

A new spiropyran containing a cationic 3Н-indolium substituent and methoxy groups at positions 5 and 5” of the indoline cycles was synthesized. Hydrolysis of this compound was observed during crystallization from EtOH, leading to the formation of protonated merocyanine form of spiropyran containing a free formyl group. The hydrolysis product was characterized by X-ray structural analysis, the intermolecular interactions in crystal were studied with the CrystalExplorer 21.5 software suite. Quantum-chemical modeling based on the Fukui function distribution was used to establish the preferred site for nucleophilic attack, and a mechanism for hydrolysis was proposed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The interest toward developing materials with tunable properties has been steadily growing over the previous decades. A striking example of the use of such materials are sensor systems,1,2 while one of the currently developing areas is the design of targeted drug delivery systems.3 The tuning of material properties can be provided by molecular switches, whose molecules tend to change their structure and properties in response to external factors. Such molecular systems are relevant not only as components of advanced materials: some of them are capable of functioning as molecular machines. Develoments in this area have been recognized with a Nobel prize a few years ago.4,5,6 Among molecular switches used in recent studies, spiropyrans of indoline series are one of the most common types.7,8
The synthesis of indoline spiropyrans, as a rule, has been accomplished by the condensation of Fischer bases with ohydroxyaromatic aldehydes.9,10 Their molecules can respond to various types of stimuli: electromagnetic radiation, changes in the temperature and acidity of the medium, pressure, solvent polarity, and other parameters,11,12 enabling a broad range of applications for compounds of this class. The switchable properties of spiropyrans are based on reversible isomerization between the spirocyclic (SP) and several merocyanine (MC) structures (Fig. 1).

Reversible isomerization of spiropyrans.
The molecules of spiropyrans can be incorporated into the structure of materials as a simple physical mixture or via new chemical bond formation. In the latter case, the presence of reactive groups, such as formyl, hydroxy, or carboxyl is obligatory.13,14 Recently, there have been active developments in the synthesis of cationic spiropyrans that can be used as the basis for the design of hybrid materials containing ionic type bonds.15,16,17,18 In this case, the properties of systems also depend on the cationic moiety structure and the type of anion.19,20,21 Of particular interest are derivatives containing a conjugated cationic fragment in the benzopyran moiety. Such compounds can be considered as structural analogs of cyanine dyes, exhibiting the bathochromically shifted absorption maximum of МС isomer22,23 and, in several cases, demonstrating photoluminescence in the near IR wavelengths.19,24 As a rule, the presence of a sufficiently strong electron-withdrawing group at position 6' leads to partial stabilization of the МС form in similar compounds due to partial neutralization of the negative δ– charge on the oxygen atom.23,24,25,26 Analogously, a stabilizing effect in the case of spiropyrans is due to the presence of electron-donating substituents at position 5 of indoline system, reducing the positive charge on the nitrogen and С-2,2' atoms.27
In order to examine the influence of electron-donating substituents in the indoline fragments on the structure and properties of compounds, a new spiropyran containing cationic 3Н-indolium fragment in the benzopyran moiety and methoxy groups in both indoline cycles was obtained.
Spiropyran 3 containing a conjugated cationic fragment in the 2Н-chromene moiety was synthesized according to a one-step procedure by a condensation reaction between 2 equiv of 5-methoxy-1,2,3,3-tetramethyl-3Н-indolium perchlorate 1 and 2-hydroxy-5-methylisophthalic aldehyde (2) in the presence of an organic base (Scheme 1).28

Synthesis of spiropyran 3
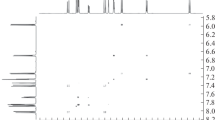
The structure of compound 3 was established from the results of NMR and high-resolution mass spectral analysis. The integrated signal intensities, chemical shifts, and the signal multiplicity in the spectrum were in complete agreement with the proposed structure. According to 1Н NMR data, compound 3 exists in CDCl3 solution as a 10:1 mixture of SP and MC isomers (Fig. 2).

1Н NMR spectrum of compound 3 в CDCl3.
1Н NMR spectrum of compound 3 contained characteristic for the SP form signals of spin-coupled protons' pair H-3' and H-4' at 5.81 and 6.86 ppm, respectively, with a spin-spin coupling constant of 10.3 Hz. The vinyl protons of 3Н-indolium moiety appeared as doublet signals at 7.98 and 7.29 ppm with a spin-spin coupling constant of 16.4 Hz, indicating a trans configuration. The protons of methyl groups at the nitrogen atoms of the cationic and the hetarene moieties appeared as three-proton singlets at 3.72 and 2.69 ppm, while the protons of methoxy groups – at 3.87 and 3.83 ppm, respectively. Taking into account the symmetric structure of the МC form, the H-12'/H-4' and H-13'/H-3' protons appeared as two-proton doublet signals with a spin-spin coupling constant of 16.5 Hz, providing evidence for a transoid configuration of the vinyl moieties. The protons of methyl groups at the indoline nitrogen atoms and the methoxy groups were observed as six-proton singlets at 4.25 ppm and 3.91 ppm, respectively.
In 13С NMR spectrum of compound 3, the signal of С-2” carbon atom appeared in the downfield region at 180.2 ppm. The signals of С-5,5”,9' carbon atoms, directly bonded to oxygen atoms, were shifted upfield relative to the signal of С-2” atom. The signal of the spiro atom appeared in its characteristic region at 107.3 ppm. It should be also noted that the signal of С-12' atom appeared at 148.3 ppm, while the С-13' atom gave a signal at 112.0 ppm. The carbon signals of МС form were not apparent due to its low concentration.
Compound 3 existed in DMSO-d6 solution entirely in its closed form, similarly to spiropyran 4 that has been studied earlier, allowing to propose that the stabilization of МС form in CDCl3 is significantly affected by the trace amounts of hydrochloric acid that are typical for this solvent and the second form observed in the spectrum was in fact protonated merocyanine. However, its concentration was too low for allowing to detect the hydroxy group protons. The respective proton signals were shifted upfield relative to those in the case of compound 4,28 due to the electron-donating effect of the methoxy group (Table 1).
Crystals were obtained by slow evaporation of ethanol solution of compound 3. The results of X-ray structural analysis unexpectedly revealed the structure of styryl salt 5 (Fig. 3). The hydrolysis performed under laboratory conditions (by heating spiropyran 3 as 4·10–3 М solution in EtOH) allowed to isolate compound 5. The experiment was repeated under argon atmosphere, again providing compound 5 as the sole product. A reference sample of compound 5 was also obtained by a two-step procedure including the synthesis of spiropyran 6 and its subsequent protonation (Scheme 2).

а) The molecular structure of compound 5 with atoms represented by thermal vibration ellipsoids of 0.5 probability. b) The molecular packing in crystal of compound 5, a view along the axis с.

The synthesis of compounds 5 and 6
1Н NMR spectrum of spiropyran 6 contained the characteristic doublet signals of H-3' and H-4' protons at 5.76 and 6.83 ppm, with a spin-spin coupling constant of 10.3 Hz. The indolium protons in 1Н NMR spectrum of spiropyran 6 appeared in the same range as the analogous protons of the SP form of compound 3, while the signal of H-7' proton was shifted upfield (7.42 ppm) compared to the SP form of compound 3 (7.53 ppm). This upfield shift can be explained by the pronounced electron-withdrawing effect of the cationic 3Н-indolium substituent.
1Н NMR spectrum of compound 5 contained a singlet signal of the hydroxy group proton at 11.70 ppm and a formyl group proton signal at 10.08 ppm. The vinyl fragment proton' signals appeared as two doublet signals at 8.32 and 7.70 ppm, with a spin-spin coupling constant of 16.6 Hz, indicating a transoid configuration of the С=С double bond. The protons of methyl group at the quaternary indolium nitrogen atom appeared as a three-proton singlet signal at 4.08 ppm, while the methoxy group protons gave a signal at 3.86 ppm. The proton signal of gem-methyl groups in the indolium moiety appeared as a six-proton singlet at 1.73 ppm, due to their magnetic equivalence.
According to the results of X-ray structural analysis, the molecule of compound 5 had a nonplanar structure: the angle between the mean square planes of rings С(9')–С(10') and С(8)–С(9) was equal to 11.66°, while the values of the torsion angles N(1)–C(2)–C(3')–C(4'), C(2)–C(3')–C(4')–C(10'), and C(3')–C(4')–C(10')–С(9') were –173.0, 173.1, and–174.4°, respectively, indicating a ТТТ configuration of the vinyl-3Н-indolium moiety. The pyrrolidine ring assumed an envelope conformation, with the С(2) atom deviating from the plane of the other ring carbon atoms by 0.09 Å. The sum of valence angles at the indolium nitrogen atom was 360°, while the length of the N(1)–C(2) bond is 1.32 Å, which corresponds to the С=N bond multiplicity equal to two29 and indicates the cationic nature of the indole fragment. The values of valence and torsion angles, as well as the main bond lengths are presented in Table 2.
Crystals of compound 5 had a layered structure where molecules were linked by electrostatic interactions with the anion and various nonspecific interactions, as shown by the analysis of Hirshfeld surfaces (Fig. 4a) constructed at the normalized contact distance dnorm. The presence of ten shortened contacts was observed for each protonated merocyanine molecule when not considering the anion and twelve contacts when analyzing the surface constructed taking into account the anion. The predominant contacts in the crystal were О···Н (39.3%), mostly occurring between the oxygen atoms of perchlorate anion and aromatic С–H protons of merocyanine molecule, as well as Н···Н contacts (39.4%) (Fig. 4b). It should be noted that O···H contacts were generally energetically much more favorable than Н···Н contacts. The enrichment coefficient ЕXY30 was equal to 1.4 and 0.96, respectively. The significant number of shortened Н···Н contacts in the given case was caused by the relatively tight packing of molecules in the crystal. Each anion interacted with three cationic organic functional groups, while each cationic site – with four anions. The structure also contained an intramolecular О(1')–Н(1')···О(2') hydrogen bond (l 1.892 Å, with the angle О(1')–Н(1')···О(2') equal to 146.9°). The shortened contacts observed in the crystal are listed in Table 3.

a) The Hirshfeld surface for compound 5, constructed by normalized contact distance dnorm. b) The fingerprint surfaces for the shortened contacts О···Н, Н···Н, and С···Н.
A wide range of styryl salts of indoline-substituted derivatives have been previously obtained by Seiler and coworkers via protonation of the corresponding spiropyrans.31 These compounds were also formed as intermediates during the synthesis of some other molecules.32 In the given case, compound 5 was generated by the hydrolysis of cationic spiropyran 3 in EtOH. It is well known that the condensation reaction of ortho-hydroxy aromatic aldehydes with Fischer bases is reversible. This fact has enabled, for example, the substitution of benzopyran moiety of a molecule incorporated in a polyphosphate chain.33 Certain representatives of spiropyrans can undergo hydrolysis in aqueous media,33,34 which creates obstacles to some applications. At the same time, the possibility of spiropyran cleavage by hydrolysis or ozonolysis35 with isolation of the respective aldehyde allows to use spiro structures as effective protecting groups. Even though cationic spiropyrans related to compound 3 are usually quite resistant to hydrolysis,19,26,28 in some cases there are precedents where the cationic group has been cleaved36 or the hetarene system has been substituted,37 using a molecule of Fischer base in the role of a nucleophile.
Compound 3 showed the presence of MC form in EtOH solution, as evidenced by the characteristic broadened absorption band with a maximum at 668 nm in the absorption spectrum. Under analogous conditions in aprotonic MeCN, the longest wavelength absorption maximum was at 433 nm, corresponding to the SP form of compound containing a conjugated cationic vinyl-3Н-indolium motif. Since polar and, in particular, protic solvents such as EtOH are known to stabilize the МС form of spiropyrans,38 the nucleophilic attack by H2O during the hydrolysis occurred most likely at one of the two sites – at the С-4' or С-12' atom (Scheme 3). We have performed a quantum-chemical study in order to gain a more detailed understanding of the hydrolysis process.

Isomerization and hydrolysis of compound 3 in EtOH
First, the structure of product 5 was used as the reference for proposing a structure for the starting merocyanine 3-МС. Since the stable merocyanine forms for the majority of spiropyrans are isomers with ТТС and ТТТ configurations of the vinylindolium fragment,39,40 we considered both of these conformations and selected the configuration of the second fragment on the basis of X-ray structural analysis data. The distribution of Fukui function, determined as the difference of electron density distribution between the starting molecule and ions obtained by adding or removing a certain number of electrons, was evaluated as a criterion for the reactivity estimation.41 The spatial distribution of Fukui function f + for two forms of 3-МС characterized by different configuration of the vinylindolium fragment obtained via spirocycle cleavage, is shown in Figure 5a.

а) The distribution of Fukui function f + and b) frontier molecular orbitals in merocyanine 3-МС.
Quantitative description of the electrophilicity or nucleophilicity of a potential reactive site, as a rule, can be obtained by comparing the atomic populations calculated by the Hirschfeld method.42,43 A higher value of f + (equation (1)) points to the preferred site of nucleophilic attack, while f – – to the preferred site of electrophilic attack (equation (2)). The calculated values of f + for 3-МС were equal to 0.049 and 0.056 for the С-4' and С-12' atoms, respectively, in the ТТС–ТТТ form or 0.054 and 0.056 for the ТТТ–ТТТ form. Despite the fact that in the case of symmetric ТТТ–ТТТ isomer the activity of the two considered sites became practically equivalent, the calculation of Gibbs free energy indicated that the isomer ТТС–ТТТ was more stable by 0.4 kcal·mol–1. At the same time, this isomer was slightly more reactive according to the analysis of frontier orbitals' energies (Fig. 5b). In such a case, the С-12' atom became the preferred site of nucleophilic attack, while the mechanism likely included a quaternization of the previously neutral indoline fragment through redistribution of electron density in the molecule
upon hydrogen addition to the phenolate oxygen atom and the transfer of anion to it (Scheme 4). This process was enabled by the localization of extended conjugation chain in this area of the molecule (Fig. 5b).

The proposed mechanism for the hydrolysis of compound 3-МС
Thus, within the scope of this work, we obtained a new cationic spiropyran containing methoxy groups in its indolium rings. According to NMR spectra acquired in CDCl3, the merocyanine form of the compound was partially stabilized. During the attempts to grow crystals from EtOH, hydrolysis of the compound was observed, resulting in the formation of protonated merocyanine containing a free formyl group. According to the calculations of Fukui function values, the preferred site of nucleophilic attack by a molecule of H2O was the С-12' atom of the cationic vinyl-3Н-indolium moiety.
Experimental
IR spectra were recorded on a Varian Excalibur 3100 FT-IR spectrometer using the ATR technique. 1Н and 13С NMR spectra were acquired on a Bruker AVANCE 600 instrument (at 600 and 151 MHz, respectively). The residual signals of deuterated solvents were used as internal standards (7.26 ppm (CDCl3) and 2.50 ppm (DMSO-d6) for 1Н nuclei; 77.2 ppm (CDCl3) and 39.5 ppm (DMSO-d6) for 13C nuclei). High-resolution mass spectra were recorded on a Bruker maXis mass spectrometer, electrospray ionization, HCO2Na–HCO2H system was used for calibration. Melting points were determined on a Fisher–Johns apparatus (Thermo Fisher Scientific, USA).
5-Methoxy-1,2,3,3-tetramethyl-3 Н- indolium iodide (7). A solution of p-methoxyhydrazine hydrochloride (3.49 g, 20 mmol) in EtOH (20 ml) was treated by adding 3-methylbutan-2-one (2.15 ml, 20 mmol) and concd HClO4 (1.2 ml, 20 mmol). The mixture was heated at reflux for 3 h then 2/3 of the solvent were distilled off, the residue was neutralized with 20% NaOH solution, and the product was extracted with CHCl3. The extract was washed with distilled H2O, dried over anhydrous Na2SO4, and the solvent was removed by evaporation. Yield 3.15 g (83%), brown oil with a characteristic odor. The product was used in the next step without additional purification.
A solution of 5-methoxy-3,3-dimethyl-2-methyleneindole (3.15 g, 16.7 mmol) and MeI (1.56 ml, 25 mmol) in MeCN (10 ml) was heated at reflux for 3.5 h. The solution was then cooled, the precipitate that formed was filtered off and washed with cold MeCN. Yield 2.36 g (43%), pale-pink powder, mp 231–232°С. IR spectrum, ν, cm−1: 1617 (C=C), 1290 (C–N Ar), 1019 (С–O–C Ar). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.80 (1H, d, J = 8.8, H-7); 7.45 (1H, d, J = 2.5, H-4); 7.13 (1H, dd, J = 8.8, J = 2.5, Н-6); 3.91 (3H, s, N+–CH3); 3.84 (3H, s, OCH3); 2.68 (3H, s, C(2)–CH3); 1.49 (6H, s, 2С(3)–СН3). Found, %: С 47.17; Н 5.51; N 4.18; I 38.35. C13H18INO. Calculated, %: С 47.13; Н 5.44; N 4.23; I 38.37.
5-Methoxy-1,2,3,3-tetramethyl-3 Н- indolium perchlorate (1). Aqueous lead(II) perchlorate solution (498 mg, 1.23 mmol, 6 ml of solution) and an excess of HClO4 were gradually added with stirring to a solution of indolium iodide (812 mg, 2.5 mmol) in MeCN (10 ml). The precipitate of lead iodide was removed by filtration, the solvent was partially evaporated, and the solution was left until the precipitation of product. Yield 362 mg (48%), bordeauxcolored crystals, mp 178–179°С. IR spectrum, ν, cm−1: 1608 (C=C), 1293 (C–N Ar), 1016 (С–O–C Ar), 1069 (ClO4−). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.79 (1H, d, J = 8.8, H-7); 7.45 (1H, d, J = 2.4, H-4); 7.13 (1H, dd, J = 8.8, J = 2.5, H-6); 3.91 (3H, s, N+–CH3); 3.84 (3H, s, OCH3); 2.67 (3H, s, C(2)–CH3); 1.48 (6H, s, 2С(3)–СН3). Found, %: С 51.37; Н 5.95; N 4.58; Cl 11.63. C13H18INO5. Calculated, %: С 51.40; Н 5.93; N 4.61; Cl 11.70.
( E )-5-Methoxy-2-[2-(5-methoxy-1,3,3,6'-tetramethylspiro[chromene-2',2-indolin]-8'-yl)vinyl]-1,3,3-trimethyl-3 H -indolium perchlorate (3). 2-Hydroxy-5-methylisophthalic aldehyde 2 (0.164 g, 1 mmol) was dissolved in i-PrOH (10 ml), indolium perchlorate 1 (0.605 g, 2 mmol) was added and then Et3N (0.1 ml, 1 mmol) was added dropwise. The mixture was heated at reflux for 4 h. The precipitate was recrystallized from EtOH and washed with cold EtOH. Yield 0.415 g (65%), orange-red powder, mp 247–248°С. IR spectrum, ν, cm−1: 1605 (C=C), 1297 (C–N Ar), 1100 (ClO4−), 932 (C–О). Found, m/z: 535.2947 [M]+. C35H39N2O3. Calculated, m/z: 535.2955. Found, %: С 66.10; Н 6.19; N 4.38; Cl 5.51. C35H39ClN2O7. Calculated, %: С 66.19; Н 6.15; N 4.41; Cl 5.59.
SP form of compound 3. 1H NMR spectrum (СDCl3), δ, ppm (J, Hz): 7.98 (1H, d, J = 16.4, Н-12'); 7.53 (1H, s, H-7'); 7.47 (1H, d, J = 8.8, H-7”); 7.29 (1H, d, J = 16.4, H-13'); 7.06 (1H, s, H-5'); 7.01 (1H, dd, J = 8.8, J = 2.3, H-6”); 6.90 (1H, d, J = 2.2, H-4”); 6.86 (1H, d, J = 10.3, H-4'); 6.77 (1Н, dd, J = 8.4, J = 2.3, Н-6); 6.74 (1H, d, J = 2.4, H-4); 6.51 (1H, d, J = 8.3, H-7); 5.81 (1H, d, J = 10.3, H-3'); 3.87 (3H, s, OCH3); 3.83 (3H, s, OCH3); 3.72 (3H, s, N+–CH3); 2.69 (3H, s, N–СН3); 2.33 (3H, s, CH3); 1.40 (3H, s, С(3,3”)–СН3); 1.37 (3H, s, С(3,3”)–СН3); 1.27 (3H, s, С(3,3”)–СН3); 1.21 (3H, s, С(3,3”)–СН3). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.94 (1H, d, J = 16.5, H-12'); 7.78 (1H, d, J = 1.2, Н-7'); 7.75 (1H, d, J = 8.8, Н-7”); 7.35 (1H, d, J = 16.5, Н-13'); 7.29 (2H, m, Н-5',4”); 7.13 (1H, dd, J = 8.8, J = 2.5, Н-6”); 7.08 (1H, d, J = 10.3, Н-4'); 6.85 (1H, d, J = 2.6, Н-4); 6.77 (1H, dd, J = 8.4, J = 2.6, Н-6); 6.57 (1H, d, J = 8.4, Н-7); 5.94 (1H, d, J = 10.3, Н-3'); 3.87 (3H, s, OCH3); 3.78 (3H, s, OCH3); 3.64 (3H, s, N+–CH3); 2.63 (3H, s, N–СН3); 2.29 (3H, s, CH3); 1.35 (3Н, s, С(3,3”)–СН3); 1.34 (3H, s, С(3,3”)–СН3); 1.23 (3H, s, С(3,3”)–СН3); 1.15 (3H, s, С(3,3”)–СН3). 13C NMR spectrum (СDCl3), δ, ppm: 180.2 (C-2”); 161.4 (C-5”); 154.4 (C-5); 153.6 (C-9'); 148.3 (С-12'); 144.9 (C-9”); 142.2 (C-8); 138.2 (C-9); 134.9 (C-8”); 133.5 (С-5'); 131.6 (С-7'); 130.8 (C-6'); 129.4 (С-4'); 120.1 (C-8'); 119.8 (С-3', C-10'); 115.8 (С-7”); 114.3 (С-6”); 112.0 (С-13'); 111.9 (С-6); 109.6 (С-4); 108.8 (С-4”); 107.8 (С-7); 107.3 (C-2,(2')); 56.1 (OCH3); 52.3 (C-3”); 51.8 (C-3); 34.1 (N+–CH3); 29.5 (N–CH3); 26.8 (С(3,3”)–СН3); 26.6 (С(3,3”)–СН3); 25.4 (С(3,3”)–СН3); 20.2 (CH3); 20.0 (С(3,3”)–СН3).
MC form of compound 3. 1H NMR spectrum (СDCl3), δ, ppm (J, Hz): 8.47 (2H, d, J = 16.5, H-4',12'); 8.33 (2H, s, H Ar); 7.70 (2H, d, J = 16.5, Н-3',13'); 7.56 (2H, s, H Ar); 7.51 (2H, d, J = 8.8, H Ar); 7.03 (2H, d, J = 2.2, H Ar); 4.25 (6H, s, 2N+–CH3); 3.91 (6H, s, 2OCH3); 1.98 (3H, s, CH3); 1.79 (12H, s, 2С(3,3”)–СН3).
5'-Methoxy-1',3',3',6-tetramethylspiro[chromene-2',2-indoline]-8-carbaldehyde (6). 5-Methoxy-1,2,3,3-tetramethylindolium iodide 7 (331 mg, 1 mmol) was added to a solution of 2,6-diformyl-4-methylphenol 2 (164 mg, 1 mmol) in i-PrOH, followed by slow dropwise addition of Et3N (0.1 ml, 1 mmol). The mixture was heated at reflux for 4.5 h, then cooled. The product was extracted with CHCl3 and dried over anhydrous Na2SO4. After the residual solvent was evaporated, the crude product was separated by silica gel column chromatography using CHCl3 as eluent. The product was recrystallized from petroleum ether. Yield 0.018 g (5%), yellow crystals, mp 152–153°С. IR spectrum, ν, cm−1: 1675 (C=O), 1296 (C–N Ar), 919 (C–О). 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 10.10 (1H, s, CHO); 7.42 (1H, d, J = 1.9, Н-7); 7.07 (1H, d, J = 1.9, Н-5); 6.83 (1H, d, J = 10.3, Н-4); 6.67 (2H, m, Н-4',6'); 6.40 (1H, d, J = 8.0, Н-7'); 5.76 (1H, d, J = 10.3, Н-3); 3.77 (3H, s, ОСН3); 2.67 (3H, s, N–СН3); 2.25 (3H, s, CH3); 1.29 (3H, s, С(3)–СН3); 1.18 (3H, s, С(3)–СН3). 13C NMR spectrum (CDCl3), δ ppm: 188.4; 155.1; 153.5; 141.5; 137.4; 132.8; 128.6; 128.2; 126.8; 121.7; 119.7; 119.5; 110.8; 109.0; 106.5; 105.3; 55.4; 51.6; 28.8; 25.1; 19.7 (2С). Found, %: С 75.57; Н 6.59; N 3.95. C35H39ClN2O7. Calculated, %: С 75.62; Н 6.63; N 4.01.
( E )-2-[2-(3-Formyl-2-hydroxy-5-methylphenyl)vinyl]-5-methoxy-1,3,3-trimethyl-3 H -indolium perchlorate (5). A solution of spiropyran 6 (18 mg, 5·10–5 mol) in CHCl3 was treated with 1 drop (~0.01 ml) of concd HClO4. The solvent was evaporated under ambient atmosphere. Yield 0.015 g (65%), orange crystals, mp 244–245°С. IR spectrum, ν, cm−1: 1642 (C=O), 1601 (C=C), 1294 (C–N Ar), 1065 (ClO4−). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 11.70 (1Н, s, ОН); 10.08 (1H, s, СНО); 8.32 (1H, d, J = 16.6, Н-12'); 8.29 (1H, d, J = 1.9, H-7'); 7.81 (2H, d, J = 8.8, H-5',7”); 7.70 (1H, d, J = 16.6, H-13'); 7.49 (1H, d, J = 2.4, Н-4”); 7.16 (1H, dd, J = 8.8, J = 2.4, Н-6”); 4.08 (3H, s, N+–CH3); 3.86 (3H, s, ОСН3); 2.37 (3H, s, CH3); 1.73 (6H, s, 2С(3)–СН3). 13C NMR spectrum (DMSO-d6), δ, ppm: 196.1 (СНО); 179.4 (С-2”); 161.0 (С-5”); 158.1 (С-9'); 145.6 (С-10'); 143.5 (С-12'); 137.6 (С-5'); 136.6 (С-7'); 135.1 (C-8”); 129.7 (С-6'); 122.9 (C-8'); 122.1 (C-9”); 116.4 (С-7”); 114.8 (С-6”); 114.1 (С-13'); 108.6 (С-4”); 56.1 (ОСН3); 52.0 (C-3”); 34.6 (N+–CH3); 25.6 (С(3)–СН3); 19.6 (CH3). Found, %: С 58.62; Н 5.78; N 3.05; Cl 7.64. C22H24ClNO7. Calculated, %: С 58.73; Н 5.34; N 3.11; Cl 7.90.
X-ray structural analysis of compound 5. The unit cell parameters for the crystal and the three-dimensional array of intensities were determined on an Xcalibur automatic diffractometer (MoKα radiation, graphite monochromator, EOS detector). Absorption was accounted for empirically according to the Multiscan procedure. The structure was solved by direct method and refined according to fullmatrix method of least squares by F2 using the SHELXTL program in anisotropic approximation for non-hydrogen atoms. The majority of H atoms were located in the crystal structure by difference Fourier synthesis of the electron density, then the coordinates and isotropic temperature parameters of all Н atoms were calculated by the method of least squares using the riding model.44 The analysis and visualization of X-ray crystallography results were performed by using the Mercury software suite.45 The complete crystallographic dataset was deposited at the Cambridge Crystallographic Data Center (deposit CCDC 2182739).
The Hirschfeld surfaces were calculated at the DFTB3LYP level with 6-311G(d,p) basis set, using the CrystalExplorer v21.5 program.46
Quantum-chemical modeling was performed with the Orca 5.0.3 software47,48 at the density functional theory level, using a three-parameter Lee–Yang–Parr functional (B3LYP)49 and 6-311G(d,p) basis set.50 The C-PCM solvation model was applied (EtOH). The character of the identified critical points was established by analytical calculation of the matrix of constants. The Hirschfeld analysis of atomic populations was performed with the Multiwfn software.51 Visualization of the results was achieved with the Chemcraft program.52
Supplementary information file containing 1Н and 13С NMR, COSY, 1Н–13С HSQC, 1Н–13С HMBC spectra, as well as the high-resolution mass spectrum of compound 3 and X-ray structural analysis results for compound 5 is available at the journal website http://springerlink.bibliotecabuap.elogim.com/journal/10593.
References
Zhao, W.; Quan, M.; Cao, Z.; Zhang, Y.; Wen, J.; Pan, D.; Dong, Z.; Yang, Z.; Wang, D.; Cao, H.; He, W. Colloids Surf., A 2018, 554, 93.
Xia, Z.; Alphonse, V. D.; Trigg, D. B.; Harrigan, T. P.; Paulson, J. M.; Luong, Q. T.; Lloyd, E. P.; Barbee, M. H.; Craig, S. L. Molecules 2019, 24, 542.
Bispo, M.; van Dijl, J. M.; Szymanski, W. In Molecular Photoswitches: Chemistry, Properties, and Applications; Pianowski, Z. L., Ed.; WILEY-VCH GmbH: Weinheim, 2022, p. 843.
Sauvage, J.-P. Angew. Chem., Int. Ed. 2017, 56, 11080.
Feringa, B. L. Angew. Chem., Int. Ed. 2017, 56, 11060.
Stoddart, J. F. Angew. Chem., Int. Ed. 2017, 56, 11094.
Klajn, R. Chem. Soc. Rev. 2014, 43, 148.
Kozlenko, A. S.; Pugachev, A. D.; Ozhogin, I. V.; El-Sewify, I. M.; Lukyanov, B. S. Chem. Heterocycl. Compd. 2021, 57, 984.
Lukyanov, B. S.; Lukyanova, M. B. Chem. Heterocycl. Compd. 2005, 41, 281.
Wizinger, R.; Wenning, H. Helv. Chim. Acta 1940, 43, 247.
Kortekaas, L.; Browne, W. R. Chem. Soc. Rev. 2019, 48, 3406.
Pugachev, A. D.; Mukhanov, E. L.; Ozhogin, I. V.; Kozlenko, A. S.; Metelitsa, A. V.; Lukyanov, B. S. Chem. Heterocycl. Compd. 2021, 57, 122.
Laptev, A. V.; Lukin, A. Y.; Belikov, N. E.; Zvezdin, K. V.; Demina, O. V.; Barachevsky, V. A.; Varfolomeev, S. D.; Khodonov, A. A.; Shvets, V. I. Russ. Chem. Bull. 2014, 63, 2026.
Nikolaeva, O. G.; Metelitsa, A. V.; Cheprasov, A. S.; Karlutova, O. Y.; Starikov, A. G.; Dubonosov, A. D.; Bren', V. A.; Minkin, V. I. Russ. Chem. Bull. 2016, 65, 944.
Frolova, L. A.; Rezvanova, A. A.; Lukyanov, B. S.; Sanina, N. A.; Troshin, P. A.; Aldoshin, S. M. J. Mater. Chem. C 2015, 3, 11675.
Bénard, S.; Rivière, E.; Yu, P.; Nakatani, K.; Delouis, J. F. Chem. Mater. 2001, 13, 159.
Bazzan, I.; Bolle, P.; Oms, O.; Salmi-Mani, H.; Aubry-Barroca, N.; Dolbecq, A.; Serier-Brault, H.; Dessapt, R.; Philippe R.; Mialane, P. J. Mater. Chem. C 2017, 5, 6343.
Kida, N.; Hikita, M.; Kashima, I.; Okubo, M.; Itoi, M.; Enomoto, M.; Kato, K.; Takata, M.; Kojima, N. J. Am. Chem. Soc. 2009, 131, 212.
Pugachev, A. D.; Ozhogin, I. V.; Lukyanova, M. B.; Lukyanov, B. S.; Rostovtseva, I. A.; Dorogan, I. V.; Makarova, N. I.; Tkachev, V. V.; Metelitsa, A. V.; Aldoshin, S. M. Spectrochim. Acta, Part A 2020, 230, 118041.
Khalanskiy, K. N.; Alekseenko, Y. S.; Lukyanov, B. S.; Borodkin, G. S.; Bezuglyi, S. O. Chem. Heterocycl. Compd. 2012, 48, 1090.
Funasako, Y.; Miyazaki, H.; Sasaki, T.; Goshima, K.; Inokuchi, M. J. Phys. Chem. B 2020, 124, 7251.
Pugachev, A. D.; Lukyanova, M. B.; Lukyanov, B. S.; Ozhogin, I. V.; Kozlenko, A. S.; Rostovtseva, I. A.; Makarova, N. I.; Tkachev, V. V.; Aksenov, N. A. J. Mol. Struct. 2019, 1178, 590.
Kozlenko, A. S.; Makarova, N. I.; Ozhogin, I. V.; Pugachev, A. D.; Lukyanova, M. B.; Rostovtseva, I. A.; Borodkin, G. S.; Stankevich, N. V.; Metelitsa, A. V.; Lukyanov, B. S. Mendeleev Commun. 2021, 31, 403.
Pugachev, A. D.; Ozhogin, I. V.; Makarova, N. I.; Rostovtseva, I. A.; Lukyanova, M. B.; Kozlenko, A. S.; Borodkin, G. S.; Tkachev, V. V.; El-Sewify, I. M.; Dorogan, I. V.; Metelitsa, A. V.; Aldoshin, S. M.; Lukyanov, B. S. Dyes Pigm. 2022, 199, 110043.
Pugachev, A. D.; Ozhogin, I. V.; Lukyanova, M. B.; Lukyanov, B. S.; Kozlenko, A. S.; Rostovtseva, I. A.; Makarova, N. I.; Tkachev, V. V.; Aldoshin, S. M.; Metelitsa, A. V. J. Mol. Struct. 2021, 1229, 129615.
Lukyanova, M. B.; Tkachev, V. V.; Lukyanov, B. S.; Pugachev, A. D.; Ozhogin, I. V.; Komissarova, O. A.; Aldoshin, S. M.; Minkin, V. I. J. Struct. Chem. 2018, 59, 565.
Balmond, E. I.; Tautges, B. K.; Faulkner, A. L.; Or, V. W.; Hodur, B. M.; Shaw, J. T.; Louie, A. Y. J. Org. Chem. 2016, 81, 8744.
Tkachev, V. V.; Lukyanova, M. B.; Lukyanov, B. S.; Pugachev, A. D.; Aldoshin, S. M.; Minkin, V. I. J. Struct. Chem. 2016, 57, 1270.
Allen, F. H.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R. In International Tables for Crystallography. Volume C: Mathematical, Physical and Chemical Tables; Prince, E., Ed.; Springer: Dordrecht, 2006, p. 803.
Jelsch, C.; Ejsmont, K.; Huder, L. IUCrJ 2014, 1, 119.
Seiler, V. K.; Callebaut, K.; Robeyns, K.; Tumanov, N.; Wouters, J.; Champagne, B.; Leyssens, T. CrystEngComm 2018, 20, 3318.
Brieke, C.; Heckel, A. Chem.–Eur. J. 2013, 19, 15726.
Hammarson, M.; Nilsson, J. R.; Li, S.; Beke-Somfai, T.; Andréasson, J. J. Phys. Chem. B 2013, 117, 13561.
Stafforst, T.; Hilvert, D. Chem. Commun. 2009, 3, 287.
Cho, Y. J.; Lee, S. H.; Bae, J. W.; Pyun, H.-J.; Yoon, C. M. Tetrahedron Lett. 2000, 41, 3915.
Luk'yanova, M. B.; Pugachev, A. D.; Tkachev, V. V.; Luk'yanov, B. S.; Shilov, G. V.; Kozlenko, A. S.; Rostovtseva, I. A.; Minkin, V. I.; Aldoshin, S. M. Dokl. Chem. 2018, 482, 220.
Pugachev, A. D.; Lukyanova, M. B.; Lukyanov, B. S.; Ozhogin, I. V.; Kozlenko, A. S.; Tkachev, V. V.; Chepurnoi, P. B.; Shilov, G. V.; Minkin, V. I.; Aldosin, S. M. Dokl. Chem. 2020, 492, 76.
Tian, W.; Tian, J. Dyes Pigm. 2014, 105, 66.
Kim, D.; Zhang, Z.; Xu, K. J. Am. Chem. Soc. 2017, 139, 9447.
Sheng, Y.; Leszczynski, J.; Garcia, A. A.; Rosario, R.; Gust, D.; Springer, J. J. Phys. Chem. B 2004, 108, 16233.
Yang, W.; Parr, R. G.; Pucci, R. J. Chem. Phys. 1984, 81, 2862.
Hirshfeld, F. L. Theor. Chim. Acta 1977, 44, 129.
Wang, B.; Rong, C.; Chattaraj, P. K.; Liu, S. Theor. Chem. Acc. 2019, 138, 123.
Sheldrick, G. M. SHELXTL v. 6.14, Structure Determination Software Suite; Bruker AXS: Madison, 2000.
Macrae, C. F.; Sovago, I.; Cottrell, S. J.; Galek, P. T. A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G. P.; Stevens, J. S.; Towler, M.; Wood, P. A. J. Appl. Crystallogr. 2020, 53, 226.
Spackman, P. R.; Turner, M. J.; McKinnon, J. J.; Wolff, S. K.; Grimwood, D. J.; Jayatilaka, D.; Spackman, M. A. J. Appl. Crystallogr. 2021, 54, 1006.
Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. J. Chem. Phys. 2020, 152, 224108.
Neese, F. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73.
Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
Hehre, W. J.; Random, L.; Schleyer, P. v. R.; Pople, J. A. Ab Initio Molecular Orbital Theory; Wiley: New York, 1986.
Lu, T.; Chen, F. J. Comput. Chem. 2012, 33, 580.
Chemcraft – Graphical Software for Visualization of Quantum Chemistry Computations. https://www.chemcraftprog.com
This study was supported by a grant from the Ministry of Science and Higher Education of the Russian Federation within the framework of State Contract for scientific research (Southern Federal University, No. 0852-2020-00-19). The X-ray structural analysis was performed in accordance with State Contract, registry No. АААА-А19-119092390076-7 (V. V. Tkachev, S. M. Aldoshin).
1Н and 13С NMR spectra were acquired at the Molecular Spectroscopy Collective Use Center of the Southern Federal University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2022, 58(12), 712–720
Supplementary Information
ESM 1
(PDF 801 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kozlenko, A.S., Pugachev, A.D., Ozhogin, I.V. et al. Synthesis and structural characterization of new spiropyran containing conjugated vinyl-3Н-indolium moiety and its hydrolysis product. Chem Heterocycl Comp 58, 712–720 (2022). https://doi.org/10.1007/s10593-023-03147-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-023-03147-5