
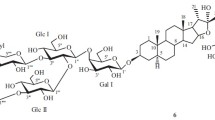
Three sesquiterpene glycosides were isolated from the MeOH (70%) extract of flowers of Yucca gloriosa L. Their chemical structures were established using spectral analyses, i.e., 1D and 2D NMR (1H, 13C, HSQC, HMBC, COSY) and mass spectroscopy (ESI-MS). They all were new glycoside derivatives of nerolidol and a new class for the genus Yucca. Glycoside 1 had the structure (1E,3S,5R,6E,9E)-5-O-β-D-glucopyranosyl-(1_6)-O-β-D-glucopyranosyl-3,5,11-trihydroxy-3,7,11-trimethyldodeca-1,6,9-triene; glycoside 2, (1E,3S,5R,6E,9E)-5-O-β-D-glucopyranosyl-(1→6)-O-β-D-glucopyranosyl-3,5,11,12-tetrahydroxy-3,7,11-trimethyldodeca-1,6,9-triene; glycoside 3, (1E,3S,5R,6E,10E)-(12-O-β-D-glucopyranosyl)-5-O-β-Dglucopyranosyl-(1→6)-O-β-D-glucopyranosyl-3,5,12-trihydroxy-3,7,11-trimethyldodeca-1,6,10-triene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The chemical composition of Yucca gloriosa L. has been the subject of our research for many years. Y. gloriosa is a tree-like, evergreen, bushy, and drought- and frost-resistant plant that is indigenous to central and southern North America and cultivated in many countries as an excellent decorative plant.
The steroidal sapogenin tigogenin from the plant leaves was recognized as an economical raw material for synthesizing steroidal hormone preparations [1, 2]. Plantations of Y. gloriosa were cultivated in eastern Georgia on an area of 150 ha to ensure a supply of tigogenin from plant raw material. Leaves, flowers, fruit, stems, and rhizomes yielded greater than 60 spirostanol, furostanol, and cholestanol glycosides, 20 of which were new. The proposed fungicidal preparation Gloriofucin that consisted mainly of spirostanol glycosides was prepared from total glycosides of dried leaves on the lower tier of the living plant [3]. The effective growth and development stimulator for agricultural crops Aleksin was developed from total steroidal glycosides from flowers [4].
Y. gloriosa flowers profusely twice per year, in the spring and autumn. The plants are in full flowering in spring and form large inflorescences with numerous flowers [5]. Plants flowering for the first time develop one inflorescence; in subsequent years, 3–4 of them.
Previously, six spirostanol and two furostanol glycosides, three flavonoids, and three phenolic carboxylic acids were isolated from the flowers and identified by us [6, 7].
In continuation of studies of the chemical composition of Y. gloriosa, we isolated from the MeOH (70%) extract of the flowers and identified three sesquiterpene glycosides (1–3). They all were new compounds and a new class for the genus Yucca.

The PMR spectrum of 1 was consistent with a vinyl ABX system as a doublet of doublets at δ 5.98 (1H, J = 17.3, 10.7 Hz), 5.28 (1H, J = 17.3, 1.5 Hz), 5.06 (1H, J = 10.7, 1.5 Hz), and 5.61 (1H, J = 15.6, 6.6 Hz) and two doublets at δ 5.63 (1H, J = 15.6 Hz) and 5.13 (1H, J = 9.4 Hz); four singlets for methyls at δ 1.68, 1.29, 1.29, and 1.28; one doublet of doublets of doublets for a hydroxymethine proton at δ 4.84 (1H, J = 9.4, 8.4, 3.7 Hz), two methylenes at δ 2.73 (2H, d, J = 6.6 Hz), 1.96 (1H, dd, J = 14.5, 8.4), and 1.63 (1H, dd, J = 14.5, 3.7); and two anomeric protons at δ 4.36 (1H, d, J = 7.8 Hz) and 4.23 (1H, d, J = 7.8 Hz) (Table 1). A COSY experiment showed cross peaks between vinyl protons [(δ 5.28↔5.06) ↔5.98; δ 5.63↔5.61] and between an olefinic proton at δ 5.13 (H-6) and a proton with δ 4.84 (H-5), the latter of which correlated with methylene protons at δ 1.96 (H-4a) and 1.63 (H-4b). An olefinic proton at δ 5.61 (H-9) correlated with a methylene doublet at δ 2.73 (H-8). The 13C NMR spectrum showed 27 C atoms that were identified using DEPT and HSQC experiments as four methyls at δ 29.9, 29.9, 28.5, and 16.9; four olefinic methines at δ 145.9, 141.2, 126.2, and 125.5; one carbinol methine at δ 72.2; three methylenes at δ 112.1, 47.9, and 43.1, one of which was olefinic (δ 112.1); three quaternary atoms at δ 140.0, 73.9, and 71.1; and twelve carbohydrate atoms (Table 2). All spectral data were indicative of a nerolidol-type sesquiterpene glycosides [8]. The configuration of C-5 (β-H form) was established from chemical shifts and J values [9, 10]. The structure of 1 was finalized using an HMBC experiment that showed significant correlation peaks between the anomeric protons and glycosylated C atoms [δ 4.23 (H-1′)/δ 72.2 (C-5) and [δ 4.36 (H-1′′)/δ 69.8 (C-6′)]. Both sugars were β-D-glucoses according to the PMR and 13C NMR spectra and J values. The results indicated that 1 was (1E,3S,5R,6E,9E)-5-O-β-D-Glcp-(1_6)-O-β-D-Glcp-3,5,11-trihydroxy-3,7,11-trimethyldodeca-1,6,9-triene.
Compound 2 differed from 1 by the presence of a C-11 exomethylene at δ 3.38 (d, J = 11.8 Hz) and 3.28 (d, J = 11.8 Hz) instead of a methyl (Table 1). Thus, compound 2 was characterized as (1E,3S,5R,6E,9E)-5-O-β-D-Glcp-(1↔6)-O-β-D-Glcp-3,5,11,12-tetrahydroxy-3,7,11-trimethyldodeca-1,6,9-triene.
The PMR spectrum of 3 showed olefinic protons of an ABX system at δ 5.97 (1H, dd, J = 17.3, 10.7 Hz), 5.39 (1H, t, J = 7.1 Hz), 5.28 (1H, dd, J = 17.3, 1.5 Hz), 5.13 (1H, d, J = 9.4 Hz), and 5.08 (1H, dd, J = 10.7, 1.5 Hz); three methyls at δ 1.80, 1.69, and 1.28 (3H each, s); four methylenes at δ 4.32 (1H, d, J = 11.5 Hz), 4.23 (1H, d, J = 11.5 Hz), 2.26 (1H, m), 2.20 (1H, m), 2.07 (2H, m), 1.96 (1H, dd, J = 14.5, 8.4 Hz), and 1.62 (1H, dd, J = 14.5, 3.7 Hz); one hydroxymethine at δ 4.84 (1H, ddd, J = 9.4, 8.4, 3.7 Hz); and three anomeric protons at δ 4.36 (1H, d, J = 7.8 Hz), 4.24 (1H, d, J = 7.9 Hz), and 4.23 (1H, d, J = 7.8 Hz)] (Table 1). The 13C NMR spectrum revealed three methyls at δ 28.5, 21.8, and 16.9; four methylenes at δ 67.8, 47.9, 40.5, and 26.6; one oxygenated tertiary at δ 72.1 and one quaternary C atom at δ 73.9 (C-3); six olefinic at δ 145.9, 140.7, 132.7, 130.5, 126.2, and 112.2; and three anomeric C atoms at δ 104.6, 102.6, and 100.3 (Table 2). All three sugars were glucose according to the PMR and 13C NMR spectra. The substitution sequence was established using the 2D HMBC spectrum, which showed significant correlation peaks between ′′′ 4.24 (H-1′′′) and 67.8 (C-12), 4.36 (H-1′′) an 69.9 (C-6′), and 4.23 (H-1′) and 72.1 (C-5). As a result, the structure of 3 was established as (1E,3S,5R,6E,10E)-(12-O-β-D-Glcp)-5-O-β-D-Glcp-(1→6)-O-β-D-Glcp-3,5,12-trihydroxy-3,7,11-trimethyldodeca-1,6,10-triene.
Experimental
Optical rotation was measured on a PerkinElmer 341 polarimeter.
UV spectra were recorded on a Shimadzu UV-1800 spectrophotometer. IR spectra were taken on a Bruker ALPHA FTIR spectrometer. NMR experiments used an Avance II 600 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany). All 1D and 2D NMR spectra were recorded in CDCl3 (99.95%, Sigma-Aldrich). NMR data were processed using MestRe-C UXNMR software (Santiago de Compostela, Spain).
Exact masses were measured using a microOTOF-QII mass spectrometer (Bruker). Samples were analyzed by highresolution time-of-flight (HR-TOF) mass spectrometry with electrospray ionization (ESI) in positive-ion mode. Exact masses were calibrated on a daily basis using an ESI-L low concentration tuning mix (Agilent, Santa Clara, USA).
Column chromatography was performed over silica gel (0.030–0.200 and 0.040–0.063 mm, Merck), Sephadex LH-20 (Sigma-Aldrich), and Diaion HP-20 resin polymer (Mitsubishi, Japan). TLC used silica gel 60 F254 plates (Merck) and solvent system CHCl3–MeOH–H2O (26:14:3). All solvents for extraction and chromatography were analytical grade (Merck, Darmstadt, Germany).
Extraction and Isolation. Flowers of Y. gloriosa were collected at the Tbilisi Test Plot of Medicinal Plants, I. G. Kutateladze Institute of Pharmacochemistry, in June 2014.
Raw material (200 g) was extracted with refluxing MeOH (70%). The MeOH was distilled off. The aqueous liquid was purified with CHCl3. The aqueous phase was transferred to a column of Diaion HP-20 and eluted by a gradient of H2O–MeOH (10:0→0:10) to produce four fractions (500 mL each), i.e., H2O (61 g), MeOH (30%) (2.5 g), MeOH (80%) (11 g), and MeOH (1.5 g). Chromatography of the 30% MeOH fraction over columns of Sephadex LH-20 and silica gel isolated 1 (2.3 mg), 2 (1.9 mg), and 3 (2.7 mg).
(1 E ,3 S ,5 R ,6 E ,9 E )-5- O - β -D-Glc p -(1_6)-O-β-D-Glcp-3,5,11-Trihydroxy-3,7,11-trimethyldodeca-1,6,9-triene (1). C27H46O13, amorphous white powder, μM – 15.5° (c 0.10, MeOH). UV spectrum (MeOH, λmax, nm) (log ε): 210 (3.00), 300 (0.95). IR spectrum (MeOH, νmax, cm–1): 3353, 2925, 1660, 1039. HR-ESI-TOF-MS m/z 579.3511 [M + H]+, MS/MSn m/z 417 [M + H – 162]+, 255 [M + H – 162 × 2]+.
(1 E ,3 S ,5 R ,6 E ,9 E )-5- O - β -D-Glc p -(1_6)-O-β-D-Glcp-3,5,11,12-Tetrahydroxy-3,7,11-trimethyldodeca-1,6,9-triene (2). C27H46O14, amorphous white powder, \( {\left[\upalpha \right]}_{\mathrm{D}}^{22} \) –14.2° (c 0.10, MeOH). UV spectrum (MeOH, λmax, nm) (log ε): 200 (2.80). IR spectrum (MeOH, νmax, cm–1): 3334, 2925, 1694, 1017. HR-ESI-TOF-MS m/z 595.2361 [M + H]+, MS/MSn m/z 433 [M + H – 162]+, 271 [M + H – 162 × 2]+.
(1 E ,3 S ,5 R ,6 E ,10 E )-(12- O - β -D-Glc p )-5- O - β -D-Glc p -(1→6)- O - β -D-Glc p -3,5,12-Trihydroxy-3,7,11- trimethyldodeca-1,6,10-triene (3). C33H56O18, amorphous white powder, \( {\left[\upalpha \right]}_{\mathrm{D}}^{22} \)_ (c 0.10, MeOH). UV spectrum (MeOH, λmax, nm) (log ε): 210 (2.90), 300 (1.0). IR spectrum (MeOH, νmax, cm–1): 3359, 2924, 1713, 1039. HR-ESI-TOF-MS m/z 741.2432 [M + H]+, MS/MSn m/z 579 [M + H – 162]+, 417 [M + H – 162 × 2]+, 255 [M + H – 162 × 3]+.
References
E. P. Kemertelidze and T. A. Pkheidze, Khim.-farm. Zh., 6 (12), 44 (1972).
N. P. Men’shova, N. P. Sorokina, G. S. Grinenko, N. N. Suvorov, Yu. R. Gurevich, E. P. Kemertelidze, and T. A. Pkheidze, Khim.-farm. Zh., 8 (7), 15 (1974).
E. P. Kemertelidze, M. M. Benidze, and A. V. Skhirtladze, Khim.-farm. Zh., 43 (1), 45 (2009).
E. Kemertelidze and M. Benidze, Bull. Georgian Acad. Sci., 164, 91 (2001).
A. Ya. Shtromberg and A. S. Muradbekova, Rastit. Resur., 12 (1), 15 (1976).
P. Montoro, A. Skhirtladze, A. Perrone, M. Benidze, E. Kemertelidze, and S. Piacente, J. Pharm. Biomed. Anal., 52, 791 (2010).
E. Kemertelidze, M. Benidze, and A. Skhirtladze, Bull. Georgian Acad. Sci., 5 (1), 158 (2011).
M. C. Yang, S. M. Kim, K. H. Lee, K. H. Kim, and K. R. Lee, Molecules, 12, 2270 (2007).
B. D’Abrosca, P. De Maria, M. DellaGreca, A. Fiorentino, A. Golino, A. Izzo, and P. Monaco, Tetrahedron, 62, 640 (2006).
A. Fiorentino, M. DellaGreca, B. D’Abrosca, A. Golino, S. Pacifico, A. Izzo, and P. Monaco, Tetrahedron, 62, 8952 (2006).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Prirodnykh Soedinenii, No. 1, January–February, 2018, pp. 66–69.
Rights and permissions
About this article

Cite this article
Benidze, M.M., Nebieridze, V.G., Ganzera, M. et al. Sesquiterpene Glycosides from Flowers of Yucca gloriosa. Chem Nat Compd 54, 73–76 (2018). https://doi.org/10.1007/s10600-018-2262-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-018-2262-x