
Abstract
A one-pot synthesis and structure study of a new salt spiropyran of the indoline series containing 2H-chromene vinyl-3H-indolium moiety as a substituent at the 8'-position are described. The structure was confirmed by 1H and 13C NMR and IR spectroscopy and high-resolution mass spectrometry. The single crystals of the compound were investigated by X-ray diffraction analysis. The structure was compared with the previously known isostructural analog.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Among the wide range of organic photochromic compounds, spiropyrans are the most promising and intensively studied classes because of their photosensitivity, structural transformations and relatively simple ability of structure modification [1–3].
For medical applications, the most important requirement to photochromic compounds is the possibility to operate in the biological window (650–1450 nm), because biological liquids and tissues are the most transparent in this wavelength range [4, 5]. Therefore, the synthesis of new spiropyrans with absorption in the range 650–1000 nm is an urgent field of research. Salt spiropyrans are also promising for designing molecular photomagnets when a complex magnetic anion is involved in the crystal structure [6].
As a rule, nonzero absorption in the range 650–1000 nm is typical of spiropyrans based on thia- and selenapyrans and benzoselenazole [7], which are difficult to obtain. We have previously shown that a salt spiropyran of the indoline series with the vinyl-3H-indolium substituent at the 2H-chromene moiety are also characterized by a long-wavelength (more than 700 nm) absorption maximum of the open form [8, 9]. The synthesis of the latter is more economic, safer, and simpler to realize.
In this work, we describe the synthesis and characteristics of a new salt spiropyran containing chlorine atoms in the 5-position of the hetarene moiety and in the 5''-position of the cationic moiety of the molecule.
The synthesis of 1,3,3,6'-tetramethyl-5-chloro-8'[(E)-2-(1'',3'',3''-trimethyl-5''-chloro-3H-indolium-2''-yl)vinyl]spiro[indoline-2,2'-2H-chromene] perchlorate (3) was conducted by a one-pot method (Scheme 1).

Scheme 1.
We failed to obtain and isolate dichloro-substituted salt spiropyran 3 by the condensation of spiropyran 4 and 1,2,3,3-tetramethyl-5-chloro-3H-indolium perchlorate (1) at the formyl group (Scheme 2). This is likely to be due to the lower reactivity of the formyl group of spiropyran 4 than in 2-hydroxy-3-formyl-5-methylbenzaldehyde (2).

Scheme 2.
The structure of compound 3 was confirmed by the data of elemental analysis, IR, 1H and 13C NMR spectroscopy, and mass spectrometry. Molecular structure was refined by single-crystal X-ray diffraction.
The IR spectrum of spiropyran 3 shows characteristic absorption bands corresponding to the stretching vibrations of C=C (1597 cm–1), C–N (1300 and 1251 cm–1), Cl=O of perchlorate anion (1099 cm–1), Cspiro–O (922 cm–1), and C–Cl bonds (736 cm–1).
The 1H NMR spectrum of compound 3 shows a six-proton singlet signal from the gem-dimethyl groups of the cationic fragment at 1.37 ppm. The gem-dimethyl groups of the hetarene moiety displays more distinct magnetic nonequivalence; therefore, they appear as two three-proton singlet signals (1.17 and 1.23 ppm). Characteristic signals of the protons at the 3'- and 4'-positions appear at 5.97 and 7.12 ppm as doublet signals with a spin-spin coupling constant of 10.3 Hz indicating the cis configuration of the C3'=C4' vinyl fragment. The spectrum shows a three-proton signal of the N–CH group at 2.68 ppm and that of the N+–CH3 group at 3.71 ppm.
For more correct signal assignment, we performed additional study using 2D 1H–1H COSY NMR spectroscopy, which allowed us to determine the positions of all aromatic protons (Fig. 1). The protons of the vinyl group of the cationic substituent appeared as doublet signals at 7.40 and 8.02 ppm with spin-spin coupling constants of 16.5 Hz indicating the trans configuration. It is noteworthy that the presence of chlorine atom in the 5- and 5''-positions caused the splitting of the signals of neighboring hydrogen atoms. 13C NMR spectrum shows the signals of all carbon atoms.

1H–1H COSY NMR spectrum of compound 3. The region of aromatic protons.
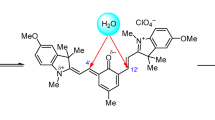
Figure 2 shows the structure of salt spiropyran 3 according to X-ray diffraction data. The N(1) and C(3)–C(9) atoms are in the same plane with a deviation not larger than 0.15 Å to form an angle of 31.05° with the plane of the N(1), C(3), and С(2'2) atoms; the sum of the angles at the N(1) nitrogen atom is 343.73°. In the right-hand moiety relative to the C(2'2) spiro atom, the O(1') and C(3')–C(12') atoms are out of the plane by no more than 0.024 Å producing an angle of 16.43° with the plane of the O(1'), C(2'2), and C(3') atoms. The indoline ring of the cationic moiety is flat, while the sum of the angles at the N(1'') atom is almost 360°.The positive charge is localized on N(1''), while the negative charge is on the perchlorate anion.

Molecular structure of compound 3 according to X-ray diffraction data.
As compared with the molecular structure of previously studied 1,3,3,6'-tetramethyl-8'[(E)-2-(1'',3'',3''-trimethyl-3H-indolium-2''-yl)vinyl]spiro[indoline-2,2'-2H-chromene] perchlorate (5) [10] (Scheme 3), compound 3 contains a hydrogen bond between the O(1') and H(12') atoms with a bond length of 2.314 Å rather than between the O(1') and H(13') atoms (bond length in 5 is 2.205 Å).

Scheme 3.
This feature leads to the fact that the 3H-indolium moiety is turned toward the hetarene moiety by the gem-dimethyl groups at the C(3'') atom rather than by the methyl group C(10'') at the nitrogen atom like in molecule 5.
Table 1 shows selected bond lengths and bond angles in compounds 3 and 5. These data display that the chlorine atoms in the 5- and 5''-positions have a slight effect on the structure of the nearest molecule moieties.
The crystals of these compounds contain a parquet-like packing of molecules (Fig. 3). Ethanol molecules are also present in the crystal lattice. Molecules in the crystal are bound by electrostatic interaction between the counterions and via 30 intermolecular short contacts.

Crystal packing of compound 3 molecules.
Thus, we obtained by one-pot synthesis and studied the new salt spiropyran of indoline series suitable for medical application and designing molecular photomagnets. We compared its structure with known analog 5 containing no chlorine atoms in the 5- and 5''-positions.
EXPERIMENTAL
The NMR spectra were recorded on a Bruker AVANCE-600 spectrometer operating at 600 MHz. Signal position was determined on the δ scale using the residual signals of deuterated solvent DMSO-d6 (2.49 ppm) as an internal reference.
The IR spectra were obtained on a Varian Excalibrum 3100 FT-I spectrophotometer by an attenuated total reflection technique.
Elemental analysis was performed by the classical microanalysis method [11]. Melting points were determined on a Fisher Scientific Fisher–Jones device.
The high-resolution mass spectra were recorded on a Bruker UHR-TOF MaxisTM Impact spectrometer.
X-ray diffraction analysis. Single crystals of the compound were grown from an ethanol solution. The unit cell parameters of crystal 3 and the three-dimensional set of reflection intensities were obtained at 150 K on an Xcalibur Eos automated diffractometer (MoKα radiation, graphite monochromator). Compound 3: orange single crystals C33H33Cl2N2O+ ClO\(_{4}^{ - }\), monoclinic, space group P21/n, with unit cell parameters a = 9.6136(7) Å, b = 14.8574(12) Å, с = 11.9380(9) Å, β = 91.008(7)°, V = 1704.88 Å3, Z = 2; М = 643.50; ρcalcd = 1.338 cm3, µ(МоKα) = 0.316 mm–1. Intensities of 7299 reflections were measured in the range 2θ ≤ 50.0° by ω-scanning using a single crystal of 0.35 × 0.30 × 0.28 mm in size.
An empirical absorption correction was applied using the Multiscan procedure. After exclusion of systematic absences and averaging intensities of equivalent reflections, the array of measured F2(hkl) and σ(F2) consisted of 5085 independent reflections, of which 2938 were with F2 > 2σ(F2). The structure was solved by direct methods and refined by full-matrix least squares on F2 using the SHELXTL program in the anisotropic approximation for non-hydrogen atoms. In crystal structures, the majority of H atoms were located in a difference Fourier synthesis, atomic coordinates and isotropic thermal parameters for all H atoms were refined by least squares with a riding model [12]; in the final cycle of full-matrix refinement, the absolute shifts of all 415 variable structure parameters were less than 0.001σ, and the final R value was 0.0865.
1,3,3,6'-Tetramethyl-5-chloro-8'[(E)-2-(1'',3'',3''-trimethyl-5''-chloro-3H-indolium-2''-yl)vinyl]spiro[indoline-2,2'-2Н-chromene] perchlorate (3). One molar equivalent of triethylamine (0.28 mL) was added dropwise on heating to a mixture of 0.328 g (0.002 mol) of 2-hydroxy-3-formyl-5-methylbenzaldehyde 2 and 1.232 g (0.002 mol) of 1,2,3,3-tetramethyl-5-chloro-3H-indolium perchlorate 1 in 15 mL of isopropanol. The reaction mixture was heated under reflux for 30 min and cooled. Next day, the precipitate formed was separated by filtration and recrystallized from acetonitrile. Tm = 253°C. Yield 0.375 g (29.1%).
IR (ν, cm–1): 1597 (C=C); 1300, 1251 (C–N); 1099 (ClO4–); 922 (Cspiro–O); 736 (C–Cl).
1H NMR (δ, ppm): 8.02 (d, 1H, J = 16.5 Hz, H-12'); 7.89 (d, 1H, J = 1.9 Hz, H-4''); 7.86 (d, 1H, J = 8.6 Hz, H-7''), 7.84 (s, 1H, H-7'), 7.67 (dd, 1H, J = 8.6, 2.0 Hz; H-6''), 7.40 (d, 1H, J = 16.5 Hz, H-13'), 7.34 (s, 1H, H-5'), 7.25 (dd, 2H, J = 5.6, 2.1 Hz; H-6, H-4), 7.12 (d, 1H, J = 10.3 Hz, H-4'), 6.69 (d, 1H, J = 8.9 Hz, H-7), 5.97 (d, 1H, J = 10.3 Hz, H-3'), 3.71 (s, 3H, N+–CH3), 2.68 (s, 3H, N–CH3), 2.30 (s, 3H, C–CH3), 1.37 (s, 6H, gem-C–CH3), 1.23 (s, 3H, gem-C–CH3), 1.17 (s, 3H, gem-C–CH3).
13C NMR (δ, ppm): 181.92 (C-2''), 152.56, 147.14, 146.52, 144.90, 140.60, 138.46, 133.95, 133.33, 130.63, 130.00, 129.46, 129.04, 127.40, 123.32, 122.99, 122.14, 119.75, 119.58, 119.37, 116.61, 112.76, 108.60, 106.29 (C-2'2), 51.76 (C-3''), 51.70 (C-3), 33.92(C-10''), 28.86 (C-10), 25.40 (C-12''), 25.13 (C-11''), 24.63 (C-12), 19.83 (C-11), 19.33 (C-11').
MS (C33H33Cl2N2O): m/z 543.1966 ([M–ClO\(_{4}^{ - }\)]+, calcd. 543.1964).
For C33H33Cl2N2O anal. calcd. (%): C, 61.54; H, 5.13; Cl, 16.55; N, 4.38.
Found (%): C, 61.62; H, 5.12; Cl, 16.41; N, 4.33.
REFERENCES
Klajn, R., Chem. Soc. Rev., 2014, vol. 43, no. 1, pp. 148–184.
Szymanski, W., Beierle, J.M., Kistemaker, H.A.V., et al., Chem. Rev., 2013, vol. 113, no. 8, pp. 6114–6178.
Bouas-Laurent, H. and Durr, H., Pure Appl. Chem., 2001, vol. 73, no. 4, pp. 639–665.
Olejniczak, J., Carling, C.-J., and Almutairi, A., J. Control Release, 2015, vol. 219, pp. 18–30.
Xie, N., Feng, K., Chen, B., et al., J. Mater. Chem. B, 2014, vol. 2, pp. 502–510.
Bénard, S., Rivière, E., Yu, P., et al., Chem. Mater., 2001, vol. 13, no. 1, pp. 159–162.
Benniston, A.C. and Fortage, J., Tetrahedron Lett., 2008, vol. 49, pp. 4292–4295.
Luk'yanova, M.B., Tkachev, V V., Pugachev, A.D., et al., Dokl. Chem., 2018, vol. 480, part 1, pp. 81–84.
Pugachev, A.D., Lukyanova, M.B., Lukyanov, B.S., et al., J. Mol. Struct., 2019, vol. 1178, pp. 590–598.
Tkachev, V.V., Luk’yanov, B.S., Luk’yanova, M.B., et al., Zh. Strukt. Khim., 2016, vol. 57, no. 6, pp. 1334–1335.
Gel’man, N.E., Terent’eva, E.A., Shanina, T.M., and Kiparenko, L.M., Metody kolichestvennogo organi-cheskogo elementnogo analiza (Methods of Quantitative Organic Elemental Analysis), Moscow: Khimiya, 1987.
Sheldrick, G.M., SHELXTL, Bruker AXS, Inc., Madison, Wisconsin, USA, 2000.
Funding
This work was supported by the Ministry of Education and Science of the Russian Federation (State Contract, project no. 4.6088.2017/8.9), X-ray diffraction study was performed under State Contract no. 0089–2019–0011 (V.V. Tkachev, G.V. Shilov, and S.M. Aldoshin).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by I. Kudryavtsev
Rights and permissions
About this article

Cite this article
Pugachev, A.D., Kozlenko, A.S., Luk’yanova, M.B. et al. One-Pot Synthesis and Structure Study of a New Indoline Spiropyran with Cationic Substituent. Dokl Chem 488, 252–256 (2019). https://doi.org/10.1134/S0012500819100021
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0012500819100021