
Abstract
The rRNA genes of mouse and human encode the three major RNAs of the ribosome and as such are essential for growth and development. These genes are present in high copy numbers and arranged as direct repeats at the Nucleolar Organizer Regions on multiple chromosomes. Not all the rRNA genes are transcriptionally active, but the molecular mechanisms that determine activity are complex and still poorly understood. Recent studies applying a novel Deconvolution Chromatin Immunoprecipitation (DChIP-Seq) technique in conjunction with conditional gene inactivation provide new insights into the structure of the active rRNA genes and question previous assumptions on the role of chromatin and histone modifications. We suggest an alternative model for the active rRNA gene chromatin and discuss how this structure is determined and maintained.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ribosomal RNA (rRNA) genes encode the three major RNAs of the ribosome, and as such, play an essential role in cell and organism growth, development, and homeostasis. In eukaryotic genomes, these genes are generally present in high copy numbers and arranged as direct repeats at one or several genetic loci. These ribosomal DNA (rDNA) loci constitute the Nucleolar Organizer Regions (NORs) whose activity is responsible for the formation of nucleoli and ribosome synthesis. However, not all rRNA genes within a cell are transcriptionally active and inactivity or silencing has more than a single molecular basis. Recent studies have applied a novel Deconvolution Chromatin Immunoprecipitation (DChIP-Seq) technique in conjunction with conditional gene inactivation in mouse to generate the first high-resolution maps of rDNA chromatin. The data have revealed important new information about the mode of transcription of the rRNA genes and have provided new insight into the structure and the establishment of active rDNA chromatin in both mouse and human. The new data have also called into question previous assertions about the role of histones and histone modifications in rDNA activity.
rRNA gene transcription
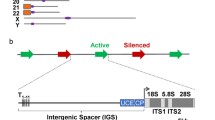
Transcription of the rRNA genes requires a distinct RNA polymerase and a unique set of basal factors (Griesenbeck et al. 2017; Grummt 2003; Moss et al. 2007). Briefly, in mouse and human, the 45/47S rRNA precursor is transcribed by RNA polymerase I (RPI/PolI) and requires at least two other basal factors, the so-called upstream binding factor UBF and the TBP-TAFI complex SL1, also known as TIF1B. The detailed mapping of these basal factors across the mouse rDNA in embryonic fibroblasts (MEFs) determined by DChIP-Seq is shown in Fig. 1a (Hamdane et al. 2014; Herdman et al. 2017; Mars et al. 2018). UBF binds across the whole 47S gene body and across the enhancer repeats, ~ 140 bp units functional in rDNA activity as transcriptional enhancers or selectors (De Winter and Moss 1987; Labhart and Reeder 1984; Moss 1983; Pikaard et al. 1990). UBF also binds at the 47S promoter and the enhancer-associated “spacer promoter,” forming discrete peaks at each. SL1 binds specifically to the gene promoter and the spacer promoter and this binding depends on the presence of UBF (Hamdane et al. 2014; Herdman et al. 2017). RPI maps continuously throughout the 47S gene body and as a peak immediately downstream of the spacer promoter. Both these interactions depend on UBF as expected from their requirement for SL1 in a pre-initiation complex formation. The distribution of factors shown in Fig. 1a is representative of the situation found in various mouse and human cell types (Mars et al. 2018; Zentner et al. 2011), and is shown diagrammatically in Fig. 1b. A third factor, RRN3 also known as TIF1A, associates with RPI to permit its recruitment to the promoter via interactions with the SL1 complex but is released during early elongation (Herdman et al. 2017). RRN3 is absolutely required for the initiation of RPI transcription, but its deletion neither affects the recruitment of UBF nor the recruitment of SL1 and formation of the pre-initiation complex (Hamdane et al. 2014; Herdman et al. 2017). Transcription termination factor 1 (TTF1) is most probably responsible for the termination of the primary rRNA transcript, as are its orthologs Nsi1p/Reb1p in yeast (Evers and Grummt 1995; Merkl et al. 2014; Reiter et al. 2012). TTF1 binds to several sites downstream of the RPI-transcribed domain, but also interacts with sites immediately upstream of the 47S promoter and immediately downstream of the Spacer Promoter. As will be seen, these latter sites have potential importance for both gene activity and silencing. Further, TTF1 is probably responsible for stalling RPI elongation complexes initiated at the spacer promoter, resulting in the peak of RPI upstream of the enhancer repeats (Fig. 1b and c). As we will discuss below, this could play roles in rDNA silencing, in enhancer functions, and also in gene activation.

The distribution of RPI basal factors across the mouse rRNA gene repeat of wild-type MEFs. a The deconvolution ChIP-Seq (DChIP-Seq) mapping data for RPI, SL1(TAF95), UBF, and TTF1 (Herdman et al. 2017). The data are available from the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under E-MTAB-5839 and was displayed using IGV (Integrative Genomics Viewer 2.3, Broad Institute). b, c A summary of the data in a in diagrammatic and enlarged forms. In each panel, the different rRNA gene subregions are indicated diagrammatically. d An example of the psoralen accessibility assay performed during the depletion of UBF. The data were obtained from the 47S transcribed region of MEFs conditional for an inactivating deletion in the UBF gene (Hamdane et al. 2014; Herdman et al. 2017)
Active and inactive rDNA and psoralen accessibility
Only a fraction of the hundreds of rRNA genes present in a mouse and human are transcriptionally active. This mixture of active and silent genes was first observed in mammals using the technique of differential psoralen crosslinking accessibility (Conconi et al. 1989), see Fig. 1d for example. This technique has since been used to demonstrate that both active and inactive genes coexist in a broad range of eukaryotes including plants, insects, and fungi. In mammals, silencing is due in part to methylation of CpG dinucleotides (meCpG) within the rRNA genes (Grummt and Pikaard 2003). However, meCpG is not the sole explanation for rDNA silencing. After inactivation of all CpG methylation, human cells continue to maintain both active and silent rDNA populations as judged by the psoralen technique (Gagnon-Kugler et al. 2009), and yeast, which naturally lacks any DNA methylation, also displays both active and silent rDNA populations (Dammann et al. 1993). Unfortunately, there is often a tacit but erroneous assumption in the literature that the psoralen technique distinguishes silent CpG-methylated rDNA from transcriptionally active unmethylated rDNA. In fact, as previously noted (Hamperl et al. 2013), the existing literature clearly demonstrates that the rRNA genes of mammals exist in one of at least four different populations, (1) silenced via CpG methylation and probably constitutively heterochromatic, (2) inactive and nucleosomal but not silenced via CpG methylation, (3) transcriptionally inactive but non-nucleosomal and poised for activity, and (4) transcriptionally engaged. The psoralen technique broadly differentiates the rDNA based on its chromatin state, that is whether the rDNA is nucleosomal or not. In fact, as will be discussed later, it separates the rDNA into an “active” fraction that is specifically bound by the UBF basal factor but may or may not be engaged in transcription, and a “silent” nucleosomal rDNA fraction that may or may not be silenced via CpG methylation (Hamdane et al. 2014; Herdman et al. 2017). We will argue that the “active” fraction is nucleosome-free, and most probably also histone-free, within the UBF interaction domain (Fig. 1a). There is a further common assumption in the literature that CpG methylation necessarily indicates silencing. Our unpublished data clearly show that the correlation is not so simple, and “active” rRNA gene repeats can carry significant meCpG levels, especially within the non-coding intergenic spacer (IGS).
In summary, the psoralen technique separates rDNA fractions based on chromatin state and not rDNA methylation levels or transcriptional activity. The presence of meCpG also does not necessarily indicate that the rDNA is transcriptionally silent, this depends on the level and distribution of the methylation.
Constitutive silencing of rDNA arrays
Despite the technical limitations in characterizing the silent and active rDNA fractions, it is clear that site-specific CpG methylation of a subpopulation of rRNA genes determines their silencing (Santoro and Grummt 2001; Stancheva et al. 1997). This subpopulation probably exists as condensed heterochromatin and has more recently been shown to be established by a mechanism akin to the X-chromosome inactivation that involves the recruitment of chromatin remodeling complexes via non-coding RNAs (lncRNAs) (Santoro et al. 2002; Savic et al. 2014; Schmitz et al. 2010). The lncRNAs are generated from the rDNA itself, but their regulation and whether they act in cis or in trans is still unclear. However, it is likely that the spacer promoter generates the lncRNA responsible for rDNA silencing, suggesting that TTF1 binding to the adjacent site regulates production of this lncRNA, and hence regulates this form of silencing (Fig. 1c).
There is some confusion in the literature as to whether meCpG-driven silencing acts at the single-gene level within otherwise active rDNA loci or at the level of whole NORs. The haploid mouse and human genomes contain around 200 rRNA genes predominantly arranged in large tandem arrays at the Nucleolar Organizer Regions (NORs) of five distinct chromosomes (Chr12, 15, 16, 18, and 19 in mouse and Chr13, 14, 15, 21, and 22 in human) (Henderson et al. 1972; Rowe et al. 1996; Schmickel 1973). It has been argued that a mosaic pattern of silencing can occur within each NOR and that this might regulate rRNA gene activity (Nemeth and Langst 2008). However, this means of regulation has been questioned (French et al. 2003; Stefanovsky and Moss 2006), and an extensive literature shows that whole NORs are generally either constitutively silent or available for transcription. NORs, active in a previous cell cycle, appear as silver-stain positive secondary constrictions (AgNORs) on metaphase chromosomes, while inactive NORs are highly condensed and not visible as chromosomal constrictions (Dev et al. 1977). AgNOR mapping in cells from different mouse strains showed patterns of NOR activity and inactivity that were characteristic of each strain, clearly demonstrating that NOR silencing is stable over many mouse generations (Kurihara et al. 1994). Inherited patterns of NOR activity/inactivity have also been observed in human cells (Heliot et al. 2000; Roussel et al. 1996; Smirnov et al. 2006). Given the role of meCpG in maintaining heterochromatic silencing (Henikoff 2000; Nishibuchi and Dejardin 2017), it is very probable that the constitutively silent NORs correspond to the CpG hyper-methylated rDNA fraction. Thus it is unlikely that mosaic meCpG silencing of the rDNA is significant, and even previously strong protagonists of mosaic silencing have come to a similar conclusion (Zillner et al. 2015).
The role of UBF in defining the active rDNA
As seen in Fig. 1, UBF associates with the full-length of the gene body encoding the 47S rRNA precursor. Its interaction extends downstream as far as the 3′-terminus of the 47S coding region and both UBF and RPI interactions end abruptly at the downstream TTF1 interaction sites. However, as described above, UBF also binds throughout the enhancer repeats and in discrete peaks at both spacer and 47S promoters, and the same pattern of UBF binding is observed across the human rDNA (Mars et al. 2018; Zentner et al. 2011).
Loss of UBF in MEFs causes complete collapse of the “active” rDNA band in the psoralen accessibility assay, e.g., see Fig. 1d, and the rDNA becomes fully nucleosomal (Hamdane et al. 2014). In contrast, inactivation of RPI transcription by loss of the initiation factor RRN3 has no effect on UBF binding and in the psoralen assay the “active” rDNA fraction remains unaffected (Herdman et al. 2017). Thus, it appears that UBF binding, not transcription, is responsible for generating the “active” rDNA fraction in the psoralen assay. Consistent with this conclusion, the double-banding pattern seen in the psoralen assay is specific to the UBF-bound rDNA domain and is not observed for IGS sequences or indeed for any other genomic loci (Gagnon-Kugler et al. 2009). This is also true for Hmo1, the probable UBF ortholog in yeast, where like UBF, it defines a non-nucleosomal domain on the rDNA (Merz et al. 2008).
These findings strongly suggest that UBF replaces nucleosomes across the rRNA gene to form a specialized chromatin specifically adapted to RPI transcription. Indeed, UBF forms a nucleoprotein structure that somewhat resembles the nucleosome in size and DNA content. This structure, referred to as the “enhance some,” consists of a dimer of UBF bound to about 140 bp of DNA that is looped into a single 360° turn (Bazett-Jones et al. 1994; Stefanovsky et al. 1996; Stefanovsky et al. 2001). UBF has very little DNA sequence selectivity and so, like the nucleosome, the enhance some can form on most DNA sequences, explaining its ability to interact throughout the rRNA genes where it regulates RPI elongation (Stefanovsky et al. 2006). However, the strict exclusion of UBF from the rRNA gene flanking IGS sequences is very striking and at variance with its poor DNA sequence specificity. This strongly suggests that the precise boundaries of UBF recruitment require other components. As we will argue, these components appear to be related to the formation of nucleosomal boundaries and that UBF occupies and stabilizes a nucleosome-free region (NFR) on the rDNA.
What high-resolution maps tell us about active rDNA chromatin and histones
Recent mapping data using DChIP-Seq has also revealed some unexpected facts about the organization of the active rDNA chromatin. Though to date, the maps have been generated from data for MEFs, which harbor both active and inactive rDNA, we believe they reveal details of the chromatin status of each of these forms. MEFs consistently show greater than 60% of genes in the active state, as defined by the psoralen technique (Hamdane et al. 2014; Herdman et al. 2017), e.g., see Fig. 1d. The “active” histone marks such as H3K4me2–3, H2A.Z, and H2A.Zac (Fig. 2) are most probably associated with this active rDNA state. However, the presence of active marks on the inactive rDNA cannot be excluded and logically could also correlate with a potentially active or poised state. Indeed, the deletion of UBF inactivates all rDNA copies and leads to its replacement by nucleosomes. But rather than this diminishing the active chromatin marks, they are in fact enhanced (Hamdane et al. 2014; Herdman et al. 2017). DChIP-Seq analyses of available data for histone modifications such as H3K9me3 and H3K27me3 (Bilodeau and Young 2011; Bulut-Karslioglu et al. 2014; Herdman et al. 2017; Kauzlaric et al. 2017) that are generally correlated with inactive rDNA, display only very low level, broadly distributed ChIP enrichments indistinguishable from the distribution of unmodified H3 (Fig. 2a and b). The data suggest that such “inactive” marks may also be present across the IGS of active gene repeats, but their density in active gene bodies is extremely low or nonexistent. Despite these incertitudes, we believe the distribution of active histone marks revealed in the DChIP-Seq maps (Fig. 2) is representative of the active rDNA, and the distribution of the inactive marks is representative of the silent rDNA. Comparison with existing data for the human rDNA leads to the same conclusion (Mars et al. 2018; Yu et al. 2015; Zentner et al. 2011).

The distribution of histone, histone modifications, CTCF, and DNA accessibility (DNase-Seq) across the mouse rRNA gene repeat of wild-type MEFs. a–c The mapping data at different enlargements. The vertical scale in each panel shows the enrichment of each component as compared to the input DNA data set as described previously (Mars et al. 2018). For histones and histone variants, enrichment (> 1) is shown in green or grey, while depletion (< 1) is shown in red. As in Fig. 1, the data are available from the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under E-MTAB-5839 and was displayed using IGV (Integrative Genomics Viewer 2.3, Broad Institute). In each panel, the different rRNA gene subregions are indicated diagrammatically
The most striking feature of the DChIP-Seq maps is the concentration of all “active” histone modifications at a single site immediately upstream of the spacer promoter (Fig. 2a) (Herdman et al. 2017). H3K4me2 and me3 are seen only within a 600 to 800 bp region that DNase-Seq suggests contains three or four-phased nucleosomes (Fig. 2c). Alignment and deconvolution of public mapping data further shows that H3K9ac, H3K27ac, and H3K36me3 are also present only within these four nucleosomes, as is the histone variant H2A.Z and its acetylated form H2A.Zac. N-terminally acetylated H4 (H4ac) may also be present, but at a very low level (Fig. 2a and c). When the distribution of total histone is mapped, as represented here by H3, we find low levels of enrichment throughout the IGS, but depletion throughout the rRNA gene body and the enhancer repeats (Fig. 2a and b). This region of histone depletion coincides exactly with the UBF-bound domain. (It should be remembered that ~ 30% of the genes in these maps are inactive and hence nucleosomal. We should then expect to detect histone within the gene body of these inactive genes). These data strongly suggesting that the UBF-bound domain is essentially histone-free but is flanked at its upstream boundary by three or four highly modified nucleosomes, a structure we have called the enhancer boundary complex, and by an otherwise predominantly unmodified and nuclesomal IGS (Herdman et al. 2017).
The enhancer boundary complex forms immediately adjacent to a site bound by the genome domain and loop-defining factors CTCF and cohesin (Herdman et al. 2017; Ong and Corces 2014; Skibbens 2015; van de Nobelen et al. 2010) (Fig. 2c), and is part of a larger structure that includes histone remodeling and modification modules such as SWI/SNF and the histone acetyltransferase (HAT) CPB/p300, known to directly bind and acetylate UBF (Herdman et al. 2017; Pelletier et al. 2000). Thus, the enhancer boundary complex is very probably a key player in defining the upstream extent of the UBF-binding domain. Consistent with this, the existence of the boundary complex is independent of the presence of UBF, since it is maintained after UBF deletion and after the re-establishment of nucleosomal chromatin across the whole rDNA repeat (Herdman et al. 2017). The establishment of the enhancer boundary complex, therefore, most probably precedes UBF recruitment, opening the question of whether it already exists on one or more of the inactive rDNA populations, perhaps marking them for potential activation.
A role for chromatin remodeling in rDNA activity
A significant mass of data has been assembled arguing that the activation of the rDNA is subject to regulation by histone modification and chromatin remodeling. These data suggest a scenario in which histone modifications and nucleosome positioning specifically at the 47S promoter determine rDNA activity (e.g., Längst et al. 1998; Li et al. 2006; Shen et al. 2013; Vintermist et al. 2011; Xie et al. 2012; Zhao et al. 2016), see (Birch and Zomerdijk 2008; Nemeth and Langst 2008) for reviews. It was then very surprising that the recent high-resolution maps of mouse rDNA revealed neither significant levels of histone modifications at the 47S promoter, nor within the enhancer repeats and rRNA gene body (Herdman et al. 2017), see Fig. 2a and c. It is important to note that these maps are in full agreement with the previous ChIP-Seq and ChIP-qPCR studies of the mouse rDNA (Hamdane et al. 2014; Nemeth et al. 2008; Zentner et al. 2014), and the human rDNA (Yu et al. 2015; Zentner et al. 2011). Each of these studies reveals the existence of a single peak of activating histone modification adjacent to the unique CTCF/cohesin enhancer boundary complex and immediately upstream of the spacer promoter, which, as we have indicated, displays an arrested RPI complex in both mouse and human (Fig. 1a–c, Fig. 2a and c), (Mars et al. 2018; Zentner et al. 2011). Further, these studies and those from yeast argue that neither nucleosomes nor histones are present to any significant degree across the active rRNA genes, making the existence of histone modifications at the 47S promoter highly questionable (Herdman et al. 2017; Wittner et al. 2011).
An important question is, therefore, how the previous literature should be interpreted in the light of the new rDNA maps, and what is the origin of the discrepancy between the two data sets? In this context, it is most significant that none of the studies arguing for a role of histone modifications at the 47S promoter have actually determined the level of these modifications at or near the enhancer boundary complex. Thus, the levels of histone modification assayed in these studies were clearly insignificant in comparison to those at the major site revealed by ChIP-Seq. It is possible that these studies simply detected small changes in low levels of histone modification–associated inactive nucleosomal rDNA.
Nucleosome positioning across the 47S gene promoter has also been suggested to be a mechanism of regulating access of the RPI transcriptional machinery to the 47S promoter, and TTF1 in combination with chromatin-modifying complexes has been implicated in determining this (Längst et al. 1998; Li et al. 2006). Much of this data comes from in vitro studies of reconstituted histone chromatin, though some derives from restriction enzyme accessibility of endogenous chromatin using linker-mediated PCR. But here again, the lack of histones and the presence of UBF and SL1 on the active in vivo promoter makes this form of regulation unlikely, unless as a step in converting inactive nucleosomal rDNA into an active UBF-bound state. However, the UBF-bound state of the mouse rDNA is stable over many hours even in the absence of RPI transcription and the same is true for yeast Hmo1 (Herdman et al. 2017; Wittner et al. 2011). Hence, it is unlikely that it is in rapid equilibrium with a population of inactive nucleosomal rRNA genes.
A model for active rDNA chromatin
Based on the presently available data, a plausible model for the mouse and human rRNA genes is presented in Fig. 3. The model essentially represents the fraction of rDNA identified by the psoralen technique as “active.” It is important to note that, as discussed above, this fraction may or may not be actively engaged in transcription. Briefly, the IGS that flanks the enhancer and rRNA gene body is classically nucleosomal and, as we have definitively shown, displays little or no active histone modifications. An extended nucleosome-free region (NFR) stretching from the upstream CTCF site through the enhancer repeats, spacer, and 47S promoters and 47S rRNA gene body to the TTF1 termination sites is maintained by the presence of UBF. This factor forms an alternative chromatin-like structure that prevents the reformation of nucleosomes throughout the region. We have shown that UBF deletion causes nucleosome reformation throughout this NFR, but the data from mouse and yeast conditional alleles suggest this is a slow event that probably only occurs during rDNA chromatin replication (Herdman et al. 2017; Wittner et al. 2011). The Enhancer Boundary Complex forms the upstream boundary and is very reminiscent of the chromatin structures observed at enhancers of RPII/PolII genes. H2A.Z is often found near CTCF and typically adjacent to NFRs and is often associated with other “active” histone modifications and with the maintenance of gene activity (Billon and Cote 2012; Bruce et al. 2005; Brunelle et al. 2015; Marques et al. 2010; Ranjan et al. 2013). The UBF-maintained NFR terminates at the downstream TTF1 termination sites, but whether this is determined by the presence of TTF1 itself or some other mechanism is still unknown.

A model for the chromatin structure of the “active” rRNA gene repeat, showing the histone modifications, genome boundary proteins CTCF and cohesin and chromatin remodeling factors for which mapping data is available relative to UBF and the other RPI basal factors. The different rRNA gene subregions are indicated below the representation of factor distributions
How UBF is brought to the rDNA and how it is so precisely distributed is still a matter for conjecture. One possibility is that UBF is laid down during rDNA replication; however, yeast data also suggest a requirement for some degree of concomitant RPI transcription. In this context, the activity of the spacer promoter and chromatin remodeling complexes associated with the enhancer boundary complex could be important. Given their lengths and sequence composition, the enhancer repeats may themselves be poor substrates for nucleosome formation and so may intrinsically favor UBF binding, as is the case for the enhancer repeats of the Xenopus rDNA (Mais et al. 2005). Perhaps the arrested RPI elongation complex associated with the spacer promoter could also be released during rDNA replication and aid in maintaining the enhancer repeats nucleosome-free. At the same time, this would generate the lncRNA needed to maintain in trans CpG methylation of the silent NORs, linking rDNA activity and silencing in a common mechanism. The establishment of conditional mutants for the key RPI factors and reliable and detailed maps of rDNA chromatin have provided the essential understanding that will allow us to test these various possibilities.
Abbreviations
- RPI:
-
RNA polymerase I, POLR1, POL1
- RPII:
-
RNA polymerase II, POLR2, POL2
- RRN3:
-
RPI associated factor
- rRNA:
-
Ribosomal RNA
- rDNA:
-
Ribosomal DNA
- SL1:
-
RNA polymerase 1-specific TBP-TAF complex
- UBF:
-
Upstream binding factor, UBTF
References
Bazett-Jones DP, Leblanc B, Herfort M, Moss T (1994) Short-range DNA looping by the Xenopus HMG-box transcription factor, xUBF. Science 264:1134–1137
Billon P, Cote J (2012) Precise deposition of histone H2A.Z in chromatin for genome expression and maintenance. Biochim Biophys Acta 1819:290–302. https://doi.org/10.1016/j.bbagrm.2011.10.004
Bilodeau S, Young RA (2011) ChIP-Seq data for histone marks in mouse embryonic fibroblasts. (data accessible at NCBI GEO database, accession GSE26657)
Birch JL, Zomerdijk JC (2008) Structure and function of ribosomal RNA gene chromatin. Biochem Soc Trans 36:619–624. https://doi.org/10.1042/BST0360619
Bruce K, Myers FA, Mantouvalou E, Lefevre P, Greaves I, Bonifer C, Tremethick DJ, Thorne AW, Crane-Robinson C (2005) The replacement histone H2A.Z in a hyperacetylated form is a feature of active genes in the chicken. Nucleic Acids Res 33:5633–5639. https://doi.org/10.1093/nar/gki874
Brunelle M, Nordell Markovits A, Rodrigue S, Lupien M, Jacques P-É, Gévry N (2015) The histone variant H2AZ is an important regulator of enhancer activity. Nucleic Acids Res 43:9742–9756. https://doi.org/10.1093/nar/gkv825
Bulut-Karslioglu A, de la Rosa-Velázquez IA, Ramirez F, Barenboim M, Onishi-Seebacher M, Arand J, Galán C, Winter GE, Engist B, Gerle B, O’Sullivan RJ, Martens JHA, Walter J, Manke T, Lachner M, Jenuwein T (2014) Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol Cell 55:277–290. https://doi.org/10.1016/j.molcel.2014.05.029
Conconi A, Widmer RM, Koller T, Sogo JM (1989) Two different chromatin structures coexist in ribosomal RNA genes throughout the cell cycle. Cell 57:753–761
Dammann R, Lucchini R, Koller T, Sogo JM (1993) Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res 21:2331–2338
De Winter RF, Moss T (1987) A complex array of sequences enhances ribosomal transcription in Xenopus laevis. J Mol Biol 196:813–827
Dev VG, Tantravahi R, Miller DA, Miller OJ (1977) Nucleolus organizers in Mus musculus subspecies and in the RAG mouse cell line. Genetics 86:389–398
Evers R, Grummt I (1995) Molecular coevolution of mammalian ribosomal gene terminator sequences and the transcription termination factor TTF-I. Proc Natl Acad Sci U S A 92:5827–5831
French SL, Osheim YN, Cioci F, Nomura M, Beyer AL (2003) In exponentially growing Saccharomyces cerevisiae cells, rRNA synthesis is determined by the summed RNA polymerase I loading rate rather than by the number of active genes. Mol Cell Biol 23:1558–1568
Gagnon-Kugler T, Langlois F, Stefanovsky V, Lessard F, Moss T (2009) Loss of human ribosomal gene CpG methylation enhances cryptic RNA polymerase II transcription and disrupts ribosomal RNA processing. Mol Cell 35:414–425. https://doi.org/10.1016/j.molcel.2009.07.008
Griesenbeck J, Tschochner H, Grohmann D (2017) Structure and function of RNA polymerases and the transcription machineries. Subcell Biochem 83:225–270. https://doi.org/10.1007/978-3-319-46503-6_9
Grummt I (2003) Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Genes Dev 17:1691–1702
Grummt I, Pikaard CS (2003) Epigenetic silencing of RNA polymerase I transcription. Nat Rev Mol Cell Biol 4:641–649
Hamdane N, Stefanovsky VY, Tremblay MG, Németh A, Paquet E, Lessard F, Sanij E, Hannan R, Moss T (2014) Conditional inactivation of upstream binding factor reveals its epigenetic functions and the existence of a somatic nucleolar precursor body. PLoS Genet 10:e1004505. https://doi.org/10.1371/journal.pgen.1004505
Hamperl S, Wittner M, Babl V, Perez-Fernandez J, Tschochner H, Griesenbeck J (2013) Chromatin states at ribosomal DNA loci. Biochim Biophys Acta. https://doi.org/10.1016/j.bbagrm.2012.12.007
Heliot L, Mongelard F, Klein C, O'Donohue MF, Chassery JM, Robert-Nicoud M, Usson Y (2000) Nonrandom distribution of metaphase AgNOR staining patterns on human acrocentric chromosomes. J Histochem Cytochem 48:13–20
Henderson AS, Warburton D, Atwood KC (1972) Location of ribosomal DNA in the human chromosome complement. Proc Natl Acad Sci U S A 69:3394–3398
Henikoff S (2000) Heterochromatin function in complex genomes. Biochim Biophys Acta 1470:O1–O8
Herdman C, Mars JC, Stefanovsky VY, Tremblay MG, Sabourin-Felix M, Lindsay H, Robinson MD, Moss T (2017) A unique enhancer boundary complex on the mouse ribosomal RNA genes persists after loss of Rrn3 or UBF and the inactivation of RNA polymerase I transcription. PLoS Genet 13:e1006899. https://doi.org/10.1371/journal.pgen.1006899
Kauzlaric A, Ecco G, Cassano M, Duc J, Imbeault M, Trono D (2017) The mouse genome displays highly dynamic populations of KRAB-zinc finger protein genes and related genetic units. PLoS One 12:e0173746. https://doi.org/10.1371/journal.pone.0173746
Kurihara Y, Suh DS, Suzuki H, Moriwaki K (1994) Chromosomal locations of Ag-NORs and clusters of ribosomal DNA in laboratory strains of mice. Mamm Genome 5:225–228
Labhart P, Reeder RH (1984) Enhancer-like properties of the 60/81 bp elements in the ribosomal gene spacer of Xenopus laevis. Cell 37:285–289
Längst G, Becker PB, Grummt I (1998) TTF-I determines the chromatin architecture of the active rDNA promoter. EMBO J 17:3135–3145
Li J, Langst G, Grummt I (2006) NoRC-dependent nucleosome positioning silences rRNA genes. EMBO J 25:5735–5741
Mais C, Wright JE, Prieto JL, Raggett SL, McStay B (2005) UBF-binding site arrays form pseudo-NORs and sequester the RNA polymerase I transcription machinery. Genes Dev 19:50–64
Marques M, Laflamme L, Gervais AL, Gaudreau L (2010) Reconciling the positive and negative roles of histone H2A.Z in gene transcription. Epigenetics 5:267–272
Mars JC, Sabourin-Felix M, Tremblay MG, Moss T (2018) A deconvolution protocol for chip-seq reveals analogous enhancer structures on the mouse and human ribosomal RNA genes. G3 (Bethesda) 8:303–314. https://doi.org/10.1534/g3.117.300225
Merkl P, Perez-Fernandez J, Pilsl M, Reiter A, Williams L, Gerber J, Bohm M, Deutzmann R, Griesenbeck J, Milkereit P, Tschochner H (2014) Binding of the termination factor Nsi1 to its cognate DNA site is sufficient to terminate RNA polymerase I transcription in vitro and to induce termination in vivo. Mol Cell Biol 34:3817–3827. https://doi.org/10.1128/mcb.00395-14
Merz K, Hondele M, Goetze H, Gmelch K, Stoeckl U, Griesenbeck J (2008) Actively transcribed rRNA genes in S. cerevisiae are organized in a specialized chromatin associated with the high-mobility group protein Hmo1 and are largely devoid of histone molecules. Genes Dev 22:1190–1204. https://doi.org/10.1101/gad.466908
Moss T (1983) A transcriptional function for the repetitive ribosomal spacer in Xenopus laevis. Nature 302:223–228
Moss T, Langlois F, Gagnon-Kugler T, Stefanovsky V (2007) A housekeeper with power of attorney: the rRNA genes in ribosome biogenesis. Cell Mol Life Sci 64:29–49
Nemeth A, Langst G (2008) Chromatin organization of active ribosomal RNA genes. Epigenetics 3:243–245
Nemeth A, Guibert S, Tiwari VK, Ohlsson R, Langst G (2008) Epigenetic regulation of TTF-I-mediated promoter-terminator interactions of rRNA genes. Embo J 27:1255–1265. https://doi.org/10.1038/emboj.2008.57
Nishibuchi G, Dejardin J (2017) The molecular basis of the organization of repetitive DNA-containing constitutive heterochromatin in mammals. Chromosom Res 25:77–87. https://doi.org/10.1007/s10577-016-9547-3
Ong CT, Corces VG (2014) CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15:234–246. https://doi.org/10.1038/nrg3663
Pelletier G, Stefanovsky VY, Faubladier M, Hirschler-Laszkiewicz I, Savard J, Rothblum LI, Côté J, Moss T (2000) Competitive recruitment of CBP and Rb-HDAC regulates UBF acetylation and ribosomal transcription. Mol Cell 6:1059–1066
Pikaard CS, Pape LK, Henderson SL, Ryan K, Paalman MH, Lopata MA, Reeder RH, Sollner-Webb B (1990) Enhancers for RNA polymerase I in mouse ribosomal DNA. Mol Cell Biol 10:4816–4825
Ranjan A, Mizuguchi G, FitzGerald PC, Wei D, Wang F, Huang Y, Luk E, Woodcock CL, Wu C (2013) Nucleosome-free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell 154:1232–1245. https://doi.org/10.1016/j.cell.2013.08.005
Reiter A, Hamperl S, Seitz H, Merkl P, Perez-Fernandez J, Williams L, Gerber J, Németh A, Léger I, Gadal O, Milkereit P, Griesenbeck J, Tschochner H (2012) The Reb1-homologue Ydr026c/Nsi1 is required for efficient RNA polymerase I termination in yeast. EMBO J 31:3480–3493. https://doi.org/10.1038/emboj.2012.185
Roussel P, Andre C, Comai L, Hernandez-Verdun D (1996) The rDNA transcription machinery is assembled during mitosis in active NORs and absent in inactive NORs. J Cell Biol 133:235–246
Rowe LB, Janaswami PM, Barter ME, Birkenmeier EH (1996) Genetic mapping of 18S ribosomal RNA-related loci to mouse chromosomes 5, 6, 9, 12, 17, 18, 19, and X. Mamm Genome 7:886–889
Santoro R, Grummt I (2001) Molecular mechanisms mediating methylation-dependent silencing of ribosomal gene transcription. Mol Cell 8:719–725
Santoro R, Li J, Grummt I (2002) The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat Genet 32:393–396
Savic N et al (2014) lncRNA maturation to initiate heterochromatin formation in the nucleolus is required for exit from pluripotency in ESCs. Cell Stem Cell 15:720–734. https://doi.org/10.1016/j.stem.2014.10.005
Schmickel RD (1973) Quantitation of human ribosomal DNA: hybridization of human DNA with ribosomal RNA for quantitation and fractionation. Pediatr Res 7:5–12
Schmitz KM, Mayer C, Postepska A, Grummt I (2010) Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev 24:2264–2269. https://doi.org/10.1101/gad.590910
Shen M, Zhou T, Xie W, Ling T, Zhu Q, Zong L, Lyu G, Gao Q, Zhang F, Tao W (2013) The chromatin remodeling factor CSB recruits histone acetyltransferase PCAF to rRNA gene promoters in active state for transcription initiation. PLoS One 8:e62668. https://doi.org/10.1371/journal.pone.0062668
Skibbens RV (2015) Cell biology: cohesin rings leave loose ends. Curr Biol 25:R108–R110. https://doi.org/10.1016/j.cub.2014.12.015
Smirnov E et al (2006) NORs and their transcription competence during the cell cycle. Folia Biol (Praha) 52:59–70
Stancheva I, Lucchini R, Koller T, Sogo JM (1997) Chromatin structure and methylation of rat rRNA genes studied by formaldehyde fixation and psoralen cross-linking. Nucleic Acids Res 25:1727–1735
Stefanovsky VY, Moss T (2006) Regulation of rRNA synthesis in human and mouse cells is not determined by changes in active gene count. Cell Cycle 5:735–739
Stefanovsky VY, Bazett-Jones DP, Pelletier G, Moss T (1996) The DNA supercoiling architecture induced by the transcription factor xUBF requires three of its five HMG-boxes. Nucleic Acids Res 24:3208–3215
Stefanovsky VY, Pelletier G, Bazett-Jones DP, Crane-Robinson C, Moss T (2001) DNA looping in the RNA polymerase I enhancesome is the result of non-cooperative in-phase bending by two UBF molecules. Nucleic Acids Res 29:3241–3247
Stefanovsky V, Langlois F, Gagnon-Kugler T, Rothblum LI, Moss T (2006) Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol Cell 21:629–639. https://doi.org/10.1016/j.molcel.2006.01.023
van de Nobelen S, Rosa-Garrido M, Leers J, Heath H, Soochit W, Joosen L, Jonkers I, Demmers J, van der Reijden M, Torrano V, Grosveld F, Delgado MD, Renkawitz R, Galjart N, Sleutels F (2010) CTCF regulates the local epigenetic state of ribosomal DNA repeats. Epigenetics Chromatin 3:19. https://doi.org/10.1186/1756-8935-3-19
Vintermist A, Bohm S, Sadeghifar F, Louvet E, Mansen A, Percipalle P, Ostlund Farrants AK (2011) The chromatin remodelling complex B-WICH changes the chromatin structure and recruits histone acetyl-transferases to active rRNA genes. PLoS One 6:e19184. https://doi.org/10.1371/journal.pone.0019184
Wittner M, Hamperl S, Stockl U, Seufert W, Tschochner H, Milkereit P, Griesenbeck J (2011) Establishment and maintenance of alternative chromatin states at a multicopy gene locus. Cell 145:543–554. https://doi.org/10.1016/j.cell.2011.03.051
Xie W, Ling T, Zhou Y, Feng W, Zhu Q, Stunnenberg HG, Grummt I, Tao W (2012) The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc Natl Acad Sci U S A 109:8161–8166. https://doi.org/10.1073/pnas.1201262109
Yu F, Shen X, Fan L, Yu Z (2015) Analysis of histone modifications at human ribosomal DNA in liver cancer cell. Sci Rep 5:18100. https://doi.org/10.1038/srep18100
Zentner GE, Saiakhova A, Manaenkov P, Adams MD, Scacheri PC (2011) Integrative genomic analysis of human ribosomal DNA. Nucleic Acids Res 39:4949–4960. https://doi.org/10.1093/nar/gkq1326
Zentner GE, Balow SA, Scacheri PC (2014) Genomic characterization of the mouse ribosomal DNA locus. G3 (Bethesda) 4:243–254. https://doi.org/10.1534/g3.113.009290
Zhao Z, Dammert MA, Hoppe S, Bierhoff H, Grummt I (2016) Heat shock represses rRNA synthesis by inactivation of TIF-IA and lncRNA-dependent changes in nucleosome positioning. Nucleic Acids Res 44:8144–8152. https://doi.org/10.1093/nar/gkw496
Zillner K, Komatsu J, Filarsky K, Kalepu R, Bensimon A, Nemeth A (2015) Active human nucleolar organizer regions are interspersed with inactive rDNA repeats in normal and tumor cells. Epigenomics 7:363–378. https://doi.org/10.2217/epi.14.93
Author statement
TM conceived the study and wrote the manuscript, J-CM and MGT performed experimental research, and MS-F analyzed the data from ChIP-Seq. All authors read and approved the manuscript.
Funding
This work was funded by operating grants from the Canadian Institutes of Health Research (CIHR, MOP12205/PJT153266) and the National Science and Engineering Council (NSERC) of Canada. The Research Centre of the Québec University Hospital Centre (CHU de Québec) is supported by the Fonds de Recherche du Québec—Santé (FRQS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Jennifer Gerton and Lev Porokhovnik
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Moss, T., Mars, JC., Tremblay, M.G. et al. The chromatin landscape of the ribosomal RNA genes in mouse and human. Chromosome Res 27, 31–40 (2019). https://doi.org/10.1007/s10577-018-09603-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-018-09603-9