
In the current work, a novel triterpene saponin, named ursolic acid 3-O-[α-L-rhamnopyranosyl(1"→6')]-β-D-glucopyranosyl-28-O-β-D-glucuronopyranoside (1), together with two known flavonol glycosides, kaemferol 3-O-α-L-rhamnopyranoside (2) and quercetin 3-O-β-D-glucopyranoside (3), was isolated from the stem outer barks of Pinus pumila by repeated column chromatography and vacuum liquid chromatography. The chemical structures of the secondary metabolites 1–3 were characterized by their spectroscopic data and other physico-chemical evidence, as well as by comparison with the literature. The secondary metabolites 2 and 3 were found in this species for the first time.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Pinus pumila (Pall.) Regel, a small nondeciduous pine species in the Pinus genus of the Pinaceae family, is extensively grown in the high altitude regions of Japan, the Korean peninsula, China, and Siberia [1,2,3]. P. pumila, called “Yan Song” in Chinese, is also known as Siberian dwarf pine, Haimatsu, or Japanese stone pine. In China, this species is mainly distributed in forests of Greater and Small Khingan Mountains, Zhang Guang Cai Ranges, as well as Chang Bai Ridges in northeastern provinces of China [4,5,6]. Plant materials and crude extracts from P. pumila barks, cones, roots, foliated twigs, buds, and needles have long been used in traditional Chinese, Russian, Korean, and Japanese medicine for the treatment or prevention of various disorders due to their antiscorbutic, antihelmintic, antibacterial, expectorant, disinfectant, diuretic, as well as wound-healing activities [6,7,8,9]. Previous investigation of P. pumila led to the isolation of serratane triterpenes, diterpenes, monoterpenoids, and ferulate derivatives [8,9,10].
In our earlier study of pine cones of P. pumila, we have extracted and elucidated a serious of chemical constituents, including daucosterol, stigmast-7-en-3β-ol, β-sitosterol, dimethylmatairesinol, stigmasterol, stigmast-7-en-3-O-β-Dglucopyranoside, β-sitosterol palmitate, and flavonoid and aliphatic compounds [2, 5]. However, a phytochemical investigation of the stem barks of P. pumila has not been carried out systematically to date, though their biological and pharmacological properties are well utilized. In the current work, we report the isolation and characterization of a novel triterpene saponin, ursolic acid 3-O-[α-L-rhamnopyranosyl(1"→6')]-β-D-glucopyranosyl-28-O-β-D-glucuronopyranoside (1), together with two known flavonol glycosides, kaemferol 3-O-α-L-rhamnopyranoside (2) and quercetin 3-O-β-D-glucopyranoside (3), from the stem outer barks of this pine. Both flavonoids 2 and 3 and their occurrence in P. pumila are reported for the first time here.
Compound 1, with optical rotation \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –13.2° (c 0.5, MeOH), mp 169–171°C (uncorrected), was isolated as a white amorphous powder. In developing solvents A and B, the Rf values of 1 were 0.90 and 0.51. Through a TLC chromogenic procedure, compound 1 was assigned to be a triterpenoid due to the presence of a purple color when spraying with 1% sulfuric acid-ethanol reagent followed by heating [11,12,13]. In the IR spectrum, compound 1 showed characteristic absorption bands at 1030, 1065, 1730, 1925, and 3405 cm–1. The molecular formula of compound 1 was deduced to be C48H76O18 from Maldi-ToF MS spectrometric evidence ([M + H]+ at m/z 941, [M + Na]+ at m/z 963, [M + K]+ at m/z 979, and [2M + Na]+ at m/z 1903, respectively).
In 1H NMR spectrum of compound 1, the aglycone unit exhibited a trisubstituted olefinic proton signal vibrating at δH 5.34 (1H, t-like, H-12), a doublet centered at δH 2.49 (1H, J = 11.6 Hz, H-18) assignable to a methine proton, five singlets [δH 1.28 (3H, H-23), 0.99 (3H, H-24), 0.85 (3H, H-25), 1.02 (3H, H-26), and 1.21 (3H, H-27)] ascribed to the methyl protons, as well as two doublets with a coupling constant of J = 6.2 Hz due to the two methyl protons at δH 0.97 (3H, H-29) and 0.87 (3H, H-30), which characteristically confirmed the presence of an ursane-type triterpenoid [14, 15]. The 13C NMR spectrum of compound 1 additionally supported the above conclusion for C-12, 13, 18, 19, 20, and 21 of the aglycone unit, which gave typically carbon peaks at δC 127.41, 138.50, 53.59, 39.87, 39.14, and 30.95 [16]. Detailed NMR spectra data, as shown in Table 1, as well as a careful comparison with authentic compound, confirmed that the aglycone was ursolic acid [14,15,16].
In 1H NMR spectrum of compound 1, the anomeric proton at δH 5.01 (H-1′) as a doublet with coupling constant 7.2 Hz originated from a sugar moiety, along with the remaining six protons [δH 3.15–4.00 (4H, m, H-2', 3', 4', 5') and 4.15–4.55 (2H, m, H-6'a, 6'b)], which indicated the presence of a β-configuration glucose moiety [17,18,19]. The α-linked rhamnose residue was identified from its anomeric proton resonating at δH 4.50 as a doublet with coupling constant J = 1.5 Hz, and the typical methyl protons appearing at δH 1.13 (J = 6.2 Hz, d, H-6"-Me) [20,21,22]. The presence of a β-configuration glucuronic acid moiety in compound 1 was confirmed by its 1H and 13C NMR spectral data [23, 24], particularly by its characteristic anomeric proton signal at δH 5.02 (1H, d, J = 6.9 Hz, H-1"') and the typical carboxyl carbon resonance at δC 173.18 (C-6"') [25, 26].
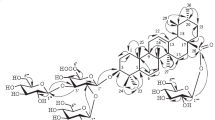
In the HMBC spectrum of compound 1, the anomeric proton signals of glucose and rhamnose showed long-range correlations with the 13C NMR signals at δC 88.65 (C-3) and 66.96 (C-6'), demonstrating glucosylation at C-3 of the triterpene aglycone and rhamnosylation at C-6' of the glucose residue (Fig. 1) [27, 28]. The 1H NMR signal at δH 5.02 (H-1"') correlated with the carbon signal at δC 176.00 (C-28), indicating that the glucuronic acid was connected to the ursolic acid at C-28 [13, 14, 16]. The DEPT experimental data, as demonstrated in Table 1, revealed 48 carbon signals in compound 1, which were sorted into eight methyls, 10 methenes, 22 methines, and eight tertiary carbons, with each peak coinciding with the corresponding site of ursolic acid 3-O-[α-L-rhamnopyranosyl(1"→6')]-β-D-glucopyranosyl-28-O-β-D-glucuronopyranoside.

The structure and key HMBC correlations of compound 1.
Based on the evidence and analysis above, the chemical structure of compound 1 was characterized as ursolic acid 3-O-[α-L-rhamnopyranosyl(1"→6')]-β-D-glucopyranosyl-28-O-β-D-glucuronopyranoside, which is a new triterpene saponin isolated for the first time here and which has not been reported from any plant previously.
The chemical structures of the known flavonol glycosides 2 and 3 were characterized as kaemferol 3-O-α-Lrhamnopyranoside and quercetin 3-O-β-D-glucopyranoside, respectively, by comparison of their NMR, MS, and other data with those in the literature [29,30,31,32]. To the best of our knowledge, the secondary metabolites 2 and 3 have never been isolated and reported from P. pumila previously.
EXPERIMENTAL
General. The optical rotation was determined with a Jasco DIP-1000 digital polarimeter (in MeOH). An Electro Thermal 9100 apparatus was used to measure the melting points (mp, uncorrected). IR spectra were obtained in KBr on a Perkin-Elmer BX FT-IR spectrometer. Maldi-TOF MS spectra were recorded on a Voyager-DE STR mass spectrometer. The NMR spectra of compounds were recorded in DMSO-d6 on a Bruker Avance DPX 400 spectrometer at frequencies of 100 and 400 MHz for 13C and 1H NMR, respectively. Silica gel and Sephadex LH-20 (Sigma) were used as packing materials for open column chromatography (OCC). ODS (50 μm, YMC) and macroporous resin D101 were used for vacuum liquid chromatography (VLC). Thin-layer chromatography (TLC) experiments were performed on DC-Plastikfolien Cellulose F plates (Merck, Darmstadt, Germany) with AcOH–H2O (3:47, solvent A) and t-BuOH–AcOH–H2O (3:1:1, solvent B) as developing solvents. TLC spots were detected by UV light exposure (254 and 365 nm) and by spraying with 1% sulfuric acid–ethanol solution followed by heating. All reagents used in this work were of analytical grade.
Plant Material. The stem outer barks of P. pumila were collected from the forest of Greater Khingan Mountains, Heilongjiang Province, P. R. China, in March of 2018, and authenticated by Prof. J. H. Wang from Research Institute of Forestry, Chinese Academy of Forestry, Beijing, P. R. China. A voucher herbarium specimen (OBPP-20180302) has been deposited in the Herbarium of Tianjin Key Laboratory of Pulp and Paper, Department of Plant Fine Chemicals, College of Light Industry Science and Technology, Tianjin University of Science and Technology, Tianjin, P. R. China.
Extraction and Isolation. Air-dried P. pumila stem outer bark materials (4.05 kg) were finely ground, then extracted in a jar (15 L) with EtOH–H2O (4:1) for more than 72 h at room temperature (three times). After evaporation under vacuum, the residues were combined and diluted with H2O, sequentially fractionated with n-hexane, CH2Cl2, EtOAc, and n-BuOH in separation funnels, then freeze dried to yield n-BuOH soluble fraction powders. A portion of the above-obtained n-BuOH soluble fraction powders (OBPPB, 40.81 g) was subjected to silica gel OCC using CH2Cl2–MeOH (99:1→1:1) as the eluting solvent system to give five major fractions (OBPPB1–OBPPB5), which were monitored and grouped by TLC experiments. Fraction OBPPB3 (25.22 g) was then subjected to VLC with ODS used as packing materials and CH2Cl2–EtOH (29:1→1:4) as washing solvents to obtain four subfractions (OBPPB31–OBPPB34). Subfraction OBPPB32 was further subjected to macroporous resin D101 VLC with MeOH–H2O (1:2) as eluting solvent to give four subfractions (OBPPB321–OBPPB324). Subfraction OBPPB323 was also purified by repeated Sephadex LH-20 column chromatography using n-hexane–EtOH (4:1→1:4) and MeOH–H2O (3:1→1:5) as gradient mobile solvents to give a white amorphous secondary metabolite (1, 40.4 mg). Subfraction OBPPB322 was further purified on a Sephadex LH-20 column using MeOH–H2O (4:1, 2:1, 1:3, and 1:5) as mobile solvent to get compounds 2 (50.3 mg) and 3 (61.5) as yellowish amorphous powders.
Ursolic Acid 3- O -[α-L-Rhamnopyranosyl(1"→6')]-β-D-glucopyranosyl-28-O-β-D-glucuronopyranoside (1). White amorphous powder; mp 169–171°C; \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \)–13.2° (c 0.5, MeOH). IR (KBr, vmax, cm–1): 3405, 1925, 1730, 1065, 1030. Rf 0.90 (solvent A) and 0.51 (solvent B); Maldi-TOF MS m/z: 941 [M + H]+, 963 [M + Na]+, 979 [M + K]+, 1903 [2M + Na]+, corresponding to molecular weight 940 and established for C48H76O18. For 1H, 13C NMR, and DEPT data (DMSO-d6), see Table 1. For key HMBC correlations (H→C), see Fig. 1.
References
Y. Amagai and G. Kudo, Alpine Bot., 129, 43 (2019).
Z. J. Li, L. Wu, C. L. Si, J. H. Wang, X. N. Yan, H. Y. Yu, R. Z. Cong, S. Y. Wang, and R. Wei, Chem. Nat. Compd., 55, 1187 (2019).
U. Zeb, R. N. Wang, P. B. Dong, N. Wang, T. T. Zhang, C. Z. Lin, and X. J. Wang, Mitochondrial DNA B, 4, 290 (2019).
N. H. Sa, N. T. Tam, N. T. H. Anh, T. D. Quan, D. D. Thien, D. T. Phong, and T. T. Thuy, Nat. Prod. Res., 32, 341 (2018).
Z. J. Li, F. Fan, C. L. Si, X. N. Yan, H. Y. Yu, J. H. Wang, and L. Wu, Chem. Nat. Compd., 56, 1128 (2020).
G. H. Chen, Y. C. Li, N. H. Lin, P. C. Kuo, and J. T. Tzen, Molecules, 23, 86 (2017).
M. Karapandzova, G. Stefkov, I. Cvetkovikj, J. P. Stanoeva, M. Stefova, and S. Kulevanova, Nat. Prod. Commun., 10, 987 (2015).
M. K. Langat, A. Helfenstein, C. Horner, P. Tammela, H. Hokkanen, D. Izotov, and D. A. Mulholland, Chem. Biodiv., 15, e1800056 (2018).
V. A. Hang, Y. V. Gatilov, Z. V. Dubovenko, and V. A. Pentegova, Chem. Nat. Compd., 16, 361 (1980).
A. V. Shpatov, S. A. Popov, O. I. Salnikova, E. N. Shmidt, S. W. Kang, S. M. Kim, and B. H. Um, Chem. Biodiv., 10, 198 (2013).
P. J. Houghton and A. Raman, Layer chromatography, Laboratory Handbook for the Fractionation of Natural Extracts, Springer, Berlin, 1998.
R. Wang, K. Wang, C. L. Si, Y. Y. Luo, W. Liu, X. X. Zhang, Y. Y. Tian, and J. H. Wang, Chem. Nat. Compd., 54, 717 (2018).
C. L. Si, Y. Gao, L. Wu, R. Liu, G. H. Wang, L. Dai, X. H. Li, and Y. M. Hong, Holzforschung, 71, 697 (2017).
W. Seebacher, N. Simic, R. Weis, R. Saf, and O. Kunert, Magn. Reson. Chem., 41, 636 (2003).
L. Wu, G. C. Wang, T. Shen, Q. Qiang, Q. Xue, M. Chen, M. Chen, J. M. Zhang, Y. Y. Luo, Y. M. Hong, C. L. Si, and W. C. Hu, Chem. Nat. Compd., 54, 210 (2018).
T. Miyase, K. I. Shiokawa, D. M. Zhang, and A. Ueno, Phytochemistry, 41, 1411 (1996).
C. L. Si, L. Wu, and Z. Y. Zhu, Biochem. Syst. Ecol., 37, 221 (2009).
Z. J. Li, J. X. Yang, H. Wang, R. Xu, Z. T. Rao, C. L. Si, J. Zhang, L. Sun, X. Y. Zhang, S. J. Han, Z. Sun, L. Wu, D. Liu, Y. Liu, and J. H. Wang, Chem. Nat. Compd., 55, 144 (2019).
N. Riaz, H. M. Rafiq, M. Saleem, B. Jabeen, M. Ashraf, I. Ahmed, T. Tahir, and R. B. Tareen, Chem. Nat. Compd., 55, 493 (2019).
C. L. Si, S. L. Chen, Z. J. Li, D. Liu, S. X. Nie, Y. Liu, L. Sun, X. Y. Zhang, and S. J. Han, Chem. Nat. Compd., 55, 252 (2019).
N. Kavtaradze, M. Alaniya, M. Masullo, A. Cerulli, and S. Piacente, Chem. Nat. Compd., 56, 70 (2020).
C. L. Si, X. H. Yang, Z. J. Li, J. S. Lu, X. Tao, J. Y. Zhang, W. Liu, and Y. S. Bae, Holzforschung, 72, 719 (2018).
W. C. Hu, X. F. Wang, L. Wu, T. Shen, L. L. Ji, X. H. Zhao, C. L. Si, Y. Y. Jiang, and G. C. Wang, Food Funct., 7, 1002 (2016).
C. L. Si, L. Wu, Z. Y. Zhu, J. K. Kim, D. Z. Kwon, and Y. S. Bae, Holzforschung, 63, 440 (2009).
M. Okawa, R. Akahoshi, K. Kawasaki, D. Nakano, R. Tsuchihashi, J. Kinjo, and T. Nohara, Chem. Pharm. Bull., 67, 159 (2019).
C. L. Si, G. H. Xu, X. F. Huang, Z. G. Du, L. Wu, and W. C. Hu, Chem. Nat. Compd., 52, 132 (2016).
C. N. Zhao, Z. L. Yao, D. Yang, J. Ke, Q. L. Wu, J. K. Li, and X. D. Zhou, Biomolecules, 10, 74 (2020).
C. L. Si, J. Z. Jiang, S. C. Liu, H. Y. Hu, X. D. Ren, G. J. Yu, and G. H. Xu, Holzforschung, 67, 357 (2013).
P. K. Agrawal, Carbon-13 NMR of Flavanoids, Elsevier, New York, 1989.
L. Q. Hu, K. Wang, G. B. Li, R. Y. Zhang, Y. Y. Luo, C. L. Si, and J. H. Wang, Holzforschung, 71, 785 (2017).
L. Wu, W. Xiong, J. W. Hu, J. Wu, Z. J. Li, Y. Gao, D. Liu, Y. Liu, W. Liu, M. Liang, C. L. Si, and Y. S. Bae, Chem. Nat. Compd., 55, 345 (2019).
J. B. Harborne and T. J. Mabry, The Flavonoids: Advances in Research, Chapman and Hall Ltd., London, 1982.
Acknowledgment
This work was financially supported by the Fundamental Research Funds for the Central Non-profit Research Institute of CAF (CAFYBB2016ZD009), Guangxi Key Laboratory of Clean Pulp & Papermaking and Pollution Control, College of Light Industry and Food Engineering, Guangxi University (KF201801-5), Key Technology Research and Development Program of Tianjin (19YFZCSN00950), and Tianjin Enterprise Technology Commissioner Project (19JCTPJC52800).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 1, January–February, 2021, pp. 96–99.
Rights and permissions
About this article

Cite this article
Liu, K., Liu, HY., Tao, X. et al. A New Triterpene Glycoside from Pinus pumila. Chem Nat Compd 57, 115–119 (2021). https://doi.org/10.1007/s10600-021-03294-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-021-03294-1