chromatin, are tightly correlated. Cedar and Razin showed more than three decades ago that inactive chromatin is enriched with hypermethylated DNA and that active chromatin is associated with hypomethylated DNA [130]. These correlations were confirmed by detailed analysis of specific genes as well as genome-wide ChIP on chip analyses. This association between the chromatin configuration and the state of DNA methylation suggests a link between the mechanisms defining DNA methylation patterns and those defining chromatin structures and obviously has important implications on the pharmacology of chromatin and DNA modulators. If indeed chromatin modification and DNA methylation are tightly linked, then the use of a histone modification drug could result in a change in DNA methylation and vice versa [194]. Since DNA demethylation plays a role in T cell activation and autoimmunity, the impact of histone-modifying drugs such as HDAC inhibitors on DNA methylation must be considered. It is now clear that the relationship between chromatin and DNA methylation is bilateral. HDAC inhibitors such TSA [195–197] and valproic acid [182] were previously shown to promote active DNA demethylation. As drug-triggered hypomethylation is strongly linked with emergence of iatrogenic lupus [198, 199], we must be extremely cautious when using HDAC inhibitors as therapeutic agents in general and as a therapy for autoimmune disease in particular. The possibility that HDAC inhibitors might induce demethylation and activation of genes involved in autoimmunity must be experimentally tested and the assessment of the safety of HDAC inhibitors should include a study of its potential impact on hypomethylation of T cells DNA. Since the clinical use of HDAC inhibitors is bound to increase in cancer, mental health, and autoimmune disease, it is important to consider the adverse effect that these therapies might have on T cells DNA and T cells autoreactivity.
Summary and prospectus
Lupus is perhaps one of the finest examples of an autoimmune disease that is causally linked to DNA hypomethylation [64, 126]. It stands to reason that DNA methylation is involved in other autoimmune states as well [4, 126]. DNA demethylation is critical for launching the changes in gene expression programming during differentiation of T cells into Th1 and Th2 cells [125]. Recent studies point to MBD2 as possibly involved in T cell DNA hypomethylation in lupus [127, 168]. It stands to reason, therefore, that one of the main targets of drug development should be inhibition of the DNA demethylation machinery. Histone-modifying enzymes that are known to activate gene expression are other possible targets such as HATs, certain histone methyltransferases, and certain histone demethylases. However, it is essential to clearly delineate those enzymes that are involved in aberrant activation of gene expression in autoimmunity in order to identify the preferred targets for drug development. We have several advances from the cancer field but the field of epigenetic pharmacology in autoimmune disease is still completely underdeveloped. HDAC inhibitors have been around for some time now and are the most advanced clinically. They were used and tested in cancer, mental health, and autoimmune disease. The use of HDAC inhibitors in autoimmunity seems to be, however, counterintuitive as they should lead to DNA demethylation and activation of genes involved in driving autoimmunity. The use of HDAC inhibitors in autoimmunity should be, therefore, carefully examined. Finally, an editor's note for completeness, in addition to this special issue on epigenetics, there have been several recent publications which have focused not only on epigenetics and autoimmunity, but epigenetics as a developmental origin of a variety of human diseases [200–210]. Notwithstanding these difficulties and uncertainties and the rather primitive state of epigenetic pharmacology, the data supports a strong epigenetic involvement in autoimmunity and, therefore, the potential is there for epigenetic pharmacology to offer a new way for approaching autoimmunity.
References
Lu Q, Kaplan M, Ray D et al (2002) Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum 46:1282–1291
Mi XB, Zeng FQ (2008) Hypomethylation of interleukin-4 and -6 promoters in T cells from systemic lupus erythematosus patients. Acta Pharmacol Sin 29:105–112
Aoki K, Sato N, Yamaguchi A, Kaminuma O, Hosozawa T, Miyatake S (2009) Regulation of DNA demethylation during maturation of CD4+ naive T cells by the conserved noncoding sequence 1. J Immunol 182:7698–7707
Richardson B (2003) DNA methylation and autoimmune disease. Clin Immunol 109:72–79
White GP, Hollams EM, Yerkovich ST et al (2006) CpG methylation patterns in the IFNgamma promoter in naive T cells: variations during Th1 and Th2 differentiation and between atopics and non-atopics. Pediatr Allergy Immunol 17:557–564
Mastronardi FG, Noor A, Wood DD, Paton T, Moscarello MA (2007) Peptidyl argininedeiminase 2 CpG island in multiple sclerosis white matter is hypomethylated. J Neurosci Res 85:2006–2016
Hu N, Qiu X, Luo Y et al (2008) Abnormal histone modification patterns in lupus CD4+ T cells. J Rheumatol 35:804–810
Szyf M (2009) Epigenetics, DNA methylation, and chromatin modifying drugs. Annu Rev Pharmacol Toxicol 49:243–263
Ateeq B, Unterberger A, Szyf M, Rabbani SA (2008) Pharmacological inhibition of DNA methylation induces proinvasive and prometastatic genes in vitro and in vivo. Neoplasia 10:266–278
Razin A (1998) CpG methylation, chromatin structure and gene silencing—a three-way connection. EMBO J 17:4905–4908
Groudine M, Eisenman R, Gelinas R, Weintraub H (1983) Developmental aspects of chromatin structure and gene expression. Prog Clin Biol Res 134:159–182
Marks PA, Sheffery M, Rifkind RA (1985) Modulation of gene expression during terminal cell differentiation. Prog Clin Biol Res 191:185–203
Ramain P, Bourouis M, Dretzen G, Richards G, Sobkowiak A, Bellard M (1986) Changes in the chromatin structure of Drosophila glue genes accompany developmental cessation of transcription in wild type and transformed strains. Cell 45:545–553
Grunstein M (1997) Histone acetylation in chromatin structure and transcription. Nature 389:349–352
Varga-Weisz PD, Becker PB (2006) Regulation of higher-order chromatin structures by nucleosome-remodelling factors. Curr Opin Genet Dev 16:151–156
Kwon SH, Workman JL (2008) The heterochromatin protein 1 (HP1) family: put away a bias toward HP1. Mol Cell 26:217–227
Bergmann A, Lane ME (2003) HIDden targets of microRNAs for growth control. Trends Biochem Sci 28:461–463
Zhang B, Pan X, Cobb GP, Anderson TA (2007) MicroRNAs as oncogenes and tumor suppressors. Dev Biol 302:1–12
Xiao C, Rajewsky K (2009) MicroRNA control in the immune system: basic principles. Cell 136:26–36
Pauley KM, Cha S, Chan EK (2009) MicroRNA in autoimmunity and autoimmune diseases. J Autoimmun 32:189–194
Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, Bluestone JA (2008) Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med 205:1983–1991
Lau NC, Seto AG, Kim J et al (2006) Characterization of the piRNA complex from rat testes. Science 313:363–367
Vu TH, Jirtle RL, Hoffman AR (2006) Cross-species clues of an epigenetic imprinting regulatory code for the IGF2R gene. Cytogenet Genome Res 113:202–208
Lee JT, Strauss WM, Dausman JA, Jaenisch R (1996) A 450 kb transgene displays properties of the mammalian X-inactivation center. Cell 86:83–94
Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP (2003) Vertebrate microRNA genes. Science 299:1540
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Saito Y, Jones PA (2006) Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle 5:2220–2222
Chuang JC, Jones PA (2007) Epigenetics and microRNAs. Pediatr Res 61:24R–29R
Verdel A, Vavasseur A, Le Gorrec M, Touat-Todeschini L (2009) Common themes in siRNA-mediated epigenetic silencing pathways. Int J Dev Biol 53:245–257
Hawkins PG, Santoso S, Adams C, Anest V, Morris KV (2009) Promoter targeted small RNAs induce long-term transcriptional gene silencing in human cells. Nucleic Acids Res 37:2984–2995
Camelo S, Iglesias AH, Hwang D et al (2005) Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol 164:10–21
Halili MA, Andrews MR, Sweet MJ, Fairlie DP (2009) Histone deacetylase inhibitors in inflammatory disease. Curr Top Med Chem 9:309–319
Finch JT, Lutter LC, Rhodes D et al (1977) Structure of nucleosome core particles of chromatin. Nature 269:29–36
Sarma K, Reinberg D (2005) Histone variants meet their match. Nat Rev Mol Cell Biol 6:139–149
Jenuwein T (2001) Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol 11:266–273
Wade PA, Pruss D, Wolffe AP (1997) Histone acetylation: chromatin in action. Trends Biochem Sci 22:128–132
Shiio Y, Eisenman RN (2003) Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci U S A 100:13225–13230
Shilatifard A (2006) Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem 2432:243–269
Wang Z, Zang C, Rosenfeld JA (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40:897–903
Henikoff S, McKittrick E, Ahmad K (2004) Epigenetics, histone H3 variants, and the inheritance of chromatin states. Cold Spring Harb Symp Quant Biol 69:235–243
Kusch T, Workman JL (2007) Histone variants and complexes involved in their exchange. Subcell Biochem 41:91–109
Wolffe AP (1996) Histone deacetylase: a regulator of transcription. Science 272:371–372
Wotton D, Lo RS, Lee S, Massague J (1999) A Smad transcriptional corepressor. Cell 97:29–39
Kuo MH, Allis CD (1998) Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 20:615–626
Lund AH, van Lohuizen M (2004) Epigenetics and cancer. Genes Dev 18:2315–2335
Perry M, Chalkley R (1982) Histone acetylation increases the solubility of chromatin and occurs sequentially over most of the chromatin. A novel model for the biological role of histone acetylation. J Biol Chem 257:7336–7347
Lee DY, Hayes JJ, Pruss D, Wolffe AP (1993) A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 72:73–84
Sinha I, Wiren M, Ekwall K (2006) Genome-wide patterns of histone modifications in fission yeast. Chromosome Res 14:95–105
Roh TY, Zhao K (2007) High-resolution, genome-wide mapping of chromatin modifications by GMAT. Methods Mol Biol 387:95–108
Roh TY, Zhao K (2008) High-resolution, genome-wide mapping of chromatin modifications by GMAT. Methods Mol Biol 387:95–108
Lu ZP, Ju ZL, Shi GY, Zhang JW, Sun J (2005) Histone deacetylase inhibitor trichostatin A reduces anti-DNA autoantibody production and represses IgH gene transcription. Biochem Biophys Res Commun 330:204–209
Gray SG, Dangond F (2006) Rationale for the use of histone deacetylase inhibitors as a dual therapeutic modality in multiple sclerosis. Epigenetics 1:67–75
Tao R, Hancock WW (2007) Regulating regulatory T cells to achieve transplant tolerance. Hepatobiliary Pancreat Dis Int 6:348–357
Li N, Zhao D, Kirschbaum M et al (2008) HDAC inhibitor reduces cytokine storm and facilitates induction of chimerism that reverses lupus in anti-CD3 conditioning regimen. Proc Natl Acad Sci U S A 105:4796–4801
Kuwatsuka Y, Ogawa F, Iwata Y et al (2009) Decreased levels of autoantibody against histone deacetylase 3 in patients with systemic sclerosis. Autoimmunity 42:120–125
Verdone L, Caserta M, Di Mauro E (2005) Role of histone acetylation in the control of gene expression. Biochem Cell Biol 83:344–353
Borrow J, Stanton VP Jr, Andresen JM et al (1996) The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet 14:33–41
van Attikum H, Gasser SM (2009) Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 19:207–217
Ikura T, Ogryzko VV, Grigoriev M et al (2000) Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102:463–473
Sun Y, Jiang X, Chen S, Fernandes N, Price BD (2005) A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A 102:13182–13187
Tang Y, Luo J, Zhang W, Gu W (2006) Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 24:827–839
Fu M, Wang C, Zhang X, Pestell RG (2004) Acetylation of nuclear receptors in cellular growth and apoptosis. Biochem Pharmacol 68:1199–1208
Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B (1988) Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol 140:2197–2200
Yung RL, Richardson BC (1994) Role of T cell DNA methylation in lupus syndromes. Lupus 3:487–491
Yung RL, Quddus J, Chrisp CE, Johnson KJ, Richardson BC (1995) Mechanism of drug-induced lupus. I. Cloned Th2 cells modified with DNA methylation inhibitors in vitro cause autoimmunity in vivo. J Immunol 154:3025–3035
Manzo F, Tambaro FP, Mai A, Altucci L (2009) Histone acetyltransferase inhibitors and preclinical studies. Expert Opin Ther Pat 19:761–774
Mai A, Rotili D, Tarantino D et al (2009) Identification of 4-hydroxyquinolines inhibitors of p300/CBP histone acetyltransferases. Bioorg Med Chem Lett 19:1132–1135
Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK (2003) Small molecule modulators of histone acetyltransferase p300. J Biol Chem 278:19134–19140
Souto JA, Conte M, Alvarez R et al (2008) Synthesis of benzamides related to anacardic acid and their histone acetyltransferase (HAT) inhibitory activities. ChemMedChem 3:1435–1442
Arif M, Pradhan SK, Thanuja GR et al (2009) Mechanism of p300 specific histone acetyltransferase inhibition by small molecules. J Med Chem 52:267–277
Lee YH, Jung MG, Kang HB et al (2008) Effect of anti-histone acetyltransferase activity from Rosa rugosa Thunb. (Rosaceae) extracts on androgen receptor-mediated transcriptional regulation. J Ethnopharmacol 118:412–417
Buczek-Thomas JA, Hsia E, Rich CB, Foster JA, Nugent MA (2008) Inhibition of histone acetyltransferase by glycosaminoglycans. J Cell Biochem 105:108–120
Sun Y, Jiang X, Chen S, Price BD (2006) Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett 580:4353–4356
Holbert MA, Marmorstein R (2005) Structure and activity of enzymes that remove histone modifications. Curr Opin Struct Biol 15:673–680
Glaser KB, Li J, Staver MJ, Wei RQ, Albert DH, Davidsen SK (2003) Role of class I and class II histone deacetylases in carcinoma cells using siRNA. Biochem Biophys Res Commun 310:529–536
Zimmermann S, Kiefer F, Prudenziati M (2007) Reduced body size and decreased intestinal tumor rates in HDAC2-mutant mice. Cancer Res 67:9047–9054
Weichert W, Roske A, Niesporek S et al (2008) Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res 14:1669–1677
Nakagawa M, Oda Y, Eguchi T et al (2007) Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep 18:769–774
Mottet D, Bellahcene A, Pirotte S et al (2007) Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res 101:1237–1246
Gan L (2007) Therapeutic potential of sirtuin-activating compounds in Alzheimer's disease. Drug News Perspect 20:233–239
Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J (2004) Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res 95:971–980
Tao R, de Zoeten EF, Ozkaynak E et al (2007) Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 13:1299–1307
Gartenberg MR (2000) The Sir proteins of Saccharomyces cerevisiae: mediators of transcriptional silencing and much more. Curr Opin Microbiol 3:132–137
Alfred J (2000) Counting the calories to immortality. Nat Rev Genet 1:88
Kim S, Benguria A, Lai CY, Jazwinski SM (1999) Modulation of life-span by histone deacetylase genes in Saccharomyces cerevisiae. Mol Biol Cell 10:3125–3136
Peixoto P, Lansiaux A (2006) Histone-deacetylases inhibitors: from TSA to SAHA. Bull Cancer 93:27–36
Richon VM, Sandhoff TW, Rifkind RA, Marks PA (2000) Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A 97:10014–10019
Santini V, Gozzini A, Ferrari G (2007) Histone deacetylase inhibitors: molecular and biological activity as a premise to clinical application. Curr Drug Metab 8:383–393
Fournel M, Bonfils C, Hou Y et al (2008) MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther 7:759–768
Xu WS, Parmigiani RB, Marks PA (2007) Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26:5541–5552
Duvic M, Vu J (2007) Vorinostat in cutaneous T-cell lymphoma. Drugs Today (Barc) 43:585–599
Siu LL, Pili R, Duran I et al (2008) Phase I study of MGCD0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. J Clin Oncol 26:1940–1947
Kell J (2007) Drug evaluation: MGCD-0103, a histone deacetylase inhibitor for the treatment of cancer. Curr Opin Investig Drugs 8:485–492
Rasheed WK, Johnstone RW, Prince HM (2007) Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig Drugs 16:659–678
Shankar S, Srivastava RK (2008) Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv Exp Med Biol 615:261–298
Sambucetti LC, Fischer DD, Zabludoff S et al (1999) Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem 274:34940–34947
Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS (2003) Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest 111:539–552
Seo SB, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D (2001) Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell 104:119–130
Hublitz P, Kunowska N, Mayer UP et al (2005) NIR is a novel INHAT repressor that modulates the transcriptional activity of p53. Genes Dev 19:2912–2924
Wiencke JK, Zheng S, Morrison Z, Yeh RF (2008) Differentially expressed genes are marked by histone 3 lysine 9 trimethylation in human cancer cells. Oncogene 27:2412–2421
Miao F, Natarajan R (2005) Mapping global histone methylation patterns in the coding regions of human genes. Mol Cell Biol 25:4650–4661
Santos-Rosa H, Schneider R, Bannister AJ et al (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419:407–411
Akan P, Sahlen M, Deloukas P (2009) A histone map of human chromosome 20q13.12. PLoS ONE 4:4479
Bernstein BE, Mikkelsen TS, Xie X et al (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326
Zhao XD, Han X, Chew JL et al (2007) Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1:286–298
Rea S, Eisenhaber F, O'Carroll D et al (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406:593–599
Peters AH, Schubeler D (2005) Methylation of histones: playing memory with DNA. Curr Opin Cell Biol 17:230–238
Cheng X, Collins RE, Zhang X (2005) Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct 34:267–294
Nguyen CT, Weisenberger DJ, Velicescu M et al (2002) Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res 62:6456–6461
Coombes MM, Briggs KL, Bone JR, Clayman GL, El-Naggar AK, Dent SY (2003) Resetting the histone code at CDKN2A in HNSCC by inhibition of DNA methylation. Oncogene 22:8902–8911
Meng CF, Zhu XJ, Peng G, Dai DQ (2007) Re-expression of methylation-induced tumor suppressor gene silencing is associated with the state of histone modification in gastric cancer cell lines. World J Gastroenterol 13:6166–6171
Schlesinger Y, Straussman R, Keshet I et al (2007) Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 39:232–236
Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A (2005) Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat Chem Biol 1:143–145
Kubicek S, O'Sullivan RJ, August EM et al (2007) Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell 25:473–481
Zhang X, Bernatavichute YV, Cokus S, Pellegrini M, Jacobsen SE (2009) Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol 10:R62
Shi Y, Lan F, Matson C et al (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119:941–953
Metzger E, Wissmann M, Yin N et al (2005) LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437:436–439
Klose RJ, Yamane K, Bae Y et al (2006) The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 442:312–316
Lee MG, Wynder C, Cooch N, Shiekhattar R (2005) An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437:432–435
Wang J, Scully K, Zhu X et al (2007) Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446:882–887
Tsukada Y, Fang J, Erdjument-Bromage H et al (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439:811–816
Huang Y, Greene E, Murray Stewart T et al (2007) Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci U S A 104:8023–8028
Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R (2006) Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol 13:563–567
Lee DU, Agarwal S, Rao A (2002) Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity 16:649–660
Santangelo S, Cousins DJ, Winkelmann NE, Staynov DZ (2002) DNA methylation changes at human Th2 cytokine genes coincide with DNase I hypersensitive site formation during CD4(+) T cell differentiation. J Immunol 169:1893–1903
Richardson B (2007) Primer: epigenetics of autoimmunity. Nat Clin Pract Rheumatol 3:521–527
Balada E, Ordi-Ros J, Vilardell-Tarres M (2007) DNA methylation and systemic lupus erythematosus. Ann N Y Acad Sci 1108:127–136
Quddus J, Johnson KJ, Gavalchin J et al (1993) Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J Clin Invest 92:38–53
Yoshida H, Yoshida M, Merino R, Shibata T, Izui S (1990) 5-Azacytidine inhibits the lpr gene-induced lymphadenopathy and acceleration of lupus-like syndrome in MRL/MpJ-lpr/lpr mice. Eur J Immunol 20:1989–1993
Razin A, Cedar H (1977) Distribution of 5-methylcytosine in chromatin. Proc Natl Acad Sci U S A 74:2725–2728
Okano M, Xie S, Li E (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases [letter]. Nat Genet 19:219–220
Vilain A, Apiou F, Dutrillaux B, Malfoy B (1998) Assignment of candidate DNA methyltransferase gene (DNMT2) to human chromosome band 10p15.1 by in situ hybridization. Cytogenet Cell Genet 82:120
Rai K, Chidester S, Zavala CV et al (2007) Dnmt2 functions in the cytoplasm to promote liver, brain, and retina development in zebrafish. Genes Dev 21:261–266
Goll MG, Kirpekar F, Maggert KA et al (2006) Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311:395–398
Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294:2536–2539
Razin A, Riggs AD (1980) DNA methylation and gene function. Science 210:604–610
Wu JC, Santi DV (1985) On the mechanism and inhibition of DNA cytosine methyltransferases. Prog Clin Biol Res 198:119–129
Ramchandani S, Bhattacharya SK, Cervoni N, Szyf M (1999) DNA methylation is a reversible biological signal. Proc Natl Acad Sci U S A 96:6107–6112
Levenson JM, Roth TL, Lubin FD et al (2006) Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem 281:15763–15773
Bruniquel D, Schwartz RH (2003) Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat Immunol 4:235–240
Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317:1760–1764
Vire E, Brenner C, Deplus R et al (2006) The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439:871–874
Di Croce L, Raker VA, Corsaro M et al (2002) Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295:1079–1082
Kersh EN, Fitzpatrick DR, Murali-Krishna K et al (2006) Rapid demethylation of the IFN-{gamma} gene occurs in memory but not naive CD8 T cells. J Immunol 176:4083–4093
Gjerset RA, Martin DW Jr (1982) Presence of a DNA demethylating activity in the nucleus of murine erythroleukemic cells. J Biol Chem 257:8581–8583
Szyf M, Theberge J, Bozovic V (1995) Ras induces a general DNA demethylation activity in mouse embryonal P19 cells. J Biol Chem 270:12690–12696
Patra SK, Patra A, Zhao H, Dahiya R (2002) DNA methyltransferase and demethylase in human prostate cancer. Mol Carcinog 33:163–171
Jost JP (1993) Nuclear extracts of chicken embryos promote an active demethylation of DNA by excision repair of 5-methyldeoxycytidine. Proc Natl Acad Sci U S A 90:4684–4688
Zhu B, Zheng Y, Hess D et al (2000) 5-methylcytosine-DNA glycosylase activity is present in a cloned G/T mismatch DNA glycosylase associated with the chicken embryo DNA demethylation complex. Proc Natl Acad Sci U S A 97:5135–5139
Barreto G, Schafer A, Marhold J et al (2007) Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 445:671–675
Jin SG, Guo C, Pfeifer GP (2008) GADD45A does not promote DNA demethylation. PLoS Genet 4:e1000013
Metivier R, Gallais R, Tiffoche C et al (2008) Cyclical DNA methylation of a transcriptionally active promoter. Nature 452:45–50
Kangaspeska S, Stride B, Metivier R et al (2008) Transient cyclical methylation of promoter DNA. Nature 452:112–115
Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR (2008) DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell 135:1201–1212
Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M (1999) A mammalian protein with specific demethylase activity for mCpG DNA [see comments]. Nature 397:579–583
Hamm S, Just G, Lacoste N, Moitessier N, Szyf M, Mamer O (2008) On the mechanism of demethylation of 5-methylcytosine in DNA. Bioorg Med Chem Lett 18:1046–1049
Kriaucionis S, Heintz N (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324:929–930
Ng HH, Zhang Y, Hendrich B et al (1999) MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet 23:58–61
Hendrich B, Guy J, Ramsahoye B, Wilson VA, Bird A (2001) Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev 15:710–723
Sansom OJ, Berger J, Bishop SM, Hendrich B, Bird A, Clarke AR (2003) Deficiency of Mbd2 suppresses intestinal tumorigenesis. Nat Genet 34:145–147
Detich N, Theberge J, Szyf M (2002) Promoter-specific activation and demethylation by MBD2/demethylase. J Biol Chem 277:35791–35794
Detich N, Bovenzi V, Szyf M (2003) Valproate induces replication-independent active DNA demethylation. J Biol Chem 278:27586–27592
Goel A, Mathupala SP, Pedersen PL (2003) Glucose metabolism in cancer. Evidence that demethylation events play a role in activating type ii hexokinase gene expression. J Biol Chem 278:15333–15340
Slack A, Bovenzi V, Bigey P et al (2002) Antisense MBD2 gene therapy inhibits tumorigenesis. J Gene Med 4:381–389
Campbell PM, Bovenzi V, Szyf M (2003) Methylated DNA binding protein 2 antisense inhibitors suppress tumorigenesis of human cancer lines in vitro and in vivo. Carcinogenesis 25:499–507
Pakneshan P, Szyf M, Rabbani SA (2004) Methylation and inhibition of uPA expression by RAS oncogene: divergence of growth control and invasion in breast cancer cells. Carcinogenesis 26:557–564
Shukeir N, Pakneshan P, Chen G, Szyf M, Rabbani SA (2006) Alteration of the methylation status of tumor-promoting genes decreases prostate cancer cell invasiveness and tumorigenesis in vitro and in vivo. Cancer Res 66:9202–9210
Balada E, Ordi-Ros J, Serrano-Acedo S, Martinez-Lostao L, Vilardell-Tarres M (2007) Transcript overexpression of the MBD2 and MBD4 genes in CD4+ T cells from systemic lupus erythematosus patients. J Leukoc Biol 81:1609–1616
Szyf M (1994) DNA methylation properties: consequences for pharmacology. Trends Pharmacol Sci 15:233–238
Jones PA, Taylor SM (1980) Cellular differentiation, cytidine analogs and DNA methylation. Cell 20:85–93
Kuendgen A, Lubbert M (2008) Current status of epigenetic treatment in myelodysplastic syndromes. Ann Hematol 87(8):601–611
Cheng JC, Matsen CB, Gonzales FA et al (2003) Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst 95:399–409
Miller CA, Sweatt JD (2007) Covalent modification of DNA regulates memory formation. Neuron 53:857–869
Ghoshal K, Datta J, Majumder S et al (2005) 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol 25:4727–4741
Juttermann R, Li E, Jaenisch R (1994) Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A 91:11797–11801
Brueckner B, Boy RG, Siedlecki P et al (2005) Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res 65:6305–6311
Oki Y, Aoki E, Issa JP (2007) Decitabine—bedside to bench. Crit Rev Oncol Hematol 61:140–152
Weiss AJ, Metter GE, Nealon TF et al (1977) Phase II study of 5-azacytidine in solid tumors. Cancer Treat Rep 61:55–58
Szyf M (2001) The role of DNA methyltransferase 1 in growth control. Front Biosci 6:D599–D609
Scheinbart LS, Johnson MA, Gross LA, Edelstein SR, Richardson BC (1991) Procainamide inhibits DNA methyltransferase in a human T cell line. J Rheumatol 18:530–534
Castellano S, Kuck D, Sala M, Novellino E, Lyko F, Sbardella G (2008) Constrained analogues of procaine as novel small molecule inhibitors of DNA methyltransferase-1. J Med Chem 51:2321–2325
Milutinovic S, D'Alessio AC, Detich N, Szyf M (2007) Valproate induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis 28:560–571
Szyf M (2007) The dynamic epigenome and its implications in toxicology. Toxicol Sci 100:7–23
Szyf M (2005) DNA methylation and demethylation as targets for anticancer therapy. Biochemistry (Mosc) 70:533–549
Szyf M (2008) The role of DNA hypermethylation and demethylation in cancer and cancer therapy. Curr Oncol 15:72–75
Campbell PM, Bovenzi V, Szyf M (2004) Methylated DNA-binding protein 2 antisense inhibitors suppress tumourigenesis of human cancer cell lines in vitro and in vivo. Carcinogenesis 25:499–507
Pakneshan P, Szyf M, Farias-Eisner R, Rabbani SA (2004) Reversal of the hypomethylation status of urokinase (uPA) promoter blocks breast cancer growth and metastasis. J Biol Chem 279:31735–31744
Detich N, Hamm S, Just G, Knox JD, Szyf M (2003) The methyl donor S-adenosylmethionine inhibits active demethylation of DNA: a candidate novel mechanism for the pharmacological effects of S-adenosylmethionine. J Biol Chem 278:20812–20820
Gorelik G, Richardson B (2009) Aberrant T cell ERK pathway signaling and chromatin structure in lupus. Autoimmun Rev 8:196–198
Carney MW, Edeh J, Bottiglieri T, Reynolds EM, Toone BK (1986) Affective illness and S-adenosyl methionine: a preliminary report. Clin Neuropharmacol 9:379–385
Gatto G, Caleri D, Michelacci S, Sicuteri F (1986) Analgesizing effect of a methyl donor (S-adenosylmethionine) in migraine: an open clinical trial. Int J Clin Pharmacol Res 6:15–17
Williams AL, Girard C, Jui D, Sabina A, Katz DL (2005) S-adenosylmethionine (SAMe) as treatment for depression: a systematic review. Clin Invest Med 28:132–139
Hosea Blewett HJ (2008) Exploring the mechanisms behind S-adenosylmethionine (SAMe) in the treatment of osteoarthritis. Crit Rev Food Sci Nutr 48:458–463
D'Alessio AC, Szyf M (2006) Epigenetic tete-a-tete: the bilateral relationship between chromatin modifications and DNA methylation. Biochem Cell Biol 84:463–476
Cervoni N, Szyf M (2001) Demethylase activity is directed by histone acetylation. J Biol Chem 276:40778–40787
Cervoni N, Detich N, Seo SB, Chakravarti D, Szyf M (2002) The oncoprotein Set/TAF-1beta, an inhibitor of histone acetyltransferase, inhibits active demethylation of DNA, integrating DNA methylation and transcriptional silencing. J Biol Chem 277:25026–25031
Weaver IC, Cervoni N, Champagne FA et al (2004) Epigenetic programming by maternal behavior. Nat Neurosci 7:847–854
Richardson BC (2002) Role of DNA methylation in the regulation of cell function: autoimmunity, aging and cancer. J Nutr 132:2401S–2405S
Sekigawa I, Okada M, Ogasawara H, Kaneko H, Hishikawa T, Hashimoto H (2003) DNA methylation in systemic lupus erythematosus. Lupus 12:79–85
Arnheim N, Calabrese P (2009) Understanding what determines the frequency and pattern of human germline mutations. Nat Rev Genet 10:478–488
Barros SP, Offenbacher S (2009) Epigenetics: connecting environment and genotype to phenotype and disease. J Dent Res 88:400–408
Figueiredo LM, Cross GA, Janzen CJ (2009) Epigenetic regulation in African trypanosomes: a new kid on the block. Nat Rev Microbiol 7:504–513
Hewagama A, Richardson B (2009) The genetics and epigenetics of autoimmune diseases. J Autoimmun 33:3–11
Invernizzi P (2009) Future directions in genetic for autoimmune diseases. J Autoimmun 33:1–2
Invernizzi P, Pasini S, Selmi C, Gershwin ME, Podda M (2009) Female predominance and X chromosome defects in autoimmune diseases. J Autoimmun 33:12–16
Larizza D, Calcaterra V, Martinetti M (2009) Autoimmune stigmata in Turner syndrome: when lacks an X chromosome. J Autoimmun 33:25–30
Persani L, Rossetti R, Cacciatore C, Bonomi M (2009) Primary ovarian insufficiency: X chromosome defects and autoimmunity. J Autoimmun 33:35–41
Sawalha AH, Harley JB, Scofield RH (2009) Autoimmunity and Klinefelter's syndrome: when men have two X chromosomes. J Autoimmun 33:31–34
Wells AD (2009) New insights into the molecular basis of T cell anergy: anergy factors, avoidance sensors, and epigenetic imprinting. J Immunol 182:7331–7341
Zernicka-Goetz M, Morris SA, Bruce AW (2009) Making a firm decision: multifaceted regulation of cell fate in the early mouse embryo. Nat Rev Genet 10:467–477
Acknowledgments
The studies in the MS laboratory were supported by the National Cancer Institute of Canada and the Canadian Institute of Health Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Szyf, M. Epigenetic Therapeutics in Autoimmune Disease. Clinic Rev Allerg Immunol 39, 62–77 (2010). https://doi.org/10.1007/s12016-009-8172-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-009-8172-8