
Abstract
In this work, the kinetic and the mechanism of isothermal degradation of local chitin (CN), chitosan (CS) and the biocomposite Bentonite/Chitosan (5%Bt/CS) were investigated by thermogravimetric analysis in air atmosphere and in the temperatures range 285–330 °C. Fourier transform-infrared, X-ray diffractogram and differential scanning calorimetry analyses were used to determine the structure of the as prepared samples. DTG curves of the samples show that CN and CS presented one peak, while those of 5%Bt/CS presented one to three peaks as the isothermal degradation increases. This difference was linked to the strong interactions between CS and Bentonite, which improve the stability of CS in the biocomposite 5%Bt/CS. The common first DTG peak appearing in each sample was treated using the Friedman method, leading to the activation energy (Ea) of Ea(CN) = 127.18 kJ/mol > Ea(CS) = 103.90 kJ/mol > Ea(5%Bt/CS) = 80.64 kJ/mol. The auto-catalytic Sestak–Berggren model was found to be qualitatively matched the isothermal degradation process of each sample. The thermodynamic parameters show the appearance of an order in the activated complex with respect to the initial state and that the isothermal degradation process is endothermic and is not spontaneous.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
CN and its derivatives, mainly CS, are part of the larger natural biopolymers that include cellulose (Ibrahim 2002; Hu et al. 2014; Wu and Farnood 2015), starch, collagen, etc. (Kumar and Majeti 2000; Rinaudo 2006; Swetha et al. 2010; Saheb and Jog 2015). Chemically, CN and CS are both linear polysaccharide containing 2-acetamido-2-deoxy-d-glucopyranose (GlcNAc) and 2-amino-2-deoxy-d-glucopyranose (GlcN) units linked by (1 → 4) glycosidic bonds. They can be distinguished by their contents as shown in Fig. 1 (Signini and Filho 1999; Lamarque et al. 2004).

Chemical structure of a CN and b CS
Their physical–chemical properties and their applications in several fields such as, biology, pharmacy, food packaging, matrix in composite materials, and medicine, depend greatly in the degree of deacetylation (DD), which determines the ratio between d-glucosamine and N-acetyl -d-glucosamine units in the polymer chains (Kumar and Majeti 2000; Khor and Lim 2003; Rinaudo 2006; Gámiz-González et al. 2017; Islam et al. 2017). DD over 50% is considered to be CS and below 50% the polymer is called CN (Pillai et al. 2009). Given the interest of these biopolymers, their obtaining from several sources, animal, fungal (mycelium walls) or vegetable (algae), has been the subject of several research (Ferraro et al. 2010). Recently CN of poriferan origin has been widely described as an attractive CN source (Wysokowski et al. 2013; Wysokowski et al. 2015; Stepniak et al. 2016; Ehrlich et al. 2017; Zółtowska-Aksamitowska et al. 2018; Żółtowska-Aksamitowska et al. 2018). However, the insolubility of CN limits its uses, unlike CS which is flexible, soluble in dilute acid acetic, HCl and other acids (Roberts 1992; Rinaudo et al. 1999; Nisticò et al. 2017). CS, is also a promising biodegradable biopolymer, with good film forming properties and immense potential as active food packaging material due to its antimicrobial activity (Zheng and Zhu 2003; Anitha et al. 2014; Baranwal et al. 2018). To obtain CS, the N-deacetylation reaction of CN is generally performed in concentrated alkaline medium and at high temperature (Sagheer et al. 2009; Teli and Sheikh 2012; Ahlafi et al. 2013).
However, in most engineering applications, there are some limitations of these polymers, due to their low mechanical, humidity resistance and thermal stability properties (Peniche-Covas et al. 1993; Barros et al. 2015; Corazzari et al. 2015). These limitations are overcome through the recent development of a new class of materials, known as biocomposites/bio-nanocomposites materials, where these biopolymers were used as a matrix (Alexandre and Dubois 2000; Kittur et al. 2002; Darder et al. 2003; Günister et al. 2007; Cárdenas et al. 2008; Chivrac et al. 2009; Le et al. 2012; Shukla et al. 2013; Xie et al. 2013). Unlike other nanofillers, biopolymer/clay bio-composites have been widely studied, and applied in several research areas, because clay materials are available, cheaper and because their intercalation chemistry is well known (Chivrac et al. 2009), etc. Among the clays, CS/Montmorillonite (MMT) is the most studied ones (Chivrac et al. 2009; Xie et al. 2013; Giannakas et al. 2014). It was demonstrated that CS/MMT displayed improved properties such as mechanical properties, gas barrier and thermal stability when compared to neat CS (Moussout et al. 2018). This was linked to the excellent dispersion of nanoclay and its strong affinity with CS matrix. However, in literature there is a lack of systematic studies about the properties of biocomposite CS/Bentonite clay (CS/Bt), in particular its thermal stability, which is an important parameter for many technological applications. Numerous studies demonstrated that this parameter can be influenced by various factors such as the origin of starting materials, DD and the concentrations of CS and the clay (Günister et al. 2007). Günister et al. (2007) observed that when the MMT amount is increased the thermal stability of CS increased. Given the different fields of application of these materials at elevated temperatures, it seems interesting to study their thermal behavior, which can be achieved by thermogravimetric technique. Indeed, various studies (Vyazovkin et al. 2011), performed under isothermal or dynamic conditions, show that thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) are useful to access the different kinetic and thermodynamic parameters involved in the thermal degradation of CN or CS. The thermal stabilities of CS and CN were evaluated as a function of their activation energy values (Ea), estimated by treating the TGA curves, recorded either under air or N2 atmosphere, using Kissinger and isoconversional methods of Friedman (1964), Coats–Redfern (modified)(Coats and Redfern 1964; 1965) and Ozawa–Flynn–Wall (O–F–W) (Wanjun et al. 2005; Arora et al. 2011; Vyazovkin et al. 2011). However, a disparity between Ea values, reported by different authors for each polymer, is noted. For example, for the thermal degradation of chitosan, Georgieva et al. (2012) found Ea = 126 kJ/mol in dynamic conditions while de Britto and Campana-Filho (2007) by applying dynamic and isothermal conditions found Ea = 149.6 kJ/mol and Ea = 153 kJ/mol, respectively. Peniche-Covas et al. (Coats and Redfern 1965) carried out their study under the same conditions and they found Ea = 181 kJ/mol and Ea = 183–227 kJ/mol, respectively. The same trend was observed in the case of chitin. Moreover, until now, the thermal stability of CS/Bt bio-nanocomposite has not been studied, with the exception of our previous work (Moussout et al. 2016; Moussout et al. 2018), performed in dynamic conditions. In that paper, the following activation energies values, Ea = 121.41 kJ/mol, Ea = 146.50 kJ/mol and Ea = 345.28 kJ/mol for CN, CS and 5%CS/Bt, were found, respectively. In addition, the mechanism of thermal degradation of CN, CS and 5%CS/Bt was deeply studied.
As a complement to our previous study (Moussout et al. 2016; Moussout et al. 2018), the thermal degradation of chitin (CN), chitosan (CS) and biocomposite 5%Bt/CS (5%Bt/CS), in isothermal conditions, was investigated in the temperature range of 285 °C ≤ T ≤ 330 °C. Simultaneous thermal analysis TGA/DTA and differential scanning calorimetry (DSC), performed under air atmosphere, were used to study the thermal stability of each sample because in real applications, under normal atmospheric conditions, oxygen plays a crucial role in the degradation process. The corresponding kinetic parameters of thermal degradation were calculated from Friedman equation. Other important thermodynamic parameters like, Gibbs energy, enthalpy and entropy of activation were also determined. Elucidation of isothermal degradation mechanism and changes in the chemical structure of these samples, during their thermal degradation, was supported by Fourier transform infrared (FTIR) and X ray diffraction (XRD) characterization technics.
Materials and methods
Samples preparations
CN was extracted from the local shrimp shells collected in the city of Meknes (Morocco). CS was obtained by deacetylation of CN in a concentrated solution of NaOH (12 N) for 6 h at 120 °C. The reaction was made in a reflux system to avoid water evaporation. The degree of deacetylation (DD) of chitosan, as determined by FTIR method (Ahlafi et al. 2013), was 83%. The 5%Bt/CS was prepared as follows: CS was dissolved in a (5%v/v) acetic acid deionized aqueous solution, which is stirred vigorously for 4 h at room temperature to form a homogeneous mixture. Then, the Bentonite solution, which was swollen in 50 mL of distilled water and sonicated for 15 min, was slowly added to the CS solution, stirred for 24 h at 60 °C. The resulting solution was centrifuged for 15 min and washed several times with deionized water until pH = 7. The obtained bio-nanocomposite was finally dried overnight at 80 °C. Bentonite was purchased from Rhône pollenc (France). The extraction of CN and the preparation of 5%Bt/CS were described in detail in our previous works (Ahlafi et al. 2013; Moussout et al. 2016).
Samples characterization
The samples were characterized by FTIR, XRD, SEM, DSC and TGA/DTA analysis:
-
XRD diffractograms were obtained using a X’PERT MPD-PRO wide angle X-ray powder diffractometer and used CuKα radiation (λ = 1.542Å) at 45 kV and 40 mA. The canning angle 2θ was in the range of 4° and 80°.
-
JASCO 4100 FTIR spectrometer was used to record FTIR spectra at a resolution of 4 cm−1 and accumulation of at least 64 scans. The samples were prepared in KBr discs in the usual way from very well dried mixture of about 4% (w/w).
-
SEM images (SEM) were obtained by Brand EIFQuanta 200 apparatus.
-
DSC analysis was carried out in an air atmosphere [25–500 °C] with a heating rate of 20 °C/min using a DSC 131Evo instrument.
-
Isothermal degradation experiments were performed on a TA60 SHIMADZU simultaneous thermal analyzer TGA/DTA. The samples were heated under air from room temperature to a desired isothermal temperature at heating rate β = 10 °C/min.
Kinetic and thermodynamic parameters
The decomposition reaction rate in the solid state can be described by the following equation:
where t is the time, T is the temperature, and \(\alpha = \tfrac{{m_{0} - m_{t} }}{{m_{0} - m_{f} }}\) is the conversion fraction, determined as a ratio of the current mass change to the total mass change. mt, m0 and mf, are the actual, initial and final masses, respectively, and \(f(\alpha )\) is the conversion function, which depends on the reaction model, and k(T) is the rate constant, giving by the Arrhenius equation:
where A (min−1) is the frequency factor, R is the gas constant, Ea is the activation energy (kJ/mol). Substituting Eq. (2) into Eq. (1) we obtain:
The differential isoconversional method or Friedman method (Friedman 1964), and it based on Eq. 3 whose logarithm is:
The slope of \(ln\left( {\frac{d\alpha }{dt}} \right)\) versus 1/T for the same value of α, gives the value of activation energy, while the frequency factor, A, can be determined from the Kissinger equation (Kissinger 1957):
where β is the heating rate (β = 10 °C/min) and Tm is the temperature related to DTG peaks at which the thermal degradation rate is maximum.
When the activation energy is calculated, the kinetic model that gives the best description of the experimental data can be determined. Indeed, two master plots functions, Z(α) and Y(α) were applied to the experimental data in order to obtain the possible kinetic model (Montserrat et al. 1998; Starink 2003). In isothermal conditions, these functions are:
The values of α at the maximum of \(Z(\alpha )\) and \(Y(\alpha )\), \(\alpha_{z}^{ * }\) and \(\alpha_{y}^{ * }\), are characteristic of the kinetic model. The shape of \(Y(\alpha )\) being formally identical to the kinetic model, the resulting experimental values of Y(α) and Z(α) are plotted versus α and compared against theoretical Y(α) and Z(α) master plots.
On the other hand, the thermodynamic activation parameters were calculated from the Eyring theory of transition state (Boonchom and Puttawong 2010; Boonchom and Thongkam 2010) of activation entropy (ΔS≠), activation enthalpy (ΔH≠) and activation Gibbs free energy (ΔG≠) were calculated for the process of thermal decomposition of the samples, using the following equations:
where e = 2.7183 is the Euler’s number, χ is the transition factor, which is unity for the monomolecular reactions, kB is the Boltzmann constant, hP is the Plank constant and Tm is the peak temperature in DTG curve.
Results and discussion
FTIR
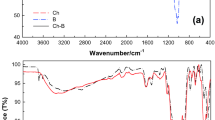
FTIR spectra of extracted CN, CS, Bentonite and 5%Bt/CS, shown in Fig. 2, were recorded in the mid IR range (400–4000 cm−1), and compared to each other. These spectra can be divided in three main spectral regions:

FTIR spectra of a CN, b CS, c Bentonite and d 5%Bt/CS
-
(a)
Region between 3700 and 2700 cm−1: the asymmetric broad band centered at 3440 cm−1 was attributed to the stretching vibrations of O–H and N–H bonds in CS and CN (spectra a and b), implicated in hydrogen bond. The bands at 2880 cm−1 and 2960 cm−1 are characteristics of anti-symmetric and symmetric stretching vibrations of C–H bond in CH2 and CH3 groups. The OH band is also observed in Bentonite spectrum. It was attributed to the adsorbed water on Bentonite (spectra c) (Fan et al. 2007). It can be noticed that all the previous bands are observed in the spectrum of the 5%Bt/CS (spectrum d).
-
(b)
Region between 1750 and 1250 cm−1: this region is characteristic of the amide I (C=O stretching in NHCOCH3), amide II (N–H blending modes in NHCOCH3) and amide III (NHCO group) in CN and CS (Branca et al. 2016). The band at 1650 cm−1 represent the bending mode of O–H groups. This band overlap with those of amide groups in 5%Bt/CS (spectra d).
-
(c)
Region between 1250 and 450 cm−1: In spectrum (c) of Bentonite, the peak at 1080 cm−1 is assigned to Si–O stretching vibrations in Si–O–Si linkage. The band at 450 and the shoulder at 900 cm−1 are assigned to bending vibrations of Si–O and Al–Al–OH, respectively (Li et al. 2009). The vibration frequencies at 1150 cm−1 correspond to asymmetric bridge oxygen stretching in glycosidic linkage C–O–C in CN and CS, and that at 1050 cm−1 is related to the asymmetric stretching of CO in the ring (spectra a and b) (Kaya et al. 2016). These bands cannot clearly distinguished in the spectrum of the 5%Bt/CS, due to the existence of the intense and large band of Si–O–Si in Bt at 1080 cm−1. The bands between 450 and 900 cm−1 in Bentonite spectrum have been assigned to typical Si–O and Si–O–Al bends modes (Li et al. 2009), respectively. The presence of all the bands described above in the spectrum of 5%Bt/CS (spectrum d) confirms the as prepared biocomposite, via the strong interactions between the positive charges of CS and the negatively charged surface of Bentonite (Li et al. 2009; Yin et al. 2011; Park et al. 2013). It was indicated that the formation of the nanocomposite Clay/CS occurs following the intercalation of CS in the interfoliaire space of clay, via electrostatic interactions between the protonated amine groups of chitosan with negatively charged clay surfaces, leading to the changes of locations and intensities of the initial bands of the started sample.
XRD
The XRD diffractograms of the samples are given in Fig. 3. The spectra (a, b) of CN and CS shows two main peaks at 2θ = 9.75° and 19.75°, which indicate that the extracted chitin from shrimps shell was in the α form (Kaya et al. 2016). XRD patterns of the Bentonite (spectrum c) shows a reflection peak at 2.θ = 5.82°, which is characteristic of its a basal spacing (d001). After incorporating 5% (wt/wt) Bentonite within chitosan the diffraction peak at 2.θ = 5.82° derived from the interlayer spacing of the Bentonite disappears, indicating that almost complete exfoliation of the silicate layers in Bentonite took place and bioocomposite structure was obtained.

XRD patterns of a CN, b CS, c Bentonite and d 5%Bt/CS
SEM
The surface morphology of synthesized samples was studied using SEM and resulting images are presented in Fig. 4. The SEM images of CN and CS are composed of lamellar fibers and exhibited rough and thick surface morphology (spectra a and b. The same morphology has previously been reported for these materials from crustaceans such as krill, Gammarus argaeus and pink shrimp (Wang et al. 2013; Kaya et al. 2016). The Bentonite surface (image c) is formed by the grains of uniform size. However, the SEM image of the 5%Bt/CS shows a compact surface and rough, with a very different morphology from those of initial materials (Bentonite and CS).

SEM images of a CN, b CS, c Bentonite and d 5%Bt/CS
DSC
Figure 5 shows the DSC thermograms for natural CN, CS, Bentonite and 5%Bt/CS. In the DSC curves of CS and 5%Bt/CS, two major thermal peaks were observed, while in the case of chitin three peaks were obtained. In all the DSC curves, the first endothermic peak corresponds to desorption of adsorbed water. The position temperatures of this peak depend on the water holding capacity and the interactions of the considered sample with water molecules (Nam et al. 2010). This suggests that 5%Bt/CS has a low water retention capacity than those of Bentonite and CS. The second exothermic peak is related to the degradation reaction of amine units (GlcN), whose exist in significant proportions in the structure of CS and 5%Bt/CS than in that of CN (DD = 83%), as proved by their peak’s areas. Thus, a shift of the exothermic peak in 5%Bt/CS to lower temperature (T = 313 °C), indicates that the thermal stability of CS was lowered when it is intercalated in the Bentonite structure. The third peak located at T = 380 °C in the DSC curve of CN is due to its deacetylation and the degradation of polysaccharide structure. Similar thermal behavior has been observed for α-chitin/chitosan samples, where the shift of the exothermic peak was attributed to the oxidative glycosidic bond, a decrease in acetyl content and the degree of polymerization (Harish Prashanth et al. 2002; Kittur et al. 2002; Guinesi and Cavalheiro 2006; Nam et al. 2010).

DSC curves of a CN, b CS, c Bentonite and d 5%Bt/CS
Kinetics of the isothermal degradation of CN, CS and 5%Bt/CS
Thermogravimetric analysis
The main purpose of this study was to determine the kinetic and the mechanism of the isothermal degradation of CN, CS and 5%Bt/CS biocomposite, using thermogravimetric analysis (TGA/DTG). The experiments were done at the isothermal temperatures of T = 285, 300, 315 and 330 °C. Figures 6, 7 and 8, shows the TGA curves for CN, CS and 5%Bt/CS, respectively, giving for each sample the mass loss and conversion fraction (α) during the degradation time. For each studied temperature, it can be seen that the degradation of each sample is slower (weight loss ≅ 10%) in the first stage (t < 15 min), and then becomes faster (t > 20 min) in the second stage (weight loss > 50%). The first common weight loss corresponds to the desorption of physically adsorbed water on the sample surfaces, while the second one is principally attributed to the thermal degradation of the biopolymers, since Bentonite is stable in the studied temperature range. The degradation rate of the samples was represented by the inset DTG curves, with their temperatures Tm at the maximum degradation rate for each isothermal temperature, shown in the Figs. 6, 7 and 8. It is appears that the Tm values, both for CN and CS are almost identical, except for the isothermal temperature T = 330 °C, where Tm(CN) = 346.90 °C > Tm(CS) = 324.50 °C. This suggest that these polymers have the same degradation kinetics and their thermal stability is linked to the difference in their degree of deacetylation and activation energy. According to the other studies, the first degradation peak, was attributed to the deacetylation of the main chain and the cleavage of glycosidic linkages of CS (Ou et al. 2010; Hao et al. 2017). Otherwise, the determined Tm values, seems to be higher than the corresponding isothermal temperature. The reason for this is that the true sample temperature lags behind that recorded by the instrument, due to the exothermic oxidative degradation in air, as supported by DTA curves given in figure S1, which shows an exothermic peak for each isothermal temperature. However, in the DTG curve of the 5%Bt/CS, one to three peaks are observed, when the isothermal temperature was increased from T = 285 to T = 330 °C. This indicated that the thermal degradation mechanism of CS matrix in the 5%Bt/CS biocomposite was a complex process, involving more than one-step, especially from T > 285 °C (Hao et al. 2017; Topcu et al. 2018). Thus, it can be concluded that the biocomposite 5%CS/Bt show higher thermal stability compared to that of neat CS. A similar behavior was observed by other authors, whose indicated that the latter stages might be caused by the thermal destruction of pyranose ring and the decomposition of the residual carbon (Wang et al. 2005; Hao et al. 2017). The results are also in agreement with our previous work (Moussout et al. 2016), where the intensities of IR bands of CS, recorded after its treatment at T = 300, 320 and 340 °C, decrease gradually.

a TGA/DrTG curves and b α versus time of CN

a TGA/DrTG curves and b α versus time of CS

a TGA/DrTG curves and b α versus time of 5% Bt/CS
Isothermal degradation kinetics
Activation energy (Ea) of isothermal degradation of CN, CS and 5%CS/Bt, was calculated using the linear Friedman’s equation (Eq. 4). This method is advantageous because it is independent of the heating program and allows the activation energy to be calculated from the slope of this equation by plotting ln(dα/dt) versus 1000/T, for each α value. The resulting straight lines are shown in Fig. 9 and the values of Ea, determined for each sample, are summarized in Table 1. It can be observed that the Ea values for the first peak (first stage of degradation) change slightly in the range 0 < α < 0.7, indicating a single reaction mechanism of the degradation for each sample. The average values of Ea of CN (Ea = 127 kJ/mol) is higher compared to those of CS (Ea = 103 kJ/mol) and of 5%CS/Bt (Ea = 80.64 kJ/mol. The stability of CN can be explained by the presence of N-deacetylation of NHCOCH3, which are dominant in CN than in CS structures. This is similar to the results reported by Gámiz-González et al. (2017) and Wanjun et al. (2005), which indicate that the activation energy depends on the deacetylation degree (DD) of CN/CS. Similarly, CS appears more stable than the biocomposite 5%Bt/CS (Ea = 80 kJ/mol), unlike the expected result. However, it has been noted above that the biocomposite degrades in three steps. The presence of Bentonite in CS probably accelerates the decomposition of species formed after the first step or there are different types of interactions between CS and clay, with different values of Ea. As the temperature increases, the mechanism of degradation might become more complex requiring higher or lower activation energy to overcome.

plot of ln(dα/dt) versus 1000/T for the isothermal degradation of a CN, b CS and c 5%Bt/CS
Kinetic model and thermodynamic parameters
Once the activation energy values have been determined, it is better to determine the kinetic model which can describe accurately the experimental data of the isothermal degradation of the studied samples. It can be done by plotting the functions Z(α) (equa.6) and Y(α) (Eq. 7) shown in Fig. 10. For each sample, the values at the maximum of these curves (\(\upalpha_{\text{y}}^{ *} \,{\text{and}}\,\upalpha_{\text{z}}^{ *}\)) are given in Table 2. It can be seen that the obtained curves and the values of αm match well the experimental curves (Fig. 10). Since, for \(0 < \alpha_{y}^{*} < \alpha_{z}^{*}\), the catalytic model SB proposed by Sesták–Berggren (1971), in which \(f\left( \alpha \right) = \alpha^{m} \left( {1 - \alpha } \right)^{n}\) (m and n are a variables parameters), seems to be the best fit. The values of these parameters in the function f(α) are given by \(p = \frac{m}{n}\) with \(p = \frac{{\alpha_{y}^{*} }}{{1 - \alpha_{y}^{*} }}\). Thus, the Eq. 4 can be expressed as (Kissinger 1957):

Experimental functions Y(α) and Z(α) for the isothermal degradation of a chitin, b chitosan and c nano 5%Bt/CS
The plot of \(Ln\left[ {\frac{d\alpha }{dt}{ \exp }\left( {\frac{{E_{a} }}{RT}} \right)} \right]\) versus \(Ln\left[ {\alpha^{p} \left( {1 - \alpha } \right)} \right]\), lead to the straight lines whose slopes equal to n. Table 2 shows the calculated values of n and m for all the samples. The resulting values of m and n tend to increase with increasing temperature.
Thus, knowing the values of Ea and A and having an expression for f(α), it is possible after its normalization, to simulate the curves f(α), which can correspond to the experimental curves dα/dt. The simulated and experimental curves corresponding to the isothermal degradation of CN, CS and 5% Bt/CS at temperatures of 285 °C, 300 °C, 315 °C and 330 °C are given in Fig. 11. The curves show a good agreement between them, indicating that the kinetic parameters and the expression of f(α) used in the simulation are entirely appropriate. In particular, it can be observed in the case of 5%Bt/CS, the appearance of two peaks in the simulated curves for T = 315 and 330 °C, according to the experimental DTG curves.

Simulated (line) and experimental (symbol) functions Y(α) for the isothermal degradation of a CN, b CS and c 5%Bt/CS
Determining the most suitable kinetic model is not an easy task and evaluating the most likely degradation mechanism is usually difficult. According to Šesták and Berggren (1971), the values of m and n can give an insight into the possible mechanism of degradation. Thus, for m > 1 and n > 1, the process is controlled by nucleation. However, it is found that the appropriate model to describe the kinetics of isothermal degradation of CN, CS and 5%Bt/CS was the autocatalytic model, introduced by Sestak and Berggren (SB (m,n)). Although there is a three major reaction models types: accelerating, decelerating, and sigmoidal (called autocatalytic) (Vyazovkin et al. 2011). However, for the isothermal data, the shape of the curve α versus t can be used to decide on the thermal degradation model, because in this case k(T) is constant. The corresponding reaction profiles of α versus t are shown Fig. 12. The comparison of these curves with those obtained experimentally, for CN (Fig. 6b), CS (Fig. 7b) and the 5%Bt/CS (Fig. 8b), confirms that the autocatalytic model is the most appropriate, whose initial and final stages demonstrate, respectively, the accelerating and decelerating behavior, in good conformity with the experimental curves, suggesting the random scission of the polymeric chain.

α versus t “reaction profiles” for accelerating, decelerating and autocatalytic models
Thermodynamic parameters
The thermodynamic parameters namely entropy ΔS#, enthalpy ΔH# and Gibbs free energy ΔG# were calculated for the formation of activated complex of CN, CS and 5%Bt/CS (Table 3) using the Eqs. 8, 9 and 10. The positive value of ΔG# indicates that the isothermal degradation process is thermodynamically not spontaneous (Stolarek and Ledakowicz 2005). However, the positive value of ΔH# shows that the degradation process is endothermic. The negative value of ΔS# shows the appearance of an order in the activated complex with respect to the initial state, and thus, the reaction is unfavorable and may be classified as slow (Hao et al. 2017).
Conclusion
The thermal degradation of CN, CS and 5%Bt/CS was studied in isothermal conditions and described using kinetic analysis. FTIR, XRD and DSC shows that the biocomposite was formed via the intercalation of CS in the interlayer space of Bentonite via the strong interaction of CS functional groups (-amino and OH) with the silicate layer of Bentonite. Based on DTG curves, it was found that the isothermal degradation mechanism of CN and CS occurs in one step (one peak), while that of biocompoiste 5%Bt/CS is complex, leading one to three DTG peaks when increasing degradation temperature. This is due to the strong interaction between CS and Bentonite, which improve the stability of the biocomposite. The activation energy values, determined for the first peak, using the Friedman method, are 127.18 kJ/mol, 103.90 kJ/mol and 80.64 kJ/mol for CN, CS and 5%Bt/CS, respectively. The stability of CN than CS is due to their deacetylation degree. It has been found that the kinetics model which qualitatively matches the experimental data issued from the isothermal degradation of the samples is that of Sestak–Berggren of the type αm(1−α) n, which correspond to the autocatalytic reaction. The thermodynamic study shows the appearance of an order in the activated complex and the isothermal degradation process is endothermic and is not spontaneous.
References
Ahlafi H, Moussout H, Boukhlifi F et al (2013) Kinetics of N-deacetylation of chitin extracted from shrimp shells collected from coastal area of Morocco. Mediterr J Chem 2:503–513. https://doi.org/10.13171/mjc.2.3.2013.22.01.20
Alexandre M, Dubois P (2000) Polymer-layered silicate nanocomposites: preparation, properties and uses of a new class of materials. Mater Sci Eng R Rep 28:1–63. https://doi.org/10.1016/S0927-796X(00)00012-7
Anitha A, Sowmya S, Kumar PTS et al (2014) Chitin and chitosan in selected biomedical applications. Prog Polym Sci 39:1644–1667. https://doi.org/10.1016/j.progpolymsci.2014.02.008
Arora S, Lal S, Kumar S et al (2011) Comparative degradation kinetic studies of three biopolymers: chitin, chitosan and cellulose. Arch Appl Sci Res 3:188–201
Baranwal A, Kumar A, Priyadharshini A et al (2018) Chitosan: an undisputed bio-fabrication material for tissue engineering and bio-sensing applications. Int J Biol Macromol 110:110–123. https://doi.org/10.1016/j.ijbiomac.2018.01.006
Barros SC, da Silva AA, Costa DB et al (2015) Thermal–mechanical behaviour of chitosan–cellulose derivative thermoreversible hydrogel films. Cellulose 22:1911–1929. https://doi.org/10.1007/s10570-015-0603-5
Boonchom B, Puttawong S (2010) Thermodynamics and kinetics of the dehydration reaction of FePO4·2H2O. Phys B Condens Matter 405:2350–2355. https://doi.org/10.1016/j.physb.2010.02.046
Boonchom B, Thongkam M (2010) Kinetics and thermodynamics of the formation of MnFeP4O12. J Chem Eng Data 55:211–216. https://doi.org/10.1021/je900310m
Branca C, D’Angelo G, Crupi C et al (2016) Role of the OH and NH vibrational groups in polysaccharide-nanocomposite interactions: a FTIR-ATR study on chitosan and chitosan/clay films. Polymer (United Kingdom) 99:614–622. https://doi.org/10.1016/j.polymer.2016.07.086
Cárdenas G, Anaya P, Von Plessing C et al (2008) Chitosan composite films. Biomedical applications. J Mater Sci Mater Med 19:2397–2405. https://doi.org/10.1007/s10856-007-3275-3
Chivrac F, Pollet E, Avérous L (2009) Progress in nano-biocomposites based on polysaccharides and nanoclays. Mater Sci Eng R Rep 67:1–17. https://doi.org/10.1016/j.mser.2009.09.002
Coats AW, Redfern JP (1964) Kinetic parameters from thermogravimetric data. Nature 201:68–69
Coats AW, Redfern JP (1965) Kinetics parameters from thermogravimetric data. II. Polym Lett 3:917–920. https://doi.org/10.1002/pol.1965.110031106
Corazzari I, Nisticò R, Turci F et al (2015) Advanced physico-chemical characterization of chitosan by means of TGA coupled on-line with FTIR and GCMS: thermal degradation and water adsorption capacity. Polym Degrad Stab 112:1–9. https://doi.org/10.1016/j.polymdegradstab.2014.12.006
Darder M, Colilla M, Ruiz-Hitzky E (2003) Biopolymer–clay nanocomposites based on chitosan intercalated in montmorillonite. Chem Mater 15:3774–3780. https://doi.org/10.1021/cm0343047
de Britto D, Campana-Filho SP (2007) Kinetics of the thermal degradation of chitosan. Thermochim Acta 465:73–82. https://doi.org/10.1016/j.tca.2007.09.008
Ehrlich H, Bazhenov VV, Debitus C et al (2017) Isolation and identification of chitin from heavy mineralized skeleton of Suberea clavata (Verongida: demospongiae: Porifera) marine demosponge. Int J Biol Macromol 104:1706–1712. https://doi.org/10.1016/j.ijbiomac.2017.01.141
Fan Q, Shan D, Xue H et al (2007) Amperometric phenol biosensor based on laponite clay–chitosan nanocomposite matrix. Biosens Bioelectron 22:816–821. https://doi.org/10.1016/j.bios.2006.03.002
Ferraro V, Cruz IB, Jorge RF et al (2010) Valorisation of natural extracts from marine source focused on marine by-products: a review. Food Res Int 43:2221–2233. https://doi.org/10.1016/j.foodres.2010.07.034
Friedman HL (1964) Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. J Polym Sci Polym Symp 6:183–195. https://doi.org/10.1002/polc.5070060121
Gámiz-González MA, Correia DM, Lanceros-Mendez S et al (2017) Kinetic study of thermal degradation of chitosan as a function of deacetylation degree. Carbohydr Polym 167:52–58. https://doi.org/10.1016/j.carbpol.2017.03.020
Georgieva V, Zvezdova D, Vlaev L (2012) Non-isothermal kinetics of thermal degradation of chitosan. Chem Cent J 6:81. https://doi.org/10.1186/1752-153X-6-81
Giannakas A, Grigoriadi K, Leontiou A et al (2014) Preparation, characterization, mechanical and barrier properties investigation of chitosan–clay nanocomposites. Carbohydr Polym 108:103–111. https://doi.org/10.1016/j.carbpol.2014.03.019
Guinesi LS, Cavalheiro ÉTG (2006) The use of DSC curves to determine the acetylation degree of chitin/chitosan samples. Thermochim Acta 444:128–133. https://doi.org/10.1016/j.tca.2006.03.003
Günister E, Pestreli D, Ünlü CH et al (2007) Synthesis and characterization of chitosan–MMT biocomposite systems. Carbohydr Polym 67:358–365. https://doi.org/10.1016/j.carbpol.2006.06.004
Hao YH, Huang Z, Ye QQ et al (2017) A comparison study on non-isothermal decomposition kinetics of chitosan with different analysis methods. J Therm Anal Calorim 128:1077–1091. https://doi.org/10.1007/s10973-016-5972-y
Harish Prashanth KV, Kittur FS, Tharanathan RN (2002) Solid state structure of chitosan prepared under different N-deacetylating conditions. Carbohydr Polym 50:27–33. https://doi.org/10.1016/S0144-8617(01)00371-X
Hu Y, Tang L, Lu Q et al (2014) Preparation of cellulose nanocrystals and carboxylated cellulose nanocrystals from borer powder of bamboo. Cellulose 21:1611–1618. https://doi.org/10.1007/s10570-014-0236-0
Ibrahim M (2002) Preparation of cellulose and cellulose derivative azo compounds. Cellulose 9:337–349. https://doi.org/10.1023/A:1021154204053
Islam S, Bhuiyan MAR, Islam MN (2017) Chitin and chitosan: structure, properties and applications in biomedical engineering. J Polym Environ 25:854–866. https://doi.org/10.1007/s10924-016-0865-5
Kaya M, Dudakli F, Asan-Ozusaglam M et al (2016) Porous and nanofiber α-chitosan obtained from blue crab (Callinectes sapidus) tested for antimicrobial and antioxidant activities. LWT Food Sci Technol 65:1109–1117. https://doi.org/10.1016/j.lwt.2015.10.001
Khor E, Lim LY (2003) Implantable applications of chitin and chitosan. Biomaterials 24:2339–2349. https://doi.org/10.1016/S0142-9612(03)00026-7
Kissinger HE (1957) Reaction kinetics in differential thermal analysis. Anal Chem 29:1702–1706. https://doi.org/10.1021/ac60131a045
Kittur FS, Harish Prashanth KV, Udaya Sankar K, Tharanathan RN (2002) Characterization of chitin, chitosan and their carboxymethyl derivatives by differential scanning calorimetry. Carbohydr Polym 49:185–193. https://doi.org/10.1016/S0144-8617(01)00320-4
Kumar R, Majeti N (2000) A review of chitin and chitosan applications. React Funct Polym 46:1–27. https://doi.org/10.1016/S1381-5148(00)00038-9
Lamarque G, Viton C, Domard A (2004) Comparative study of the first heterogeneous deacetylation of α- and β-chitins in a multistep process. Biomacromolecules 5:992–1001. https://doi.org/10.1021/bm034498j
Le HR, Qu S, Mackay RE, Rothwell R (2012) Fabrication and mechanical properties of chitosan composite membrane containing hydroxyapatite particles. J Adv Ceram 1:66–71. https://doi.org/10.1007/s40145-012-0007-z
Li JM, Meng XG, Hu CW, Du J (2009) Adsorption of phenol, p-chlorophenol and p-nitrophenol onto functional chitosan. Bioresour Technol 100:1168–1173. https://doi.org/10.1016/j.biortech.2008.09.015
Montserrat S, Málek J, Colomer P (1998) Thermal degradation kinetics of epoxy–anhydride resins: I. Thermochim Acta 313:83–95. https://doi.org/10.1016/S0040-6031(97)00482-6
Moussout H, Ahlafi H, Aazza M, Bourakhouadar M (2016) Kinetics and mechanism of the thermal degradation of biopolymers chitin and chitosan using thermogravimetric analysis. Polym Degrad Stab 130:1–9. https://doi.org/10.1016/j.polymdegradstab.2016.05.016
Moussout H, Ahlafi H, Aazza M, Amechrouq A (2018) Bentonite/chitosan nanocomposite: preparation, characterization and kinetic study of its thermal degradation. Thermochim Acta 659:191–202. https://doi.org/10.1016/j.tca.2017.11.015
Nam YS, Park WH, Ihm D, Hudson SM (2010) Effect of the degree of deacetylation on the thermal decomposition of chitin and chitosan nanofibers. Carbohydr Polym 80:291–295. https://doi.org/10.1016/j.carbpol.2009.11.030
Nisticò R, Franzoso F, Cesano F et al (2017) Chitosan-derived iron oxide systems for magnetically guided and efficient water purification processes from polycyclic aromatic hydrocarbons. ACS Sustain Chem Eng 5:793–801. https://doi.org/10.1021/acssuschemeng.6b02126
Ou C-Y, Zhang C-H, Li S-D et al (2010) Thermal degradation kinetics of chitosan–cobalt complex as studied by thermogravimetric analysis. Carbohydr Polym 82:1284–1289. https://doi.org/10.1016/j.carbpol.2010.07.010
Park H, Choi B, Nguyen J et al (2013) Anionic carbohydrate-containing chitosan scaffolds for bone regeneration. Carbohydr Polym 97:587–596. https://doi.org/10.1016/j.carbpol.2013.05.023
Peniche-Covas C, Argüelles-Monal W, San Román J (1993) A kinetic study of the thermal degradation of chitosan and a mercaptan derivative of chitosan. Polym Degrad Stab 39:21–28. https://doi.org/10.1016/0141-3910(93)90120-8
Pillai CKS, Paul W, Sharma CP (2009) Chitin and chitosan polymers: chemistry, solubility and fiber formation. Prog Polym Sci 34:641–678. https://doi.org/10.1016/j.progpolymsci.2009.04.001
Rinaudo M (2006) Chitin and chitosan: properties and applications. Prog Polym Sci 31:603–632. https://doi.org/10.1016/j.progpolymsci.2006.06.001
Rinaudo M, Pavlov G, Desbrières J (1999) Influence of acetic acid concentration on the solubilization of chitosan. Polymer (Guildf) 40:7029–7032. https://doi.org/10.1016/S0032-3861(99)00056-7
Roberts GAF (1992) Solubility and solution behaviour of Chitin and Chitosan. In: Roberts GAE (ed) Chitin chemistry. Palgrave, London, pp 274–329
Sagheer FAA, Al-Sughayer MA, Muslim S, Elsabee MZ (2009) Extraction and characterization of chitin and chitosan from marine sources in Arabian Gulf. Carbohydr Polym 77:410–419. https://doi.org/10.1016/j.carbpol.2009.01.032
Saheb N, Jog J (2015) Natural fiber polymer composites : a review. Adv Polym Technol 2329:351–363. https://doi.org/10.1002/(SICI)1098-2329(199924)18
Šesták J, Berggren G (1971) Study of the kinetics of the mechanism of solid-state reactions at increasing temperatures. Thermochim Acta 3:1–12. https://doi.org/10.1016/0040-6031(71)85051-7
Shukla SK, Mishra AK, Arotiba OA, Mamba BB (2013) Chitosan-based nanomaterials: a state-of-the-art review. Int J Biol Macromol 59:46–58. https://doi.org/10.1016/j.ijbiomac.2013.04.043
Signini R, Filho SPC (1999) On the preparation and characterization of chitosan hydrochloride. Polym Bull 42:159–166
Starink M (2003) The determination of activation energy from linear heating rate experiments: a comparison of the accuracy of isoconversion methods. Thermochim Acta 404:163–176. https://doi.org/10.1016/S0040-6031(03)00144-8
Stepniak I, Galinski MI, Nowacki K et al (2016) A novel chitosan/sponge chitin origin material as a membrane for supercapacitors-preparation and characterization. RSC Adv 6:4007–4013. https://doi.org/10.1039/c5ra22047e
Stolarek P, Ledakowicz S (2005) Pyrolysis kinetics of chitin by non-isothermal thermogravimetry. Thermochim Acta 433:200–208. https://doi.org/10.1016/j.tca.2005.03.012
Swetha M, Sahithi K, Moorthi A et al (2010) Biocomposites containing natural polymers and hydroxyapatite for bone tissue engineering. Int J Biol Macromol 47:1–4. https://doi.org/10.1016/j.ijbiomac.2010.03.015
Teli MD, Sheikh J (2012) Extraction of chitosan from shrimp shells waste and application in antibacterial finishing of bamboo rayon. Int J Biol Macromol 50:1195–1200. https://doi.org/10.1016/j.ijbiomac.2012.04.003
Topcu C, Caglar B, Onder A et al (2018) Structural characterization of chitosan–smectite nanocomposite and its application in the development of a novel potentiometric monohydrogen phosphate-selective sensor. Mater Res Bull 98:288–299. https://doi.org/10.1016/j.materresbull.2017.09.068
Vyazovkin S, Burnham AK, Criado JM et al (2011) ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim Acta 520:1–19. https://doi.org/10.1016/j.tca.2011.03.034
Wang SF, Shen L, Tong YJ et al (2005) Biopolymer chitosan/montmorillonite nanocomposites: preparation and characterization. Polym Degrad Stab 90:123–131. https://doi.org/10.1016/j.polymdegradstab.2005.03.001
Wang Y, Chang Y, Yu L et al (2013) Crystalline structure and thermal property characterization of chitin from Antarctic krill (Euphausia superba). Carbohydr Polym 92:90–97. https://doi.org/10.1016/j.carbpol.2012.09.084
Wanjun T, Cunxin W, Donghua C (2005) Kinetic studies on the pyrolysis of chitin and chitosan. Polym Degrad Stab 87:389–394. https://doi.org/10.1016/j.polymdegradstab.2004.08.006
Wu T, Farnood R (2015) A preparation method of cellulose fiber networks reinforced by glutaraldehyde-treated chitosan. Cellulose 22:1955–1961. https://doi.org/10.1007/s10570-015-0609-z
Wysokowski M, Bazhenov VV, Tsurkan MV et al (2013) Isolation and identification of chitin in three-dimensional skeleton of Aplysina fistularis marine sponge. Int J Biol Macromol 62:94–100. https://doi.org/10.1016/j.ijbiomac.2013.08.039
Wysokowski M, Petrenko I, Stelling AL et al (2015) Poriferan chitin as a versatile template for extreme biomimetics. Polymers (Basel) 7:235–265. https://doi.org/10.3390/polym7020235
Xie DF, Martino VP, Sangwan P et al (2013) Elaboration and properties of plasticised chitosan-based exfoliated nano-biocomposites. Polymer (United Kingdom) 54:3654–3662. https://doi.org/10.1016/j.polymer.2013.05.017
Yin P, Tian Y, Wang Z et al (2011) Synthesis of functionalized silica gel with poly(diethylenetriamine bis(methylene phosphonic acid)) and its adsorption properties of transition metal ions. Mater Chem Phys 129:168–175. https://doi.org/10.1016/j.matchemphys.2011.03.067
Zheng LY, Zhu JF (2003) Study on antimicrobial activity of chitosan with different molecular weights. Carbohydr Polym 54:527–530. https://doi.org/10.1016/j.carbpol.2003.07.009
Zółtowska-Aksamitowska S, Shaala LA, Youssef DTA et al (2018) First report on chitin in a non-verongiid marine demosponge: the Mycale euplectellioides case. Mar Drugs 16:1–17. https://doi.org/10.3390/md16020068
Żółtowska-Aksamitowska S, Tsurkan MV, Lim SC et al (2018) The demosponge Pseudoceratina purpurea as a new source of fibrous chitin. Int J Biol Macromol 112:1021–1028. https://doi.org/10.1016/j.ijbiomac.2018.02.071
Acknowledgments
This work was supported by MESRSFC and CNRST-Rabat-Morocco, within the framework of the PPR2 project.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Moussout, H., Ahlafi, H., Aazza, M. et al. Kinetic and mechanism studies of the isothermal degradation of local chitin, chitosan and its biocomposite bentonite/chitosan. Cellulose 25, 5593–5609 (2018). https://doi.org/10.1007/s10570-018-1999-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-018-1999-5