
It is all about ribosomes.
Lev Ovchinnikov
Abstract
This review is focused on biology of genes encoding ribosomal RNA (rRNA) in mammals. rRNA is a structural component of the most abundant cellular molecule, the ribosome. There are many copies of rRNA genes per genome that are under tight transcriptional control by epigenetic mechanisms serving to meet cellular needs in protein synthesis. Curiously, only a fraction of rRNA genes is used even in the fast-growing cells, raising a question why unused copies of these genes have not been lost during evolution. Two plausible explanations are discussed. First, there is evidence that besides their direct function in production of rRNA, ribosomal RNA genes are involved in regulation of many other genes in the nucleus by forming either temporary or persistent complexes with these genes. Second, it seems that rRNA genes also play a role in the maintenance of genome stability, where lower copy number of rRNA genes destabilizes the genome. These “additional” functions of rRNA genes make them recurrent candidate drivers of chronic human diseases and aging. Experimental support for the involvement of these genes in human diseases and potential mechanisms are also discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Ribosome production starts with the synthesis of ribosomal RNA and its assembly with ribosomal proteins that takes place in the nuclear organelles, nucleoli. This process engages multiple accessory factors and utilizes most of the cellular energy. In this review we discuss epigenetic mechanisms of regulation of ribosomal RNA genes (rDNA). Epigenetics is broadly defined as lasting changes in gene expression that can propagate though cell divisions and can even be transmitted through generations. Epigenetic changes in gene expression states are associated with the altered profiles of histone modifications and DNA methylation at the promoter, gene body, and distant regulatory elements. In this context we also discuss non-canonic role of rDNA loci in regulation of other genes and genome integrity.
STRUCTURAL ORGANIZATION OF rDNA
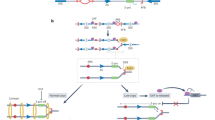
The number of rDNA repeats per genome varies between species, from few (7 in E. coli) to thousands (about 12,000 in Zea mays) [1]. In mammals, there are several hundred copies of rDNA genes per genome (300-500 in human), which are organized in several large genomic domains that also contain other sequences with essentially unknown function. The number of rDNA repeats per genome is not fixed, for example, it varies between the human individuals up to three-fold. In the nucleus, rDNA clusters are associated with the large chromosome compartments known as Nucleolar Organizer Regions (NORs) that were initially defined as regions stained with silver nitrate that are in nucleoli, compartments where rDNA genes are transcribed, ribosomal RNA (rRNA) is processed and assembled with ribosomal proteins. NORs appear as dark areas on the metaphase chromosomes that form the nucleolus during interphase. In human, for example, rDNA repeats are clustered in the NORs located on the p-arms of the five acrocentric chromosomes (Chr13, 14, 15, 21, and 22) (figure, a) [2]. Sequencing analysis provides evidence for exchanges between these chromosomal arms and reveal extensive structural variation between the chromosomes and among the individuals [3]. Distribution of rDNA copies among the chromosomes is uneven, for example, human chromosome 21 harbors the lowest number of the repeats [4, 5]. Similar organization is found in other species, including mice [6]. The number of chromosomes that harbor NORs varies between species. Due to their highly repetitive nature, NORs remain poorly explored and not annotated in the published mammalian genomes. Current NGS analysis tools filter out repetitive sequences. Therefore, rDNA clusters are largely excluded from epigenetic analysis in the ChIP-seq (chromatin immunoprecipitation followed by sequencing) or methylated-DNA-seq studies, obscuring their potential role in organism development and disease.

rDNA repeats. a) Genomic organization: small arms of five human chromosomes are dedicated to rDNA (orange boxes). b) An array or rDNA repeats with transcribed and silenced unis. Below, structure of one rDNA repeat unit. Locations of IGS, rRNA coding region (gray box), upstream control elements (UCE), and core promoter (CP) are shown. T1-11 transcription terminators. c) On active rDNA copies (left), Pol I is recruited on active rDNA copies (left) through UBF (RNA polymerase I transcription factor) to nucleosome-free template, whereas silenced state (right) is maintained by factors that condense chromatin and prevent binding of UBF
rDNA repeats in each cluster are oriented in one direction, for example, in human genome, on all five chromosomes (figure, a) they are transcribed towards the centromere. Such telomere-to-centromere transcriptional organization of rDNA is unlikely to be functional because in the species close to human, chimpanzee, rDNA is oriented in the opposite direction towards telomere [3]. Presumably, such unidirectionality of rDNA reflects ongoing recombination between the rDNA clusters that maintains uniformity of their orientation. Yet, not all rDNA repeats in the clusters are oriented in the same direction (head-to-tail, telomere-to-centromere) as revealed by the fluorescent in situ hybridization (FISH) analysis of single DNA molecules that detected a substantial fraction (up to 30%) of human rDNA repeats in non-canonical orientation (palindromes) [7]. Mutations in the WRN gene (Werner’s syndrome), member of the RecQ helicase family that cause accelerated aging, markedly increase the fraction of re-oriented rDNA repeats (up to 50%) compared to the healthy siblings and age-matched controls.
rDNA is one of the most unstable regions in the genome due to recombination between the repeats leading to dynamic changes in the number of rDNA copies per genome, as demonstrated in yeast [8]. While similar changes in the rDNA were not described in mammalian cells, wide variation in the number rDNA copies per genome between individual animals of the same species and extensive recombination between repeats within rDNA clusters [7] indicate that these genomic regions in mammals are also prone to dynamic re-arrangements.
Transcribed region of the rDNA (grey box in figure, b) encodes three rRNA species 18S, 5.8S, and 28S, and external (5′-ETS, 3′-ETS) and internal (ITS1, ITS2) transcribed sequences. Non-transcribed part of the rDNA unit is called Intergenic Spacer (IGS) (figure, b). It contains several relatively conserved regions, such as Pol I core promoter (CP in figure, b) and terminator (T1-11 in human), while other sequences are highly divergent between the mammalian species. It has been found that IGS encodes non-coding RNAs (ncRNAs) that play a regulatory role in the rDNA transcription [9]. For example, the ncRNA transcribed from the core promoter induces rDNA silencing by recruitment of DMNT3b [10] (see below). Analysis of the IGS regions encoding ncRNA with confirmed regulatory function revealed high levels of histone modifications associated with active transcription [11].
rDNA EXPRESSION
rRNAs comprise about 80% of total cellular RNA. To meet cellular need in ribosomes, production of rRNA during cell proliferation takes substantial part of all transcriptional resources. In eukaryotes, rDNA is transcribed by the specialized RNA polymerase I (Pol I) that is believed to serve exclusively at this locus. Yet, Pol I is not absolutely required for the synthesis of ribosomal RNA to support cell growth as it has been demonstrated in yeast strains, where rRNA synthesis was performed by Pol II from the rDNA constructs driven by the galactose-inducible GAL7 promoter [12]. Pol I produces a 45S precursor transcript (35S in yeast), which is processed into the mature 18S, 5.8S, and 28S (25S in yeast) rRNA species. rRNA-encoding genes are highly evolutionary conserved, yet inter- and intra-individual rDNA nucleotide variations were found in human and mice [13]. Some of these pervasive rRNA sequence heterogeneities were mapped to the functional centers of the ribosome. Tissue-specific expression of some of the variant rRNA alleles indicates that they have functional role in organism development and tissue function.
One of the main transcription factors linked to regulation of the rRNA gene locus is upstream binding factor (UBF, figure, c). UBF belongs to the family of high mobility group (HMG) proteins and has no other functions elsewhere outside the Pol I transcription. UBF binds to the rRNA gene promoter and expands throughout the entire transcribed region [14], suggesting that UBF binding contributes to the formation of the active chromatin state at the gene [15, 16]. Along with UBF, other transcription factors participate in regulation of rDNA transcription (see [17] for a review), including RRN3, that transmit external nutrient and other signals to alter chromatin states at the gene [18, 19].
Only part of the rDNA genes is transcribed in the cell. The number of rDNA repeats transcribed in the cell is determined by the external and internal signals (see below) and is dependent on the levels of the UBF transcription factor [16]. Untranscribed repeats could be either inactive (poised for transcription) or silenced and unavailable for transcription; these states utilize different factors. In mammalian cells, silencing of rDNA is marked by methylation of cytosines in CpG dinucleotides, 5-methyl-cytosine (5mC), at the promoter, which inhibits formation of Pol I transcription complex by preventing recruitment of the transcription factor UBF [20]. Silenced state is established by the nucleolar remodeling complex NoRC that includes TIP5 (TTF-I interacting protein 5) and SNF2H (also known as SMARCA5), a member of the ISWI subfamily with ATPase activity that is also involved in other complexes [21-23]. It is believed that NoRC is tethered to rDNA promoter by the long non-coding RNA transcribed from IGS bound to the transcription termination factor TTF-1, which binds to the DNA element T0 within the promoter (figure, b) [24-26]. NoRC recruits DNA methyltransferases (DNMTs) and histone modification enzymes, such as PARP1 (ADP-ribosyltransferase) and HDAC1 (histone-deacetylase), further promoting transcriptional repressive state [21, 23, 27]. Along with the DNA methylation, rDNA silencing in mammals is closely associated with high density of the repressive histone modifications such as di- or trimethylation of histone H3 at lysine 9 (H3K9me2/3), tri-methylation of lysine 27 (H3K27me3), and tri-methylation of lysine 20 of histone H4 (H4K20me3). In contrast to the nucleosome-packed compact silenced chromatin, little or no nucleosomes are present in the active copies of rDNA.
ROLE OF UNTRANSCRIBED rDNA REPEATS
Dividing cells maintain high rates of rRNA transcription to produce ribosomes in sufficient amounts, which may explain why mammals have many rDNA copies per genome. However, even in the fast-growing cells only a fraction of rDNA repeats is transcribed, while remaining repeats are silenced. The transcriptionally silent and active rDNA regions are located close to each other on the chromosomes (figure, a) [5] indicating that they can switch between the states individually rather than switching of the entire cluster. These observations raise a question why cells always keep substantial fraction of rDNA repeats in the silenced state that is not used to produce rRNA. In yeast, for example, the rDNA copy number per cell varies dynamically and the unused gene copies could be easily lost over time, however the silenced rDNA copies are faithfully maintained indicating that they serve an important function. To address this issue, yeast strains were constructed to have different amounts of rDNA repeats per genome. While the cells with reduced rDNA copy number (25 copies) grew with the same pace as the controls (150 copies), they became sensitive to the DNA damaging agents such as methyl methanesulfonate (MMS) and ultraviolet light (UV). Sensitivity to these agents was inversely correlated with the number of rDNA repeats [28]. Authors suggested that the inactive rDNA units provide space/storage for the DNA damage response enzymes (a “footing place”) needed to repair the damage. In support of this notion, inactive rDNA units are associated with the condensin protein complex that connects sister-chromatids to facilitate repair by homologous recombination. In the strain with low number of rDNA repeats, which are all occupied by the transcription machinery, condensin cannot access rDNA and any damage that might occur cannot not be swiftly repaired. In mammalian cells, the silenced rDNA copies are believed to play a similar role in the maintenance of genomic stability [29]. For example, common to malignancy DNA aberrations in some cancers are associated with the reduction of rDNA copy number [30, 31]. Thus, it is plausible that the inactive repeats serve to provide storage space for the repair machineries in eukaryotes from yeast to human. Still, it is not entirely clear why would such DNA-repair complexes stay bound to the apparently undamaged silenced rDNA copies. As an alternative explanation, one can suggest that the rDNA silencing is a transient state caused by the transcription-induced DNA damage [32, 33], i.e., intense transcription introduces torsion stress that increases probability of the double stranded breaks, which block transcription. Furthermore, the naturally abundant DNA modifications like 5-hydroxymethyl cytosine (5hmC), exacerbate the torsion stress-induced transcription block [34]. Such block needs time to be resolved by topoisomerases and nucleotide excision repair enzymes. From this point of view, transcription causes damage in the DNA, which shifts it to the silenced state for repair. Like in farming, where crop fields are left unplanted (fallow) for a cycle or two to let them recover, silencing of rDNA copies might be needed for DNA recovery after a burst of intense transcription.
In addition to the proposed role in safeguarding genome stability, silenced rDNA copies may also regulate expression of other genes, either directly or indirectly. In Drosophila, it has been shown that the deletions within the rDNA cluster on Y chromosome, that reduced the rDNA copy number, altered expression of the unlinked genes elsewhere in the genome as a result of global loss of heterochromatic component of the genome, similar to the effect of mutations in the known heterochromatin factors [35]. Importantly, these observations exclude a simple “heterochromatic sink” model, where rDNA acts as a storage for heterochromatin factors, and rDNA deletions release such factors and thus enhance heterochromatin formation elsewhere in the genome. As rDNA deletions did not alter translational capacity of the cell [35], the authors suggested that the nucleolus structure (its size, presence of silenced rDNA repeats, etc.), rather than rRNA production, was important in heterochromatin regulation. As a possible scenario of how rDNA can help heterochromatin formation at other loci, it has been shown that the inactive X chromosome in the female mice (Xi) physically binds to nucleolus during the cell cycle, whereas prevention of such interaction results in Xi reactivation [36]. In the suggested model, rDNA heterochromatic regions directly contact with Xi to maintain silenced state of the chromosome. Genes located on autosomes (other than sex chromosomes) can be similarly targeted by rDNA. In embryonic stem cell model system, heterochromatin formation at rDNA genes triggered chromatin changes at different genomic regions associated with the genes involved in differentiation and loss of pluripotency [26, 37]. This notion of physical/functional interaction between rDNA and other genes is further supported by the Hi-C analysis (a method for analyzing contacts between the distant DNA regions) of rDNA contacts with other genomic loci [38]. This study revealed thousands of contacts of rDNA with the nearby rDNA-associated regions and genes located on all chromosomes. Analysis of the rDNA contacts in different cell lines uncovered conserved and cell line-specific features. All these data support the idea that rDNA can be directly involved in regulation of multiple genes.
ASSOCIATION BETWEEN rDNA AND DISEASES
Human diseases like cancer and diabetes are complex pathologies that involve changes in gene expression, metabolism, and physiology. Being the most abundant molecules in a cell, ribosomes and their production inevitably have an impact on origins and progression of many diseases via mechanisms discussed above. First, rDNA transcription and ribosomal biogenesis take up to 70% of cellular resources [39], making it the biggest player in the control of energy and nutrient utilization balance in the cell, an issue central for metabolic diseases such as obesity and diabetes. Second, number of the silenced rDNA copies per cell is important for genomic integrity, and rDNA-dependent loss of genomic stability may lead to cancer and aging. Third, through direct and indirect mechanisms rDNA can alter expression of the disease-related genes. Below we discuss the data that support the link between rDNA and human diseases.
Chronic diseases. Risk of chronic diseases can be increased by adverse environmental exposures during the early stages of organism development [40]. One of the most famous observations that supported this notion was the Dutch Famine Study [41]. In the fall of 1944, as a reprisal to a railway strike, Germans temporarily banned all food transport resulting in supply collapse and famine during the following winter-spring. Adults had only 400-800 calories per day during that period (two-three times lower than before and after). People who had been exposed to famine in utero were compared to the unexposed people that were born before and after the famine in the same country. The exposed newborn babies had smaller birth weight, and when examined ~50 years later they had reduced glucose tolerance, increased body mass index, and higher risk of cardio-vascular diseases. Findings based on examination of human cohorts and similar studies conducted in animal models became a foundation for the “Developmental Origins of Health and Disease” (DOHaD) concept [42] stating that the risks of adult chronic diseases are pre-programmed early in life. Search for molecular mechanisms that link fetal exposures to adult diseases revealed no genes or epigenetic alterations that were common to different experimental models or animal species. What seems to be universal is that the adverse exposures are associated with global downregulation of gene expression (due to nutrient/oxygen deprivation). These changes were so strong that were detectable as a decrease in fetal cellular RNA content. This phenomenon was observed across different animal models including maternal malnutrition in pigs and mice, and placental insufficiency in sheep [43, 44]. These observations identified rDNA, which produces ~80% of cellular RNA, as one of the putative downregulated genes. In fact, rDNA was hypermethylated in the fetal tissues during malnutrition, a sign of transcription silencing. Importantly, the altered rDNA methylation states (hypomethylation) persisted in adulthood [44, 45]. These studies identified rDNA as a candidate target of fetal programing of predisposition to adult chronic diseases that is common to different animal species. While mechanisms linking rDNA hypomethylation to chronic diseases were not explored, it suggests that the rates of rDNA transcription were constitutively elevated in these animals with the consequences discussed above (i.e., changes in the balance of energy consumption, and genome destabilization).
A link between the rDNA transcription and obesity was described in the mouse model of high-fat diet induced obesity [46]. It has been found that the recruitment of nucleolar transcription repressor NML to rDNA in the obese mice is increased compared to the controls. To explore relationship between the obesity and rDNA transcription, NML gene was deleted. As expected, the NML-knockout mice had higher rDNA expression levels and reduced cellular ATP concentration. Furthermore, fat accumulation in the mutant animals was significantly lower than in the wild-type mice. Authors suggested that the NLM-mediated repression of rDNA transcription releases energy that result in the increase of fat accumulation. These findings support a link between the rates of rDNA expression and obesity mediated by alterations in cellular energy balance. It would be interesting to test the NML knockout animals for their risks of chronic diseases in the above models of maternal malnutrition.
Cancer. rDNA alterations can be detected in many cancers manifested as changes in nucleolar morphology and upregulation in rRNA transcription and ribosome biogenesis [47-49]. Size and/or number of nucleoli are increased in the tumor cells serving as an indicator of the rates of cell proliferation, a feature used as a diagnostic marker for some cancers [50]. No doubt that rDNA transcription and ribosome biogenesis are on the list of potential therapeutic targets in cancer [51, 52].
As mentioned above, rDNA is mostly excluded from the genome-wide epigenetic studies due to its repetitive nature. Therefore, relatively little is known about epigenetic changes at rDNA that are specific to cancer. Nonetheless, consistent with the changes in rDNA expression, rDNA methylation patterns are also altered in the malignant cells. However, the observed changes are not always consistent, and upregulation of rRNA expression in some tumors is associated with rDNA promoter hypomethylation [53], whereas in other tumors there is no such association [54], i.e., promoter of the over-expressed rDNA is not hypomethylated.
DNA aberrations, including point mutations and small/large scale chromosomal re-arrangements, drive tumor pathogenesis. In agreement with the suggested role of rDNA in maintaining genome stability, in some cancers rDNA copy number is reduced [30, 31], which is associated with accumulation of the genomic DNA abnormalities. The casual link between these events has not been shown yet.
Aging. Aging is defined as a gradual loss of organ functions and increasing probability of death with age. Aging is one of the key risk factors for chronic diseases and cancer. For example, the median patient age at the time of cancer diagnosis is 66 (National Cancer Institute). Not surprisingly, many laboratories tried to find a role of rDNA in aging as well. In 1970s the Strehler lab discovered the loss of ribosomal RNA gene copies in the post-mitotic animal tissues during aging [55]. However, these observations have never been replicated in other laboratories. In 1990s, the Guarente lab described accumulation of the extra-chromosomal rDNA circles as a primary cause of aging in yeast [56], which revived interest to rDNA in this field. Since then, however, no extrachromosomal rDNA circles have been found in other species.
More recent studies indicate that after all there might be a role for rDNA in aging of species other than yeast. First, rDNA methylation gradually changes with age, and it can be used as a tool to estimate age of a DNA sample, in animals and human [57]. Although exciting and useful as an “aging clock”, the observed changes in rDNA methylation might a consequence rather than the cause of aging processes. Second, studies in C. elegance revealed that all major pathways that increase lifespan (including caloric restriction and mutations in the factors mediating signal transduction from nutrients to the nucleus) inhibit nucleolar function [58]. It has been found that the nucleolar size is inversely correlated with longevity, i.e., animals that have smaller nucleoli at young age live longer than those with larger nucleoli. All previously described mutations that prolong lifespan in C. elegance, including eat-2, daf-2, glp-1, isp-1 and TOR knockdown, decreased the nucleoli size and reduced rRNA/ribosome production. Importantly, mutations in one of the major functional nucleolar components fibrillarin (FIB-1, a methyltransferase involved in pre-rRNA processing and modification), decreased nucleolar size and increased lifespan. Smaller nucleoli were also associated with longevity in other organisms, including Drosophila, mice, and humans [58]. Interestingly, the elderly human volunteers who had reduced caloric intake and performed moderate physical exercises demonstrated smaller nucleoli than the age-matched controls who had no dietary restrictions and did not exercise. Third, mutations that cause accelerated aging in human, Hutchinson–Gilford Progeria syndrome (HGPS), have also been found to dysregulate rDNA [59]. The rDNA promoter in the HGPS cells was hypomethylated, the nucleoli were enlarged and produced more pre-rRNA resulting in higher levels of mature 28S and 18S rRNA species per cell, which was also associated with elevated translation. All these observations suggest that the rate of rDNA expression might be involved in modulation of the aging pace, and that unlike in yeasts, there are no rDNA mutations/re-arrangements in mammals that accumulate with time and drive aging processes. Faster rDNA expression contributes to accelerated aging, and to live longer and healthier life one might need to keep slower rDNA expression and translation rates.
CONCLUSIONS
Multiple studies provide evidence that rDNA may have functions beyond its canonic role in production of rRNA. These non-canonical functions include maintenance of genome stability and regulation of expression of the unlinked genes through direct physical contacts. Does it mean that in eukaryotes rDNA gained additional functions that are not present in bacteria? No, there is no need to suggest that the new cellular functions were acquired by rDNA during evolutionary transition to eukaryotes. One important change that was associated with such transition was the gain of histones and other chromatin proteins that structured DNA (which made it easier to handle longer DNA molecules in a small volume). As a result, Pol I encountered more challenging topological barriers during transcription, which increased probability of the DNA damage (breaks). It is plausible that after a burst of intense Pol I-mediated transcription, the damaged rDNA attracts the DNA-damage response factors for repairs that temporarily keep this gene silenced, like farrow fields in farming practice. As silenced chromatin regions tend to clump together, large untranscribed rDNA domains aggregate with other silenced genomic regions and therefore regulate expression (maintain silenced state) of associated genes. In this model, nutrition accelerates rDNA transcription and thus decreases the time available for the repair of damaged gene copies, causing accumulation of mutations. Associated reduction of the size of the silenced rDNA domain causes derepression of the genes that rely on perinucleolar silenced chromatin to maintain their repressed state. Accumulated DNA mutations and dysregulation of gene expression may contribute to the onset and pathogenesis of human diseases and accelerate aging. Hopefully, most of such rDNA changes are driven by epigenetic mechanisms, that are reversible and can be targeted by the designed small molecule drugs to cure diseases and slow down aging.
POSTSCRIPTUM
I am grateful to Lev Ovchinnikov, my teacher and supervisor, for inspiring interest in ribosomes and molecular biology in general, and I feel lucky to start career in his lab in the atmosphere of highest academic standards that also encouraged broad freedom of choice.
Abbreviations
- IGS:
-
intergenic spacer in rDNA
- NOR:
-
nucleolus organizer region
- rDNA:
-
ribosomal RNA gene
- rRNA:
-
ribosomal RNA
- UBF:
-
RNA polymerase I transcription factor
References
Long, E. O., and Dawid, I. B. (1980) Repeated genes in eukaryotes, Annu. Rev. Biochem., 49, 727-764, https://doi.org/10.1146/annurev.bi.49.070180.003455.
Henderson, A. S., Warburton, D., and Atwood, K. C. (1972) Location of ribosomal DNA in the human chromosome complement, Proc. Natl. Acad. Sci. USA, 69, 3394-3398, https://doi.org/10.1073/pnas.69.11.3394.
Van Sluis, M., Gailin, M. O., McCarter, J. G. W., Mangan, H., Grob, A., and McStay, B. (2019) Human NORs, comprising rDNA arrays and functionally conserved distal elements, are located within dynamic chromosomal regions, Genes Dev., 33, 1688-1701, https://doi.org/10.1101/gad.331892.119.
Héliot, L., Mongelard, F., Klein, C., O’Donohue, M. F., Chassery, J. M., et al. (2000) Nonrandom distribution of metaphase AgNOR staining patterns on human acrocentric chromosomes, J. Histochem. Cytochem., 48, 13-20, https://doi.org/10.1177/002215540004800102.
Sullivan, G. J., Bridger, J. M., Cuthbert, A. P., Newbold, R. F., Bickmore, W. A., et al. (2001) Human acrocentric chromosomes with transcriptionally silent nucleolar organizer regions associate with nucleoli, EMBO J., 20, 2867-2874, https://doi.org/10.1093/emboj/20.11.2867.
Kurihara, Y., Suh, D. S., Suzuki, H., and Moriwaki, K. (1994) Chromosomal locations of Ag-NORs and clusters of ribosomal DNA in laboratory strains of mice, Mamm Genome, 5, 225-228, https://doi.org/10.1007/BF00360550.
Caburet, S., Conti, C., Schurra, C., Lebofsky, R., Edelstein, S. J., et al. (2005) Human ribosomal RNA gene arrays display a broad range of palindromic structures, Genome Res., 15, 1079-1085, https://doi.org/10.1101/gr.3970105.
Kobayashi, T. (2014) Ribosomal RNA gene repeats, their stability and cellular senescence, Proc. Jpn. Acad. Ser. B Phys. Biol. Sci., 90, 119-129, https://doi.org/10.2183/pjab.90.119.
Audas, T. E., Jacob, M. D., and Lee, S. (2012) Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA, Mol. Cell, 45, 147-157, https://doi.org/10.1016/j.molcel.2011.12.012.
Schmitz, K. M., Mayer, C., Postepska, A., and Grummt, I. (2010) Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes, Genes Dev., 24, 2264-2269, https://doi.org/10.1101/gad.590910.
Zentner, G. E., Saiakhova, A., Manaenkov, P., Adams, M. D., and Scacheri, P. C. (2011) Integrative genomic analysis of human ribosomal DNA, Nucleic Acids Res., 39, 4949-4960, https://doi.org/10.1093/nar/gkq1326.
Nogi, Y., Yano, R., and Nomura, M. (1991) Synthesis of large rRNAs by RNA polymerase II in mutants of Saccharomyces cerevisiae defective in RNA polymerase I, Proc. Natl. Acad. Sci. USA, 88, 3962-3966, https://doi.org/10.1073/pnas.88.9.3962.
Parks, M. M., Kurylo, C. M., Dass, R. A., Bojmar, L., Lyden, D., et al. (2018) Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression, Sci. Adv., 4, eaao0665, https://doi.org/10.1126/sciadv.aao0665.
O’Sullivan, A. C., Sullivan, G. J., and McStay, B. (2002) UBF binding in vivo is not restricted to regulatory sequences within the vertebrate ribosomal DNA repeat, Mol. Cell Biol., 22, 657-668, https://doi.org/10.1128/MCB.22.2.657-668.2002.
Chen, D., Belmont, A. S., and Huang, S. (2004) Upstream binding factor association induces large-scale chromatin decondensation, Proc. Natl. Acad. Sci. USA, 101, 15106-15111, https://doi.org/10.1073/pnas.0404767101.
Sanij, E., Poortinga, G., Sharkey, K., Hung, S., Holloway, T. P., et al. (2008) UBF levels determine the number of active ribosomal RNA genes in mammals, J. Cell Biol., 183, 1259-1274, https://doi.org/10.1083/jcb.200805146.
Russell, J., and Zomerdijk, J. C. (2005) RNA-polymerase-I-directed rDNA transcription, life and works, Trends Biochem. Sci., 30, 87-96, https://doi.org/10.1016/j.tibs.2004.12.008.
Blattner, C., Jennebach, S., Herzog, F., Mayer, A., Cheung, A. C., et al. (2011) Molecular basis of Rrn3-regulated RNA polymerase I initiation and cell growth, Genes Dev., 25, 2093-2105, https://doi.org/10.1101/gad.17363311.
Grummt, I., and Voit, R. (2010) Linking rDNA transcription to the cellular energy supply, Cell Cycle, 9, 225-226, https://doi.org/10.4161/cc.9.2.10614.
Santoro, R., and Grummt, I. (2001) Molecular mechanisms mediating methylation-dependent silencing of ribosomal gene transcription, Mol. Cell, 8, 719-725, https://doi.org/10.1016/s1097-2765(01)00317-3.
Santoro, R., Li, J., and Grummt, I. (2002) The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription, Nat. Genet., 32, 393-396, https://doi.org/10.1038/ng1010.
Strohner, R., Nemeth, A., Jansa, P., Hofmann-Rohrer, U., Santoro, R., et al. (2001) NoRC – a novel member of mammalian ISWI-containing chromatin remodeling machines, EMBO J., 20, 4892-4900, https://doi.org/10.1093/emboj/20.17.4892.
Zhou, Y., Santoro, R., and Grummt, I. (2002) The chromatin remodeling complex NoRC targets HDAC1 to the ribosomal gene promoter and represses RNA polymerase I transcription, EMBO J., 21, 4632-4640, https://doi.org/10.1093/emboj/cdf460.
Leone, S., Bar, D., Slabber, C. F., Dalcher, D., and Santoro, R. (2017) The RNA helicase DHX9 establishes nucleolar heterochromatin, and this activity is required for embryonic stem cell differentiation, EMBO Rep., 18, 1248-1262, https://doi.org/10.15252/embr.201744330.
Mayer, C., Schmitz, K. M., Li, J., Grummt, I., and Santoro, R. (2006) Intergenic transcripts regulate the epigenetic state of rRNA genes, Mol. Cell, 22, 351-361, https://doi.org/10.1016/j.molcel.2006.03.028.
Savić, N., Bär, D., Leone, S., Frommel, S. C., Weber, F. A., et al. (2014) lncRNA maturation to initiate heterochromatin formation in the nucleolus is required for exit from pluripotency in ESCs, Cell Stem Cell, 15, 720-734, https://doi.org/10.1016/j.stem.2014.10.005.
Guetg, C., Scheifele, F., Rosenthal, F., Hottiger, M. O., and Santoro, R. (2012) Inheritance of silent rDNA chromatin is mediated by PARP1 via noncoding RNA, Mol Cell, 45, 790-800, https://doi.org/10.1016/j.molcel.2012.01.024.
Ide, S., Miyazaki, T., Maki, H., and Kobayashi, T. (2010) Abundance of ribosomal RNA gene copies maintains genome integrity, Science, 327, 693-696, https://doi.org/10.1126/science.1179044.
Stochaj, U., and Weber, S. C. (2020) Nucleolar organization and functions in health and disease, Cells, 9, 526, https://doi.org/10.3390/cells9030526.
Udugama, M., Sanij, E., Voon, H. P. J., Son, J., Hii, L., et al. (2018) Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers, Proc. Natl. Acad. Sci. USA, 115, 4737-4742, https://doi.org/10.1073/pnas.1720391115.
Xu, B., Li, H., Perry, J. M., Singh, V. P., Unruh, J., Yu, Z., et al. (2017) Ribosomal DNA copy number loss and sequence variation in cancer, PLoS Genet., 13, e1006771, https://doi.org/10.1371/journal.pgen.1006771.
Ju, B. G., Lunyak, V. V., Perissi, V., Garcia-Bassets, I., Rose, D. W., et al. (2006) A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription, Science, 312, 1798-1802, https://doi.org/10.1126/science.1127196.
Kim, N., and Jinks-Robertson, S. (2012) Transcription as a source of genome instability, Nat. Rev. Genet., 13, 204-214, https://doi.org/10.1038/nrg3152.
Desai, R. V., Chen, X., Martin, B., Chaturvedi, S., Hwang, D. W., et al. (2021) A DNA repair pathway can regulate transcriptional noise to promote cell fate transitions, Science, 373, eabc6506, https://doi.org/10.1126/science.abc6506.
Paredes, S., and Maggert, K. A. (2009) Ribosomal DNA contributes to global chromatin regulation, Proc. Natl. Acad. Sci. USA, 106, 17829-17834, https://doi.org/10.1073/pnas.0906811106.
Zhang, L. F., Huynh, K. D., and Lee, J. T. (2007) Perinucleolar targeting of the inactive X during S phase: evidence for a role in the maintenance of silencing, Cell, 129, 693-706, https://doi.org/10.1016/j.cell.2007.03.036.
Feinberg, A. P. (2014) The nucleolus gets the silent treatment, Cell Stem Cell, 15, 675-676, https://doi.org/10.1016/j.stem.2014.11.017.
Yu, S., and Lemos, B. (2016) A portrait of ribosomal DNA contacts with Hi-C reveals 5S and 45S rDNA anchoring points in the folded human genome, Genome Biol. Evol., 8, 3545-3558, https://doi.org/10.1093/gbe/evw257.
Warner, J. R. (1999) The economics of ribosome biosynthesis in yeast, Trends Biochem. Sci., 24, 437-440, https://doi.org/10.1016/s0968-0004(99)01460-7.
Barker, D. J., and Clark, P. M. (1997) Fetal undernutrition and disease in later life, Rev. Reprod., 2, 105-112, https://doi.org/10.1530/ror.0.0020105.
Roseboom, T. J., van der Meulen, J. H., Ravelli, A. C., Osmond, C., Barker, D. J., et al. (2001) Effects of prenatal exposure to the Dutch famine on adult disease in later life: An overview, Mol. Cell. Endocrinol., 185, 93-98, https://doi.org/10.1016/S0303-7207(01)00721-3.
Barker, D. J. (1992) The effect of nutrition of the fetus and neonate on cardiovascular disease in adult life, Proc. Nutr. Soc., 51, 135-144, https://doi.org/10.1079/pns19920023.
Denisenko, O., Lin, B., Louey, S., Thornburg, K., Bomsztyk, K., et al. (2011) Maternal malnutrition and placental insufficiency induce global downregulation of gene expression in fetal kidneys, J. Dev. Origins Health Dis., 2, 124-133, https://doi.org/10.1017/S2040174410000632.
Denisenko, O., Lucas, E. S., Sun, C., Watkins, A. J., Mar, D., et al. (2016) Regulation of ribosomal RNA expression across the lifespan is fine-tuned by maternal diet before implantation, Biochim Biophys Acta, 1859, 906-913, https://doi.org/10.1016/j.bbagrm.2016.04.001.
Holland, M. L., Lowe, R., Caton, P. W., Gemma, C., Carbajosa, G., et al. (2016) Early-life nutrition modulates the epigenetic state of specific rDNA genetic variants in mice, Science, 353, 495-498, https://doi.org/10.1126/science.aaf7040.
Oie, S., Matsuzaki, K., Yokoyama, W., Tokunaga, S., Waku, T., et al. (2014) Hepatic rRNA transcription regulates high-fat-diet-induced obesity, Cell Rep., 7, 807-820, https://doi.org/10.1016/j.celrep.2014.03.038.
Derenzini, M., Trerè, D., Pession, A., Govoni, M., Sirri, V., et al. (2000) Nucleolar size indicates the rapidity of cell proliferation in cancer tissues, J. Pathol., 191, 181-186, https://doi.org/10.1002/(SICI)1096-9896(200006)191:2<181::AID-PATH607>3.0.CO;2-V.
Hein, N., Hannan, K. M., George, A. J., Sanij, E., and Hannan, R. D. (2013) The nucleolus: an emerging target for cancer therapy, Trends Mol. Med., 19, 643-654, https://doi.org/10.1016/j.molmed.2013.07.005.
Montanaro, L., Treré, D., and Derenzini, M. (2013) The emerging role of RNA polymerase I transcription machinery in human malignancy: a clinical perspective, Onco Targets Ther., 6, 909-916, https://doi.org/10.2147/OTT.S36627.
Derenzini, M., Montanaro, L., and Treré, D. (2009) What the nucleolus says to a tumour pathologist, Histopathology, 54, 753-762, https://doi.org/10.1111/j.1365-2559.2008.03168.x.
Bywater, M. J., Poortinga, G., Sanij, E., Hein, N., Peck, A., et al. (2012) Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53, Cancer Cell, 22, 51-65, https://doi.org/10.1016/j.ccr.2012.05.019.
Drygin, D., Rice, W. G., and Grummt, I. (2010) The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer, Annu. Rev. Pharmacol. Toxicol., 50, 131-156, https://doi.org/10.1146/annurev.pharmtox.010909.105844.
Shao, F., Liu, X., Zhang, X., Wang, Q., and Wang, W. (2021) Methylation of 45S ribosomal DNA (rDNA) is associated with cancer and aging in humans, Int. J. Genomics, 2021, 8818007, https://doi.org/10.1155/2021/8818007.
Uemura, M., Zheng, Q., Koh, C. M., Nelson, W. G., Yegnasubramanian, S., et al. (2012) Overexpression of ribosomal RNA in prostate cancer is common but not linked to rDNA promoter hypomethylation, Oncogene, 31, 1254-1263, https://doi.org/10.1038/onc.2011.319.
Johnson, R., and Strehler, B. L. (1972) Loss of genes coding for ribosomal RNA in ageing brain cells, Nature, 240, 412-414, https://doi.org/10.1038/240412a0.
Sinclair, D. A., and Guarente, L. (1997) Extrachromosomal rDNA circles – a cause of aging in yeast, Cell, 91, 1033-1042, https://doi.org/10.1016/S0092-8674(00)80493-6.
Wang, M., and Lemos, B. (2019) Ribosomal DNA harbors an evolutionarily conserved clock of biological aging, Genome Res., 29, 325-333, https://doi.org/10.1101/gr.241745.118.
Tiku, V., Jain, C., Raz, Y., Nakamura, S., Heestand, B., et al. (2017) Small nucleoli are a cellular hallmark of longevity, Nat. Commun., 8, 16083, https://doi.org/10.1038/ncomms16083.
Buchwalter, A., and Hetzer, M. W. (2017) Nucleolar expansion and elevated protein translation in premature aging, Nat. Commun., 8, 328, https://doi.org/10.1038/s41467-017-00322-z.
Acknowledgments
The author is grateful to Elizaveta Minina for invaluable assistance in preparing the Russian-language version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author declares no conflicts of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by the author.
Additional information
Translated from Uspekhi Biologicheskoi Khimii, 2022, Vol. 62, pp. 225-242.
Rights and permissions
About this article

Cite this article
Denisenko, O. Epigenetics of Ribosomal RNA Genes. Biochemistry Moscow 87 (Suppl 1), S103–S110 (2022). https://doi.org/10.1134/S0006297922140097
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297922140097