
Abstract
In this paper, we have studied within the molecular electron density theory the mechanism and the origin of the selectivity of the [3 + 2] cycloaddition reaction between electrophilic nitrone 1 and nucleophilic alkene, indole 2, using DFT method at the B3LYP/6-31G(d) theoretical level. The obtained results clearly indicated that this reaction is characterised by an ortho regioselectivity and an exo stereoselectivity. Inclusion of the solvent effects does not modify the obtained gas-phase selectivities but slightly increase the activation energies. The analysis of the geometries of the transition states with bond order and global electron density transfer shows that this reaction takes place via a slightly asynchronous non-polar one-step mechanism. Analysis of the electronic structure of nitrone 1 indicates that this species has a zwitterionic-type structure that enables its participation in zw-type 32CA reactions. Conceptual DFT reactivity indices analysis confirms the highly obtained activation energies and local Parr functions analysis allows us to explain the ortho regioselectivity of this 32CA reaction. NCI, ESP and QTAIM analyses indicate that the presence of several conventional and non-conventional interactions at the structure of the most favourable approach ortho–exo is the origin for the selectivity of this 32CA reaction.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Heterocyclic compounds are of paramount importance in many regards. They are present in small as well as large and complex molecules and have broad applications. Natural products such as vitamins, alkaloids, macrocycles and flavonoids are in the base of heterocycles [1,2,3]. Heterocycles are also found in a wide variety of biologically active synthetic compounds such as pharmaceuticals and agrochemicals. As a consequence of its ability to form various non-covalent interactions with the biological target, more than 90% of new drugs contain heterocycles and the interface between chemistry and biology, at which so much new scientific insight, discovery and application are taking place crossed by heterocyclic compounds [4, 5].
Isoxazolidines are five-membered saturated heterocycles containing N–O bond, and they have attracted considerable attention due to their potential biological activities and are also used as precursors, especially for β-amino alcohols through reductive cleavage of the N–O bond. Some isoxazolidines possess medicinal activities such as antibacterial, anticonvulsant, antibiotic, antitubercular and antifungal activities [6, 7].
Today, the development of new, simple and efficient synthetic procedures for enantioselective chemical transformations is still a tremendous challenge. However, these heterocycles can be obtained through one of the simplest approaches that is the 1,3-dipolar cycloaddition (32CA) of nitrones with alkenes (Scheme 1) [8].

Synthesis of isoxazolidines by 32CA reaction between nitrones and alkenes
The literature tells us that the majority of these reactions lead to the formation of a single isomer or in other cases, a mixture of two isomers, in which one is obtained in a major amount among four possible isomers [9,10,11,12].
In continuation of our previous studies [13,14,15,16], for the understanding the origin of the selectivity and the mechanism nature of cycloaddition reactions, in this work, we focused to perform a theoretical study of the regio- and stereoselectivities as well as the molecular mechanism of the 32CA performed by Dmitriev et al. [17] between ethyl 3- (4,5,6,7-tetrahydro-1H-indol-1-yl) acrylate as alkene named as indole 2 and N- (2-oxo-2)-(phenylamino) ethylidene) aniline oxide, named as nitrone 1 (Scheme 2). Nitrones are zwitterionic TACs [18]. Usually, due to the nucleophilic character of nitrones, they react with electrophilic alkenes with high meta–endo selectivity, which depends on of the polar character of the reactions [19]. Interestingly, the studied 32CA reaction between the electrophilic nitrone 1 and the nucleophilic indole 2 presents an opposite ortho–exo selectivity. Indole 2, which is a derivative of methyl acrylate, is a captodative ethylene. However, due to the poor electron-withdrawing character of the carboxylate group indole 2 acts as a strong nucleophilic species. The main goal is to shed light on the factors that controlled the regio- and stereoselectivities, as well as the molecular mechanism of this 32CA reaction.

32CA reaction between nitrone 1 and indole 2
2 Computational details
The program Gaussian 09 [20] is the software used to optimise all the structures involved in this study. The quantum DFT methods with the functional hybrid B3LYP and the 6-31G(d) basis set [21,22,23,24] are the used theoretical level for performing this study. The frequency calculations were used to confirm the nature of the stationary points, in which only one imaginary frequency corresponding to the new forming bonds was obtained in the transition states. The NBO method [25, 26] is used to analyse the electronic structures of the transition states. The effects of toluene solvent were considered through single-point calculations of the optimised gas-phase structures using the polarisable continuum model (PCM) developed by the Tomasi group. [24, 27] within the self-consistent reaction field (SCRF) [28,29,30] Values of Gibbs free energies, enthalpies and entropies were calculated using standard statistical thermodynamics at 383 K and 1 atmosphere over the optimised gas-phase structures [22].The electrophilic index ω [31] is given by the following formula, ω = (μ2/2η), and for the electronic chemical potential μ, and the overall hardness η can be calculated from the energies of the frontier molecular orbitals εHOMO and εLUMO, respectively, as μ = (εHOMO + εLUMO)/2 and η = εLUMO − εHOMO [32, 33].On the other hand, the nucleophilic index [34, 35], N, obtained from the HOMO energies through the formula N = εHOMO(Nu) − εHOMO(TCE), in which the reference is tetracyanoethylene (TCE). The \(P_{K}^{ + }\) electrophilic and \(P_{K}^{ - }\) nucleophilic Parr functions [36] which enable characterisation of the electrophilic and nucleophilic centres of a molecule were obtained by the analysis of the Mulliken atomic spin density of the radical anion and the radical cation, respectively, of the molecule studied.
Non-covalent interaction (NCI) analysis was performed by evaluating the reduced density gradient and low-gradient isosurfaces [37, 38] were performed using NCI plot [39], while ESP [40] and QTAIM [41] were performed with the Multiwfn [42] program, using the corresponding B3LYP/6-31G(d,p) monodeterminantal wave functions.
The global electron density transfer (GEDT) [43] was computed by the sum of the natural atomic charges (q), obtained by a natural population analysis (NPA) [25, 26], Topological analysis of the electron localisation function (ELF) [44] was performed with the Multiwfn [42] package using the corresponding B3LYP/6-31G(d) monodeterminantal wave functions., while the representation of the ELF basin isosurfaces was done by using the UCSF Chimera program [45].
3 Results and discussion
This part has been divided into four parts: first, the electronic structure of the nitrone 1 and the simplest nitrone is explored in terms of ELF and NPA analyses. In the second part, we have studied the reactivity and the regioselectivity of this 32CA reaction using conceptual DFT (CDFT) indices [46, 47] and local Parr indices [36], respectively. The third part is dedicated to the analysis of different energy profiles of the possible reactive pathways associated with this 32CA reaction, as well as the geometries of the transition states, bond order and GEDT. Finally, we have used ESP, NCI and QTAIM analyses in order to unravelling the origin of the exo–ortho selectivity of the studied 32CA reaction.
3.1 ELF and NPA characterisation of the electronic structure of the simplest nitrone and nitrone 1
Molecular electron density theory (MEDT) [48] studies devoted to 32CA reactions have shown a good correlation between the electronic structure of TAC such as nitrones and their reactivity towards ethylenes [49]. Therefore, from the electronic structure of the TACs, the non-polar 32CA reactions may be classified into pseudodiradical-type (pdr-type), pseudoradical-type (pmr-type), carbenoid-type (cb-type) and zwitterionic-type (zw-type) reactions [50].
An attractive procedure that has given us a direct link between the distribution of electron density and the chemical structure is the quantum chemical topological analysis of ELF established by Becke and Edgecombe [44]. Therefore, for the characterisation of the electronic structure of the nitrone 1 and its comparison with that of the simplest nitrone counterpart, we have performed a topological analysis of the ELF of both TACs.
ELF localisation domains and their attractor positions, together with the most representative valence basin populations, and the proposed ELF-based Lewis structure, together with the natural atomic charges, are given in Fig. 1.

a ELF localisation domains, represented with an isosurface value of ELF = 0.84; b ELF basin attractor positions, together with the most representative valence basin populations; c the proposed ELF-based Lewis structures, together with the natural atomic charges of the most relevant atoms; for simplest nitrone and nitrone 1. Natural atomic charges are given as average number of electrons, e
The ELF topology of the simplest nitrone 1 has been studied [51]. For our case, we have obtained the almost same results except in some negligible differences in the basin populations due to employed theoretical method.
Within the context of ELF, monosynaptic basins V(X) are corresponding to non-bonding regions, while disynaptic basins V(X,Y) are related to bonding regions [52].
From Fig. 1, the simplest nitrone presents two V(O) and V′(O) monosynaptic basins, integrating 3.02 and 2.82e, and one V(O,N) disynaptic basin with a population of 1.37e. This behaviour suggests that the O–N bonding region is strongly polarised towards the oxygen atom. In addition, the presence of one V(N,C) disynaptic basin integrating 3.92e indicates that the C–N bonding region has a strong double-bond character. Consequently, the ELF topology of the simplest nitrone clearly indicates that this TAC is able to participate only in zw-type 32CA reactions.
Thus, by relating the ELF electron populations with the Lewis bonding model [53], the V(O) and V′(O) monosynaptic basins can be associated with oxygen ‘lone pairs’, the V(N,O) disynaptic basin with an N–O single bond, and the V(C,N) disynaptic basin with a C=N double bond (see the proposed ELF-based Lewis structures in Fig. 1).
On the other hand, the ELF topology of nitrone 1 shows a similar bonding pattern to that found in the simplest nitrone. Indeed, the substitutions of the hydrogen atom linked to the nitrogen by a phenyl group and to the carbon by a phenylcarbamoyl group produce a slightly significant change in the ELF valence basin populations (see Fig. 1). Although in this case the C3 atom (see Scheme 2 for atom numbering) is characterised by the presence of monosynaptic basin, V(C3), integrating a population of 0.21e, it does not appear in the ELF localisation domains, indicating that it is not a pseudoradical centre. Therefore, according to the ELF topological analysis, nitrone 1 will behave as a zwitterionic TAC participating only in zw-type 32CA reactions such as the simplest nitrone [48]. Although the ELF topological analysis of nitrones 1 allows the establishment of a bonding pattern in these TACs, NPA indicates that neither nitrone has zwitterionic charge distribution.
Note that although the oxygen has a high negative charge, − 0.462e (simplest nitrone) and − 0.502e (nitrone 1), the nitrogen presents almost no charge, − 0.015e (simplest nitrone) and -0.025e (nitrone 1). Moreover, the carbon appears negatively charged at nitrone 1, − 0.195e, while at nitrone 1 it presents almost a null charge, − 0.045e. Thus, although ELF topological analysis provides a bonding pattern concordant with the commonly accepted Lewis structures of nitrone 1 and the simplest nitrone, the NPA is completely in disagreement with the representation of their electronic structure as 1,2-zwitterionic structure. Nevertheless, ELF topological characterisation of the electron density distribution at these nitrones accounts for their zw-type reactivity.
3.2 Analysis of the conceptual DFT indices at the ground state of the reagents
Table 1 summarises the global reactivity indices, including the electronic chemical potential (μ), the chemical hardness (η), global electrophilicity index (ω) and the nucleophilic index, N, in eV, for nitrone 1 and indole 2.
We can notice from Table 1 that the electronic chemical potential μ of the indole 2, − 3.44 eV, is higher than that of the nitrone 1, − 4.01 eV, indicating that at the transition states, the global electron density transfer (GEDT) [43] will take place from the indole 2 framework towards the nitrone 1 one. The 2.30 eV and 1.32 eV values of electrophilic index of the nitrone 1 and indole 2, respectively, indicate that these reagents are strong electrophiles and on the basis of the electrophilic scale [54]. On the other hand, the nucleophilic indices for indole 2 and nitrone 1 are 3.43 and 3.35 eV, respectively, allow us to classify them as strong nucleophiles [55]. In addition, the low electrophilicity difference [54] (Δω = 0.99 eV) and the very low nucleophilicity difference (ΔN = 0.08 eV) clearly indicate that this cycloaddition reaction has occurred between two reagents with the same electronic nature. Based on these facts, we can conclude that this 32CA reaction required high activation energy, represented by the high temperature (T = 110 °C), and explain the low values of GEDT which account for the non-polar character of the studied 32CA reaction.
The local indices calculated with Parr functions together with the maps of atomic spin density (ASD) of the ions, radical anion of nitrone 1 and the radical cation of the indole 2 are given in Fig. 2. An analysis of the electrophilic \(P_{K}^{ + }\) Parr function of the nitrone 1 indicates that the carbon atom C3 (see Scheme 2 for atom numbering) is the most electrophilic centre with, \(P_{K}^{ + }\) = 0.208. On the other hand, the nucleophilic \(P_{K}^{ - }\) Parr function of indole 2 indicates that the carbon atom C5 is the most nucleophilic centre with a value, \(P_{K}^{ - }\) = 0.116. According to the rule that states the most favourable nucleophilic/electrophilic interaction will occur between the most electrophilic centre of the electrophile and the most nucleophilic centre of the nucleophile [56], this 32CA leads to the formation of ortho regioisomers, in agreement with experimental data.

Maps of the ASD of the radical cation of indole 2 and the radical anion of nitrone 1, together with the nucleophilic \(P_{K}^{ - }\) and electrophilic \(P_{K}^{ + }\) indices
3.3 Energies and geometries
In this 32CA reaction, the reagents can follow two regioisomeric reactive channels, namely the ortho and meta pathways, and in each regioisomeric pathway, there are two possible sterioisomeric approaches of the two reagents, one to the other, the endo and exo one. Thereby, in this 32CA reaction, there are four transition states, named TSmn, TSmx, TSon and TSox, leading to the corresponding four possible cycloadducts, CAmn, CAmx, CAon and CAox, respectively, which have been located and characterised (Scheme 3). The relative energies in the gas phase and in the toluene solvent of the stationery points involved in this 32CA reaction are collected in Table 2, while the total ones are collected in Table S1 in Supporting Information. The Cartesian coordinates of all the stationary points and imaginary frequencies for the transition states involved in the studied 32CA reaction are also given in Supporting Information.

The possible reactive paths for the 32CA reaction between nitrone 1 and indole 2
From Table 2, we can notice by a comparison between the activation energies of the gas phase associated with the four competitive pathways of this 32CA that the ortho–exo (Ea = 23.60 kcal/mol) is kinetically favoured cycloadduct, in which ΔΔE = 4.58 kcal/mol than the second more favoured one (meta–exo). In addition, we notice that the pathways ortho–exo and meta–exo are irreversible (ΔE < 0), while the ortho–endo and meta–endo ones are reversible (ΔE > 0). Consequently, the kinetically product is a stable cycloadduct, and thereby the reaction is only under a kinetic control. In order to study the effect of solvent–solute interactions along the reaction pathways, further calculations were also performed taking into account the solvent toluene. From the values of relative energies of both TSs and CAs in solution phase, we notice a slight increase in the activation energies and slight decrease in the exothermic character of these pathways. These changes are due to the better solvation of the reagents than the TSs and CAs [57].
As zwitterionic species, nitrones participate in zw-type 32CA reactions with high activation energies, which decrease with the polar character of the reaction [18]. In the case of nucleophilic nitrones, the relative energies of the meta–endo TS range from -4 to 14 kcal/mol, depending on the electrophilic character of the ethylene [19]. The 32CA reaction between nitrone 1 and indole 2 presents a very high relative energy of 23.6 kcal/mol (see Table 1). This energy is even higher than that of the 32CA of azides [58], which are very unfavourable.
Figure 3 presents the geometries of the optimised structures of transition states associated with the 32CA reaction of the indole 2 with the nitrone 1 together with the lengths of new forming bonds.

Optimised structures of the TSs of the 32CA reaction of indole 2 with nitrone 1 together with the lengths of the new forming bonds (in Å). GEDT direction and values, in red and the BO in blue
The lengths of the new forming bonds O–C and C–C are 2.161 and 1.986 Å for the TSmx and 2.042 and 2.164 Å for the TSmn, respectively. For the ortho pathways, the lengths of the O–C and C–C bonds in the case of TSox are 1.924 and 2.222 Å and for the TSon are 2.112 and 2.115 Å. Taking into account that the formation of the C–C single bonds takes place in the range of 2.0–1.9 Å, while formation of the C–O single bond takes place in the range of 1.8 and 1.7Å [18], the obtained values account for a slightly asynchronous mechanism for all reactive paths, in which the formation of C–C new bond is advanced than that of the C–O one, except the more favoured TSox, in which we notice the reverse.
On the other hand, in order to confirm this mechanism nature, further, analysis using B3LYP/6-31G(d) Wiberg bond indices [59] was computed using the NBO method.
The BO values for the C–C and O–C forming bonds at the TS associated with this 32CA reaction are 0.43 and 0.33 at TSon, 0.38 and 0.43 at TSox, 0.41 and 0.38 at TSmn and 0.52 and 0.31 at TSmx. These values suggest that the bond formation process is slightly asynchronous, in which the formation of the C–C bond is slightly more advanced than that of the C–C one. Note that at the most favourable TSox, the formation of the O–C new bond is advanced than that of the C–C one.
Since most part of organic reactions occur in liquid media and required some other enhancement such as heating, and in order to take into account the experimental condition of this 32CA reaction, a further calculation of the thermodynamic properties such as enthalpy, entropy and free energy is necessary to include the effect of temperature, pressure and nature of solvent. The values of relative enthalpies, entropies and free energies are collected in Table 3, while the total ones are given in Table S2 in Supporting Information.
From Table 3, the inclusion of thermal parameters in calculation did not modify the previously obtained ortho–exo selectivity, in which the enthalpy values clearly indicate that the ortho–exo (26.32 kcalmol−1) is the most favoured. In addition, all pathways become having an endothermic character except the CAmx which possesses a little exothermic character. On the other hand, taking into account the entropic contribution, calculations gave high values of activation free energies which are increased by 18.93, 18.42, 16.98 and 21.65 with respect to the activation enthalpies for TSmn, TSmx, TSon and TSox, respectively. This increase is due to the negative values of the entropy related to the bimolecular character and the voluminous geometry of the studied system. These high values of free energies explain well the experimental conditions of this 32CA reaction and the observed regio- and stereoselectivities.
The negative sign of GEDT calculated at the nitrone 2 framework indicates that the direction of the electron density flux takes place from indole 2 towards nitrone 1. The values of the GEDT are 0.060e (TSon), 0.001e (TSox), 0.0134e (TSmn) and 0.040e (TSmx). These very low values indicate that this zw-type 32CA reaction has a non-polar character, in agreement with the obtained high activation energies [60].
3.4 Origin of the ortho–exo selectivity
In addition to the functional groups of the reagents, during the reaction, there is a GEDT from the nucleophilic framework towards the electrophilic framework, which gives positive regions and other regions negatively charged at both reagents. Therefore, the structure of the transition states as well as cycloadducts may have some non-covalent interactions that stabilised these structures. In this section, we have performed an investigation in order to confirm and to determine the presence and the nature of these interactions at the most favourable transition states.
3.4.1 Electrostatic surface potential (ESP) analysis
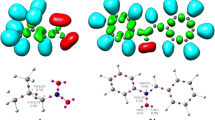
Previous studies indicate that the more favourable relative orientation of both polarised frameworks at the TSs, the stronger the electrostatic interactions [61, 62]. In addition, ESP has been widely used for the prediction of nucleophilic and electrophilic sites for a long time. It is also valuable in studying hydrogen bonds, halogen bonds, molecular recognitions and the intermolecular interaction of aromatics [40]. Figure 4 shows the ESPs of the most favoured TSox and the second most favoured TSmx. As can be seen, at both structures, the nitrone 1 framework gathers the more intense blue region (negative charge) of the ESP, indicating that it received some electron density from the indole 2 framework, while the ESP region around the indole 2 is the almost reddest (positive charge) within these molecules.

Electrostatic potential surfaces of TSox and TSmx
The structure of TSox is situated in a rather extended way that may establish many favourable electrostatic interactions. Consequently, this disposition favours the formation of electrostatic interactions between both nucleophilic and electrophilic frameworks with respect to the TSmx, thus justifying the preference for the ortho–exo stereoselectivity in the 32CA reaction between nitrone 1 and indole 2.
Analysis of the ESP of TSmx indicates that the bluest region is almost concentrated in one region of the structure gathering the most negative charge at the nitrone 1 fragment facing the reddest region at the indole 2 fragment, which may create some electrostatic interactions, but fewer than that presented at TSox. Therefore, the presence of several favourable electrostatic interactions taking place at TSox participates for the enhancing of the ortho–exo stereoselectivity of this 32CA reaction between nitrone 1 and indole 2.
3.4.2 NCI analysis
Our previous studies indicate that non-covalent interactions may be the principal factor responsible for the stereoselectivity of the 32CA reactions [63,64,65]. Thus, an analysis of the structure of the favourable approaches, TSox and TSmx, reveals that one hydrogen atom (N–H) of the amide function of nitrone 1 fragment is located towards the oxygen atom of the carbonyl function of indole 2 fragment, in which this O–H distance is 1.93 and 1.84 Å, respectively. Therefore, these hydrogen bonds (HBs) may be formed between these atoms, which enhance the stabilisation of both structures. Thus, an NCI analysis of the structure of TSox and TSmx was performed. Figure 5 shows the reduced density gradient for TSox and TSmx.

NCI gradient isosurfaces of TSox and TSmx
An analysis of Fig. 5 indicates the presence of surface with a light blue colour between the oxygen and hydrogen atoms in TSox and TSmx, which confirm the presence of the predicted previous hydrogen bonds. In addition, the presence of several surfaces with green and turquoise colours is an indicator of the presence of several weak non-covalent interactions in both structures.
In order to distinguish between the non-covalent interactions presented at both TSox and TSmx, further analysis becomes necessary. Thus, an analysis of the reduced density gradient was performed, and the low-gradient isosurfaces for TSox and TSmx are displayed in Fig. 6. The low-gradient isosurfaces of both transition states indicate the presence of a low-density gradient spike in the borderline of − 0.02 a.u, accounting for the formation of an hydrogen bond. The spikes at ca. − 0.06 and − 0.08 a.u. are attributed to the strong attractive interactions related to the formation of the new bonds. The main difference between these isosurfaces is the high number of spikes at TSox in the borderline of 0 a.u which is attributed to weak non-conventional HBs.

Plots of the reduced density gradient (RDG) versus the electron density multiplied by the sign of the second Hessian eigenvalue for TSox and TSmx
3.4.3 QTAIM analysis
QTAIM analysis of the electron density ρ provides a series of critical points (cps), which are points of the molecular space where ∇ρ(r) = 0 [41]. The presence of a (3, − 1) cp appears to provide the boundaries between the basins of neighbouring atoms, being called a bonding cp (bcp). Thus, the existence of a (3, − 1) bcp and its associated atomic interaction line indicates that electron density is accumulated between the nuclei linked in such a manner. Several studies indicated that the strength of interaction, particularly of HB, is expressed by the characteristics of its bonding critical point (bcp) [66, 67]. As proposed by Rosas et al. [66], HBs interactions could be classified according to three types, in which strong HBs are characterised by a Laplacian, ∇2ρbcp < 0 and a total electron energy density, Hbcp < 0, a medium strength HBs are characterised by a, ∇2ρbcp < 0 and Hbcp > 0, while weak strength HBs are defined by, ∇2ρbcp > 0 and Hbcp > 0.
The Laplacian of the electron density in a cp, ∇2ρbcp, is a very appealing property that determines where ρbcp is locally concentrated, ∇2ρbcp < 0, and locally depleted, ∇2ρbcp > 0. Thus, the sign of ∇2ρbcp determines which of these two competing effects is dominant, allowing the characterisation of (3, − 1) bcps associated with covalent bonds, ∇2ρbcp < 0 and high ρbcp values, and those associated with ionic bonds or weak non-covalent interactions such as hydrogen bonds (HB) or van der Waals (VDW) interactions, ∇2ρbcp > 0 and low ρbcp values.
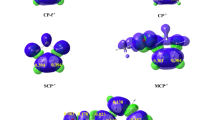
In order to confirm the presence of conventional and non-conventional HB, a QTAIM topological analysis [41] at TSox was carried out. The QTAIM parameters of the (3, − 1) critical points (cps) are collected in Table 4, while the representation of the molecular graphs of TSox obtained by QTAIM analysis of B3LYP/6-31G(d) electron density is shown in Fig. 7.

Molecular graphs of TSox obtained by QTAIM analysis of B3LYP/6-31G(d) electron density, with (3, − 1) critical points (orange sphere) bond paths (brown lines)
From Table 4, a comparison between the Laplacian of all bcps indicates that the HB between H45 and O31 is the strong non-covalent interaction which characterised by Hbcp < 0 like that of the new forming bonds. This HB is classified as conventional hydrogen bond since it occurs between oxygen atom and hydrogen atom linked to nitrogen atom. In addition to this strong stabilised HB, the positive sign of Laplacian ∇2ρbcp > 0 and Hbcp > 0 of (3, − 1) bcp indicates that there are a several other weak stabilised non-conventional HBs (Hbcp > 0), such as H–H, C–H, O–C and O–H. On the other hand, we can notice the presence of two intramolecular non-conventional stabilised HB, C–H and O–H, which are characterised by Laplacian ∇2ρbcp > 0 and Hbcp > 0. Consequently, in addition to the strong HB, the presence of several non-conventional weak interactions at TSox participates in the stabilisation of the ortho–exo approach and thereby enhances the stereoselectivity of the 32CA reaction between nitrone 1 and indole 2.
4 Conclusions
In this paper, we have performed a computational study of the regio- and stereoselectivities of the 32CA reaction between nitrone 1 and indole 2 using DFT method at the B3LYP/6-31G(d) level of theory within the MEDT, which has been established as a new theory of reactivity in organic chemistry. The main outcomes of the present study are the followings:
-
1.
ELF analysis of nitrone 1 confirmed their electronic structure as TAC and showed that the substitution present in this TAC does not change its participation in zw-type 32CA reactions.
-
2.
The molecular mechanism of this 32CA reaction is a slightly asynchronous one-step mechanism.
-
3.
The studied reaction is characterised by a complete ortho regioselectivity and total exo stereoselectivity, as observed experimentally.
-
4.
Toluene solvent stabilises the reactants than transition states and cycloadducts.
-
5.
The high activation energy explains well the high temperature and the long times experimentally observed
-
6.
Analysis of the CDFT global reactivity indices explains the high activation energy that is related to the similitude of the electronic character of both reagents.
-
7.
Local Parr reactivity indices explain the experimentally ortho regioselectivity.
-
8.
ESP, NCI and QTAIM analyses indicate that the presence of several non-covalent interactions, such as non-conventional and conventional HBs, at the TSox is the origin of the ortho–exo selectivity observed experimentally.
References
Cordell GA (2013) The alkaloids, chemistry and biology. Elsevier, Amsterdam, pp 1–348
Anderson RJ, Faulkner DJ, Chu-Heng H, Van Duyne GD, Clardy J (1985) J Am Chem Soc 107:5492
Copp BR, Ireland CM, Barrows LR (1991) J Org Chem 56:4596
Dua R, Shrivastava S, Sonwane SK, Srivastava SK (2011) Adv Biol Res 5(3):120–144
Gomtsyan A (2013) Chem Heterocycl Compd 1:12–15
Patterson W, Cheung PS, Ernest MJ (1992) J Med Chem 35:507
Wagner E, Becan L, Nowakowska E (2004) Bioorg Med Chem 12:265
Huisgen R (1984) In: Padwa A (ed) 1,3-Dipolar cycloaddition chemistry, vol 1. Wiley, New York, pp 42–47
Piotrowska Dorota G (2006) Tetrahedron Lett 47:5363
Sousa CAD, Vale MLC, Garcia-Mera X, Rodriguez-Borges JE (2012) Tetrahedron 68:1682
Saubern S, Macdonald JM, Ryan JH, Woodgate RCJ, Louie TS, Fuchter MJ, White JM, Holmes AB (2010) Tetrahedron 66:2761
Hellel D, Chafaa F, Nacereddine AK, Djerourou A, Vrancken E (2017) RSC Adv 7(48):30128–30141
Nacereddine AK, Yahia W, Bouacha S, Djerourou A (2010) Tetrahedron Lett 51:2617–2621
Nacereddine AK, Yahia W, Sobhi C, Djerourou A (2012) Tetrahedron Lett 53(43):5784–5786
Barama L, Bayoud B, Chafaa F, Nacereddine AK, Djerourou A (2018) Struct Chem 29(6):1709–1721
Yahia W, Khorief Nacereddine A, Liacha M, Djerourou A (2018) Int J Quantum Chem 118(11):e25540
Dmitriev VA, Efremova MM, Novikov AS, Zarubaev VV, Slita AV, Galochkina AV, Starova GL, Ivanov AV, Molchanov AP (2018) Tetrahedron Lett 59:2327–2331
Rios-Gutierrez M, Domingo LR (2019) Eur J Org Chem 2019:267
Domingo LR, Ríos-Gutiérrez M, Pérez P (2018) J Org Chem 83:2182
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F Jr, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox D (2009) Gaussian 09, Revision A.02. Gaussian, Wallingford, CT
Lee C, Yang W, Parr RG (1988) Phys Rev 37:785
Becke AD (1993) J Chem Phys 98:5648
Hehre WJ, Radom L, Schleyer PVR, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
Tomasi J, Persico M (1994) Chem Rev 94:2027
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899
Reed AE, Weinstock RB, Weinhold F (1985) J Chem Phys 83:735
Simkin BY, Sheikhet I (1995) Quantum chemical and statistical theory of solutions—computational approach. Ellis Horwood, London
Cances E, Mennucci B, Tomasi J (1997) J Chem Phys 107:3032
Cossi M, Barone V, Cammi R, Tomasi J (1996) Chem Phys Lett 255:327
Barone V, Cossi M, Tomasi J (1998) J Comput Chem 19:404
Parr RG, von Szentpaly L, Liu S (1922) J Am Chem Soc 1999:121
Parr RG, Pearson RG (1983) J Am Chem Soc 105:7512
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford University Press, New York
Domingo LR, Chamorro E, Pérez P (2008) J Org Chem 73:4615
Domingo LR, Pérez P (2011) Org Biomol Chem 9:7168
Domingo LR, Pérez P, Sáez JA (2013) RSC Adv 3:1486
Johnson ER, Keinan S, Mori-Sanchez P, Contreras-Garcia J, Cohen J, Yang AW (2010) J Am Chem Soc 132:6498
Lane JR, Contreras-Garcia J, Piquemal JP, Miller BJ, Kjaergaard HGJ (2013) Chem Theory Comput 9:3263
Contreras-Garcia JE, Johnson R, Keinan S, Chaudret R, Piquemal JP, Beratan DN, Yang W (2011) J Chem Theory Comput 7:625
Murray JS, Politzer P (1998) In: Schleyer PVR (ed) Encyclopedia of computational chemistry. Wiley, West Sussex, pp 912–920
Bader RFW (1990) Atoms in molecules. A quantum theory. Claredon Press, Oxford
Lu T, Chen F (2012) J Comput Chem 33:580
Domingo LR (2014) RSC Adv 4:32415–32428
Becke AD, Edgecombe KE (1990) J Chem Phys 92:5397–5403
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) J Comput Chem 25:1605–1612
Geerlings P, De Proft F, Langenaeker W (2003) Chem Rev 103:1793–1873
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Molecules 21:748
Domingo LR (2016) Molecules 21:1319
Domingo LR, Emamian SR (2014) Tetrahedron 70:1267
Domingo LR, Ríos-Gutiérrez M (2017) Molecules 22:750
Ríos-Gutiérrez M, Darù A, Tejero T, Domingo LR, Merino P (2017) Org Biomol Chem 15:1618
Silvi B (2002) J Mol Struct 614:3
Lewis GN (1916) J Am Chem Soc 38:762
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Tetrahedron 58:4417
Jaramillo P, Domingo LR, Chamorro E, Pérez P (2008) J Mol Struct (Theochem) 865:68
Domingo LR, Pérez P, Sáez JA (2012) Org Biomol Chem 10:3841
Benchouk W, Mekelleche SM, Silvi B, Aurell MJ, Domingo LR (2011) J Phys Org Chem 24:611
El Ayouchia HB, Bahsis L, Anane H, Domingo LR, Stiriba SE (2018) RSC Adv 8:7670–7678
Wiberg KB (1968) Tetrahedron 24:1083
Domingo LR, Aurell MJ, Pérez P (2014) Tetrahedron 70:4519
Ríos-Gutiérrez M, Nasri L, Khorief Nacereddine A, Djerourou A, Domingo LR (2018) J Phys Org Chem 31:e3830
Nasri L, Ríos-Gutiérrez M, Nacereddine AK, Djerourou A, Domingo LR (2017) Theor Chem Accounts 136:104
Nacereddine AK, Sobhi C, Djerourou A, Ríos-Gutiérrez M, Domingo LR (2015) RSC Adv 5:99299
Sobhi C, Khorief Nacereddine A, Djerourou A, Ríos-Gutiérrez M, Domingo LR (2017) J Phys Org Chem 30:e3637
Lachtar Z, Khorief Nacereddine A, Djerourou A (2019) Struct Chem. https://doi.org/10.1007/s11224-019-01400-2
Rozas I, Alkorta I, Elguero J (2000) J Am Chem Soc 122:11154
Grabowski SJ, Sokalski WA, Dyguda E, Leszczynski J (2006) J Phys Chem B 110:6444
Acknowledgments
This work was supported by the Ministry of Higher Education and Scientific Research of the Algerian Government (project PRFU Code: B00L01UN230120180010).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chafaa, F., Khorief Nacereddine, A. & Djerourou, A. Unravelling the mechanism and the origin of the selectivity of the [3 + 2] cycloaddition reaction between electrophilic nitrone and nucleophilic alkene. Theor Chem Acc 138, 123 (2019). https://doi.org/10.1007/s00214-019-2510-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-019-2510-6