
A new sesquiterpenoid glycoside, called speciosaoside A ( 1 ), and a known sesquiterpenoid glycoside, (1′R,3′R,5′R,8′S)-epi-dihydrophaseic acid β-D-glucoside ( 2 ), were isolated from the fruits of Chaenomeles speciosa (Sweet) Nakai (Rosaceae) using column chromatography and preparative HPLC. The structure of compound 1 was deduced from comprehensive spectroscopic analysis including IR, HR-ESI-MS, 1D NMR, and 2D NMR. The structure of compound 2 was identified by comparison of its spectral data with those reported in the literature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The dried fruit of Chaenomeles speciosa (Sweet) Nakai (Rosaceae) is a traditional medicine, widely used for the treatment of inflammatory and infectious diseases, as well as gastrointestinal symptoms, such as rheumatoid arthritis, cholera, diarrhea, dyspepsia, and gastrointestinal spasm [1]. It is also well known in China as a food consumed as an appetizer. It was reported to have a variety of biological activities such as antimicrobial, analgesic [2–5], antidiabetic [6], and antioxidant effects [7]. A previous study showed that the effective constituents of C. speciosa included mainly glycosides, flavones, phenolics, tannins, triterpenes, and organic acids [8–14]. Previously, our laboratory had reported that the 80% EtOH extract of the fruits of C. speciosa showed positive anti-inflammatory and analgesic activities in carrageenan-induced paw edema in rats [5]. The present study was undertaken to investigate the chemical constituents of the extract, which led to the isolation of a new sesquiterpenoid glycoside called speciosaoside A (1) and a known sesquiterpenoid glycoside (1′R,3′R,5′R,8′S)-epidihydrophaseic acid β-D-glucoside (2). Their structures were elucidated by chemical and spectroscopic methods. Compound 2 was isolated from the fruits of C. speciosa for the first time.
Compound 1 was obtained as a brown amorphous powder. The [M + H]+ ion peak at m/z 477.1978 in the HR-ESI-MS corresponded to a molecular formula C21H32O12, representing six unsaturated degrees. The 1H NMR spectrum (Table 1) of compound 1 in CD3OD contained abundant singlet signals of three methyl groups [δ 2.09 (3H, br.s, CH3-6), 1.10 (3H, s, CH3-7′), 1.40 (3H, s, CH3-9′)], two methylene signals [δ 1.96 (1H, d, J = 17 Hz, H-3′ax), 1.99 (1H, d, J = 17 Hz, H-3′eq), 2.12 (1H, d, J = 10 Hz, H-5′ax), 2.52 (1H, d, J = 10 Hz, H-5′eq)], and three signals of olefinic protons [δ 5.88 (1H, br.s, H-2), 6.47 (1H, d, J = 16.5 Hz, H-5), and 7.83 (1H, d, J = 16.5 Hz, H-4)]. The complex oxymethine region integrated for six protons that belong to a glucose unit [δ 3.20 (1H, dd, J = 7.8, 9.0 Hz, H-2″), 3.44 (1H, m, H-3″), 3.37 (1H, m, H-4″), 3.37 (1H, m, H-5″), 3.68 (1H, br.d, J = 12.0 Hz, H-6″a), 3.87 (1H, dd, J = 4.8, 12.0 Hz, H-6″b)], and resonance 4.46 (1H, d, J = 7.8 Hz, H-1″) was assigned to the anomeric glucose proton. The 13C NMR spectrum (Table 1) revealed 21 carbon resonances, six of which were assigned to a β-D-glucopyranosyl unit [15] and 15 to the aglycone moiety. From the 13C and DEPT NMR experiments, these 15 signals were identified as six quaternary carbons: one olefinic carbon C-3 (δ 151.3), two oxygenated carboxyls C-1 (δ 170.8) and C-8′ (δ 182.3), and three aliphatic carbons: C-1′ (δ 83.1), C-2′ (δ 53.5), and C-6′ (δ 90.9); three tertiary carbons: C-2 (δ 120.2), C-4 (δ 133.3), and C-5 (δ 130.1); two secondary carbons: C-3′ (δ 38.6) and C-5′ (δ 38.5); and three primary carbons: C-6 (δ 21.2), C-7′ (δ 14.3), and C-9′ (δ 18.3). The overall data suggested that compound 1 was a phaseic acid derivative [16]. Interestingly, compound 1 has a similar skeleton compared to the known compound: (1′R,3′R,5′R,8′S)-epi-dihydrophaseic acid β-D-glucoside (2).
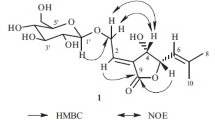
The specific structure of compound 1 was unambiguously confirmed by heteronuclear correlation experiments such as 1H–1H COSY and HMBC. The correlations (Fig. 1) of H-5 (δ 6.47) with C-4/C-3, H-4 (δ 7.83) with C-2/C-6, and H-2 (δ 5.88) with C-6/C-3/C-1 were observed in the HMBC spectrum. Besides, in the 1H–1H COSY spectrum, H-4 showed a correlation with H-5. All the correlations indicated the presence of the –C=C–C(CH3)=C–COOH moiety. Meanwhile, H-4 and H-5 showed correlations with the carbon proton C-1′ (δ 83.1), which indicated that the –C=C–C(CH3)=C–COOH moiety was at C-1′ of the cyclohexene ring. The anomeric sugar proton H-1″ (δ 4.46) was correlated to the C-4′ (δ 73.3) in the HMBC spectrum, so the sugar moiety was attached to the cyclohexene ring at C-4′. In the HMBC, H-3′ was correlated with CH3-7′ (δC 14.3), C-2′ (δ 53.5), and C-8′ (δ 182.3), which indicated that the –COOH and –CH3 were attached at C-2′ of the cyclohexene ring. Also, the protons of the methyl group CH3-9′ (δH 1.40) showed correlations with C-1′ (δ 83.1), C-5′ (δ 38.5), and C-6′ (δ 90.9), which indicated that –CH3 was at C-6′ of the cyclohexene ring. The above important correlations are shown in Fig. 1.

Key HMBC correlations of compound 1.
Selective NOE irradiation on axial proton H-5′ax (δ 2.12) resulted in strong NO effects on H-3′ax (δ 1.96), protons of methyl group CH3-9′ (δH 1.40), and the proton of olefinic H-5 (δ 6.47), clearly indicating that these interfering protons are located on the same side of the cyclohexane plane (Fig. 2). This implied an equatorial orientation for CH3-9′, and the olefinic side chain in axial orientation, hence the β-D-glucose moiety was located on the opposite side of the ring system and is also axially oriented. Also, glucosidation at C-4′ was evident from the abundant NOE resonances between H-4′ (δ 4.03) and the anomeric proton H-1″ (δ 4.46) (Fig. 2) in the opposite direction. Confirmation for the stereochemistry at H-6′ was obtained by selective irradiation of H-5′b (δH 2.52), which resulted in a NOE resonance to H-5′a (δH 2.12) but not in a through-space interaction to CH3-9′. The double-bond configurations in the olefinic side chain were visible by selective NOE enhancements: the trans-configuration of the ∆ 4-bond was due to the significant NO effect between H-5 (δ 6.47) and CH3-6 (δH 2.09); also, irradiation of CH3-6 (δH 2.09) resulted in a NOE for H-2 (δ 5.88) showing a cis-configuration of the double bond ∆ 2. Hence, the new compound was named speciosaoside A.

Structure-relevant NOE resonances in compound 1.
Besides the new sesquiterpenoid glycoside speciosaoside A (1), a known sesquiterpenoid glycoside, (1′R,3′R,5′R,8′S)- epi-dihydrophaseic acid β-D-glucoside (2) [16], was identified by comparison of its physical and spectroscopic data with those reported in the literatures. The obtained spectral data (MS, 1H and 13C NMR spectra) and optical rotation of this known compound were similar to those reported in the previous literature.
Experimental
General Procedures. Silica gel (Jiangyou Company of Yantai, 100–200 and 200–300 mesh), RP-C18 (43–60 μm; Merck, Darmstadt, Germany), Sephadex LH-20 (Pharmacia, Uppsala, Sweden), and MCI gel (Mitsubishi Chemical Corporation, Tokyo, Japan) were used for column chromatography. Preparative HPLC was performed using an Agilent (Palo Alto, CA, USA) 1200 liquid chromatography system equipped with a quaternary solvent delivery system and an ultraviolet detector. A Platisil ODS C18 column was used for analytical (4.6 mm × 250 mm; Waters, USA) and preparative (250 mm × 20 mm; Waters, USA) purposes. Infrared (IR) spectra were recorded on a Bruker Vector 22 spectrometer with a KBr pellet. NMR, including COSY, HMBC, and HSQC experiments, were recorded on a Bruker Avance 600 NMR spectrometer operating at 600 MHz (1H) and 150 MHz (13C), with chemical shifts given in ppm (δ), using tetramethylsilane (TMS) as an internal standard. HR-ESI-MS spectra were recorded on an Agilent Technologies 6538 UHD Accurate-Mass Q-TOF LC/MS spectrometer (Agilent Technologies, Santa Clara, CA with an electrospray interface) and Agilent 1290 Infinity modules (G4220A binary pump, G4212A photodiode array detector, and G4226A autosampler).
Plant Material. The dried fruits of C. speciosa were purchased from Linyi of Shandong Province, China, in July 2006. The plant was identified by Prof. Hanming Zhang at the School of Pharmacy, Second Military Medical University, China. A voucher specimen (No. TD20060720) was deposited in the Department of Pharmacognosy, Second Military Medical University, China.
Extraction and Isolation. A total of 10 kg of dried and powdered fruits of C. speciosa was soaked in 10 L of 80% ethanol for 2 days and percolated with 160 L of 80% ethanol for 7 days at room temperature. The percolate was then collected and concentrated at 60C under vacuum to obtain a crude extract. The aqueous supernatant was chromatographed on D101 macroporous resin eluted with water, 10% ethanol, 30% ethanol, 50% ethanol, and 95% ethanol in series to yield fractions 1–5.Fraction 2 was evaporated under reduced pressure, and a portion (150 g) was dissolved in H2O (300 mL), then applied on an MCI gel column chromatograph eluted with H2O, followed by increasing concentrations of MeOH (10, 20, 50, 100%) to give five fractions: Fr. 2–1 to 2–5. Fraction 2–4 was applied on an ODS column with MeOH–H2O (0:1, 2:8, 5:5, 1:0) to obtain four fractions (Fr. 2-4-1 to Fr. 2-4-4). Fractions 2-4-2 (3.6 g) and 2-4-3 (4.7 g) were repeatedly purified by Sephadex LH-20 and semipreparative HPLC to yield compounds 1 (4.8 mg) and 2 (10.8 mg).
Compound 1. Brown amorphous powder, [α]D 20 –5.0 (c 0.16, MeOH). HR-ESI-MS m/z 477.1978 [M + H]+; calcd 477.1972. For 1H and 13C NMR, see Table 1.
References
Chinese Pharmacopoeia Commission: Pharmacopoeia of the People’s Republic of China, Vol. 1, People’s Medical Publishing House, Beijing, 2005, p. 87.
X. F. Xie, X. Q. Cai, S. Y. Zhu, and G. L. Zou, Food Chem., 100, 1312 (2007).
J. S. Kong, X. H. Yang, and Lishizhen, Med. Mater. Med. Res., 20, 549 (2009).
J. N. Wang, H. Yoshio, N. Taro, and Y. J. Chen, Phytochemistry, 53, 1097 (2000).
X. Li, Y. B. Yang, Q. Yang, L. N. Sun, and W. S. Chen, J. Med. Food., 12, 1016 (2009).
S. Sancheti, S. Sancheti, and S. Y. Seo, Exp. Toxic. Pathol., 65, 55 (2013).
L. Zhang, Y. X. Cheng, A. L. Liu, H. D. Wang, Y. L. Wang, and G. H. Du, Molecules, 15, 8507 (2010).
D. Li and L. He, Chin. J. Hosp. Pharm., 25, 259 (2005).
R. L. Chen, T. J. Wu, and Y. J. Dai, West. Chin. J. Pharm. Sci., 15, 38 (2000).
M. Dai, W. Wei, Y. X. Shen, and Y. Q. Zheng, Acta Pharm. Sin., 24, 1161 (2003).
H. Y. Gong, H. Wang, Z. Xu, and G. Z. Liu, Pharmacol. Clin. Chin. Mater. Med., 2, 30 (1995).
H. C. Chen, L. S. Ding, S. L. Peng, and X. Liao, Chin. Trad. Herb. Drugs, 36, 30 (2005).
X. M. Guo, L. Zhang, S. C. Quan, Y. F. Hong, L. N. Sun, and M. Z. Liu, Chin. J. Chin. Mater. Med., 23, 546 (1998).
K. Yin, H. Y. Gao, X. N. Li, and L. J. Wu, J. Shengyang Pharm. Univ., 23, 760 (2006).
E. Breitmaier and W. Voelter, Carbon-13 NMR Spectroscopy (3rd completely revised ed.), Weinheim, VCH, 1989, 381 pp.
J. Gerold, V. Socorro, L. D. Fernando, W. Reiner, and W. Peter, Phytochemistry, 65, 955 (2004).
Acknowledgment
The authors are grateful to Prof. Gen-Jin Yang (School of Pharmacy, Second Military Medical University) for his assistance with the measurement of NMR spectra. This project was financially supported by a grant from the National Science Fund for Distinguished Young Scholars (No. 81325024) and the National Specific Project of New Drugs Innovation (No. 2011ZX09102-006-03).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 2, March–April, 2015, pp. 235–237.
Rights and permissions
About this article

Cite this article
Huang, GH., Xi, ZX., Li, JL. et al. Sesquiterpenoid Glycosides from the Fruits of Chaenomeles speciosa . Chem Nat Compd 51, 266–269 (2015). https://doi.org/10.1007/s10600-015-1258-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-015-1258-z