
Abstract
Objective
Myocardial ischemia/reperfusion (I/R) injury can cause severe cardiac damage. Aloperine is a quinolizidine alkaloid found in the leaves and seeds of Sophora alopecuroides L. It has been recognized that aloperine has organ-protective properties; however, its role in cardioprotection is poorly characterized. This study aimed to evaluate the cardioprotective effects of aloperine against myocardial I/R injury in vivo.
Methods
Adult male Sprague‒Dawley rats were randomly divided into sham-operated, control, and aloperine groups. All rats except for the sham-operated rats were subjected to 45 min of myocardial ischemia (by left anterior descending ligation) followed by 3 h of reperfusion. Aloperine (10 mg/kg) was given intravenously at the onset of reperfusion. The cardioprotective effects of aloperine were evaluated by determining infarct size, hemodynamics, histological changes, cardiac biomarkers, and cardiac apoptosis.
Results
Aloperine limited infarct size; improved hemodynamics; attenuated myocardial I/R-induced histological deterioration; decreased serum LDH, CK-MB, and α-HBDH levels; and inhibited apoptosis after myocardial I/R injury. Moreover, aloperine stimulated the phosphorylation of ventricular ERK1/2, which is a major module of MAPK signaling pathways. Furthermore, aloperine increased the ventricular expression levels of β-catenin. Pharmacological inhibition of ERK1/2 diminished aloperine-induced cardioprotection and blocked ERK1/2/β-catenin signaling.
Conclusions
These data support the cardioprotective effect of aloperine against myocardial I/R injury, which is mediated, at least in part, by the ERK1/2/β-catenin signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases (CVDs) are the leading cause of death globally and account for one-third of all deaths worldwide. Ischemic heart disease is the most prevalent cardiovascular disease and a dominant contributor to disability [1]. Myocardial ischemia occurs when an imbalance exists between myocardial oxygen supply and demand [2]. When blood flow decreases or ceases, myocardial infarction occurs. It can be lethal and cause sudden cardiac death. The major clinical interventions for the treatment of acute myocardial infarction include thrombolytic drugs, percutaneous coronary intervention (PCI) or coronary artery bypass grafting (CABG) [3]. However, all of these therapeutic strategies aimed at restoring coronary blood flow after an ischemic episode may cause severe cardiac damage, called ischemia and reperfusion (I/R) injury [4]. Therefore, cardioprotective strategies for reducing infarct size and restoring cardiac function against reperfusion injury are urgently needed.
Aloperine (ALO) is a quinolizidine alkaloid isolated from the leaves and seeds of Sophora alopecuroides L. Previous studies have demonstrated that aloperine is associated with a variety of beneficial activities, including anti-infection, antitumor, antibacterial, and antiviral activities. Moreover, it has been reported that aloperine has organ-protective effects. For example, it protects the brain, lungs, kidneys, colon and pancreas against deleterious stimuli and injury [5]. Regarding the cardiovascular system, researchers have shown that aloperine has a vasodilatory effect [6], which is KCNQ-dependent and specifically binds to the KCNQ5 R212 site [7]. Moreover, aloperine could ameliorate pulmonary artery remodeling in a rat model of monocrotaline-induced pulmonary hypertension [8]. These studies support the therapeutic effect of aloperine on vascular diseases. Notably, although studies of the effects of aloperine on heart tissue are inadequate, one report showed that aloperine improved cardiac dysfunction and inhibited cardiomyocyte apoptosis induced by coronary microembolization in rats [9]. Very recently, we found that aloperine could protect hearts against lethal ventricular arrhythmias and sudden cardiac death [10]. Although other active ingredients of Sophora alopecuroides L. could ameliorate cardiac damage and limit infarct size after myocardial I/R injury [11], the effect of aloperine in the treatment of myocardial I/R-induced myocardial injury is unclear. However, whether aloperine has infarct-sparing effects and can prevent cardiac damage due to myocardial I/R injury has not been determined. Moreover, understanding the underlying molecular mechanisms responsible for aloperine-related cardioprotection is important for identifying aloperine as a potential candidate drug for the therapeutic treatment of myocardial I/R injury-induced cardiac damage in the perioperative period.
In the present study, we investigated the hypothesis that aloperine may reduce myocardial damage and protect hearts against myocardial I/R injury using a rat model of myocardial I/R injury induced by left anterior descending coronary artery ligation. Furthermore, we aimed to explore the underlying mechanism of this aloperine-induced cardioprotection.
Materials and Methods
Animals
All the experimental procedures complied with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH Publication 8th edition, 2011) and were approved by the Institutional Animal Care and Use Committee of Sichuan University (approval number: 20211709A). Eight- to ten-week-old male Sprague Dawley rats (200–250 g body weight) were purchased from Dashuo Experimental Animal Research Center (Chengdu, China). All animals were housed in ventilated cages in specific-pathogen-free and temperature-controlled animal facilities until the time of the experiment. The rats had free access to standard laboratory chow and tap water.
Experimental Protocol
The experimental protocol is illustrated in Fig. 1. The rats were randomly assigned to (1) the sham-operated group (sham), (2) the control group (CON), or (3) the aloperine group (ALO). Rats in the sham group underwent the same surgical procedures without left anterior descending coronary artery (LAD) ligation. Rats in the CON and ALO groups received 45 min of LAD occlusion followed by 3 h of reperfusion. Aloperine (10 mg/kg; Kmaels, Shanghai, China) was dissolved in dilute acetic acid solution (saline with 5% acetic acid) and was administered to the rats via the femoral vein at the onset of LAD reperfusion. The dose of aloperine was chosen based on our previously published study [10]. Isovolumetric saline (containing 5% acetic acid) was given to the rats in the sham and CON groups. To study the effect of pharmacological inhibition of ERK1/2 on aloperine-induced cardioprotection, the ERK1/2 inhibitor U0126 (0.5 mg/kg) (MedChemExpress, Monmouth Junction, NJ, USA) was given intravenously to the rats in the CON (CON + U0126) and ALO (ALO + U0126) groups from the femoral vein 10 min before myocardial reperfusion.

Experimental protocol. All groups except for the sham-operated group were subjected to 40 min of myocardial ischemia (induced by LAD ligation) followed by 180 min of reperfusion. Aloperine was dissolved in saline with 5% acetic acid and given to the rats at the onset of reperfusion. The rats in the control and sham groups received the same volume of saline with 5% acetic acid. The ERK1/2 inhibitor U0126 was administered 10 min before reperfusion. sham, sham-operated group; CON, control; ALO, aloperine
Surgical Procedures
The surgical techniques have been described previously [12]. Briefly, sodium pentobarbital (50 mg/kg, i.p.) was used for animal anesthesia. The depth of anesthesia was assessed by the absence of corneal and toe-pinch reflexes. Tracheostomy was performed, and the trachea was cannulated with a rodent ventilator (Chengdu Taimeng Technology Co., Ltd., Chengdu, China). The ventilator settings were as follows: frequency, 70–80 times/min; tidal volume, 8 mL/kg; and expiration: inspiration ratio, 5:4. After thoracotomy, the heart was exposed, and the left anterior descending coronary artery was occluded with 6–0 silk (Ethicon, Somerville, NJ, USA) for 45 min followed by 3 h of reperfusion. Standard limb lead II electrocardiograms were used (Powerlab/8sp system, AD Instruments, Colorado Springs, CO, USA) to monitor heart rhythm throughout the experiment. Myocardial ischemia was confirmed by ST elevation and epicardial cyanosis of the ischemic area. Body temperature was maintained by a homoeothermic blanket. At the end of the experiment, all rats were intraperitoneally injected with an overdose of sodium pentobarbital (200 mg/kg) for euthanasia. Death was confirmed by visualizing the cessation of breathing and heartbeat.
Hemodynamics
A 20-G heparin-filled catheter (Spacelabs Medical, Inc., Redmond, WA, USA) was inserted through the right carotid artery into the left ventricle. A pressure transducer (Biolap 420F, Chengdu Taimeng, Chengdu, China) was connected to the catheter to record the biological signals. The left ventricular systolic pressure (LVSP), left ventricular end diastolic pressure (LVEDP), and maximum rate of increase (+ dP/dtmax) and decrease (-dP/dtmax) in left ventricular pressure were recorded and analyzed offline with BL-420F software (Chengdu Taimeng, Chengdu, China).
Serum Preparation and Tests
Blood samples were taken from the heart. The serum was collected by centrifugation for 10 min (1000 × g) at 4 °C before being stored at -80 °C until analysis. Serum concentrations of lactate dehydrogenase (LDH), creatine kinase-MB (CK-MB) and α-hydroxybutyrate dehydrogenase (α-HBDH) were determined by using an automatic biochemical analyzer (BS-120, Mindray, Shenzhen, China).
Infarct Size Determination
At the end of the experiment, infarct size was measured in the control and aloperine groups using 2,3,5-triphenyltetrazolium chloride (TTC) staining. The LAD was occluded again, and 5 mL of 1% Evans blue (Sigma Chemical Co., St. Louis, MO, USA) was injected via the left ventricle to determine the area at risk (AAR) region. Next, the heart was cut into 2 mm-thick slices from the apex, perpendicular to the ventricular long axis. Then, the heart sections were incubated at 37 °C for 20 min in a 1% phosphate-buffered solution of TTC (Sigma Chemical Co., St. Louis, MO, USA). The infarcted area was stained white, while the AAR area was stained red. The nonischaemic tissues were not stained (blue). The myocardium was separated and weighed according to the color. The infarct size was calculated as the percentage of the area at risk.
Tissue Sample Collection and Preparation
In addition, in the experimental sets, the LAD was reoccluded at the end of reperfusion, and AAR regions were identified by Evans blue staining. The myocardial tissues from the AAR regions were carefully separated and stored at -80 °C for analysis of protein expression or were kept in formalin for histological evaluation.
Histological Evaluation and TUNEL Staining
The AAR sections were fixed overnight using a 10% neutral buffered formalin solution before being embedded in paraffin and sectioned into 5 μm-thick consecutive paraffin sections. The heart sections were mounted on glass slides, dewaxed and stained with hematoxylin and eosin (H&E). A histological evaluation system was used based on a previously published scoring system according to the severity of myocardial damage [12]. Briefly, myocardial damage, including interstitial vacuolization, hemorrhage and edema, was evaluated according to the following criteria: (1) distribution: 0 indicates zero damage; 1: focal; 2: multifocal; and 3: massive damage. (2) Severity: 0 indicates zero damage; 1: mild; 2: moderate; 3: marked damage. A mean score was given for each heart slice. Hearts were evaluated by two independent researchers in a double-blind manner.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed to evaluate myocardial apoptosis according to the manufacturer’s instructions (Roche Diagnostics, Indianapolis, IN, USA). Green staining indicates TUNEL-positive nuclei, while blue indicates total nuclei (DAPI). Ten fields per heart section were randomly selected for analysis under a fluorescence microscope (Nikon Instruments, Inc.). The apoptotic index was expressed as the proportion of apoptotic cells among the total cell population. Image-Pro Plus software was used for image analysis (Media Cybernetics, Inc., Carlsbad, CA, USA). Hearts were evaluated in a double-blind manner.
Western Blotting
Myocardial AAR tissue samples were collected at the end of the experiment and homogenized in lysis buffer. The protein concentration was measured by the BCA method (Pierce, Rockford, IL, USA). The samples were loaded on 12% SDS‒PAGE gels and transferred onto nitrocellulose membranes (Pall Corporation, Pensacola, FL, USA). The membrane was then blocked in 5% nonfat milk for 45 min, followed by incubation with primary antibodies against phosphorylated P38 MAPK (Thr180/Tyr182) (p-P38 MAPK), total P38 MAPK, phosphorylated extracellular signal-regulated kinase 1/2 (ERK1/2) (Thr202/Tyr204), total ERK1/2, phosphorylated glycogen synthase kinase-3β (Ser9) (p-GSK-3β Ser9), total-GSK-3β Ser9, phospho-AKT (Ser473, p-AKT), total AKT, phosphorylated STAT3 (Tyr705) (p-STAT3), total STAT3, phosphorylated STAT5 (Tyr694) (p-STAT5), and total STAT5 (all: rabbit, 1:1000; from Cell Signaling Technology, Danvers, MA, USA). β-catenin and β-actin were purchased from Affinity (all 1:1000; Affinity Biosciences, Cincinnati, OH, USA). The membrane was incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:5000; Bio-Rad, Hercules, CA, USA) for 2 h at room temperature. ECL™ Western Blotting Detection Reagents (GE Healthcare, Little Chalfont, UK) were used for protein detection. The band densities were detected by an Amersham Imager 600 system (GE Healthcare, Little Chalfont, UK) and quantified by ImageJ (National Institutes of Health, Bethesda, MD, USA). The densities of the phosphorylated proteins were normalized to the total protein. The densities of β-catenin were normalized to those of β-actin.
Immunohistochemistry
The expression levels of Bcl-2, Bax and phosphorylated ERK1/2 were assessed via immunohistochemistry (IHC). Heart sections were dewaxed and rehydrated. The tissue sections were then microwaved in an EDTA buffer oven twice for 8 min before being incubated in hydrogen peroxide for 20 min at 37 °C. The slices were then blocked with 5% goat serum for 40 min at 37 °C. Bcl-2, Bax, β-catenin and phosphorylated ERK1/2 primary antibodies (all, 1:200; Affinity Bioscience, Cincinnati, OH, USA) were used at 4 °C overnight. The sections were then incubated with a goat anti-rabbit secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Tissues were stained with 3,3'-diaminobenzidine solution (Beijing Zhongshan Golden Bridge Biotechnology, Beijing, China) and counterstained with hematoxylin. The sections were analyzed with Image-Pro Plus software (Media Cybernetics, Inc., Carlsbad, CA, USA). Ten random views were observed for each slice. Brown staining indicating positive expression was identified in the cytoplasm of cardiac myocytes.
Statistics
Statistical analysis was performed using GraphPad Prism 8 software (version 8.0; San Diego, CA, USA) or SPSS 16.0 software for Windows (SPSS, Inc., Chicago, IL, USA). P values < 0.05 indicated statistical significance. The normality of the distribution was evaluated by the Kolmogorov‒Smirnov test. The unpaired two-tailed Student's t test was used for comparisons of two groups. Hemodynamic data were analyzed by two-way repeated-measures ANOVA. For more than three groups, one-way ANOVA followed by the Newman‒Keuls test or Dunnett's T3 test was used for comparison.
Results
Infarct Size Measurement
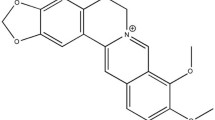
The molecular structure (Fig. 2A) and the chemical 3D structure (Fig. 2B) of aloperine (C15H24N2) are shown. Aloperine is a whitish solid (Fig. 2C) derived from the seeds of Sophora alopecuroides L. (Kudouzi) (Fig. 2D). To examine the cardioprotective effect of aloperine, a rat model of myocardial I/R injury was used in our study. Representative images of myocardial infarction after reperfusion from the control and aloperine groups are shown in Fig. 2E. The myocardial infarct size was 50.3 ± 9.2% in the control group. However, the infarct size was reduced to 36.1 ± 7.4% after aloperine treatment (P < 0.01 vs. CON; Fig. 2F). Similarly, the ratio of the AAR to the left ventricle was similar between the control (38.3 ± 3.2%) and aloperine (37.3 ± 4.0%) groups (Fig. 2G), indicating that LAD ligation affected similar areas in both groups.

Aloperine alleviated myocardial damage postreperfusion. The molecular structure (A), chemical 3D structure (B), appearance (C), and plant source (D) of aloperine. E Representative images of infarcted rat hearts. Heart tissue was harvested 3 h after reperfusion and stained by the triphenyltetrazolium chloride (TTC) method. CON, control; ALO, aloperine. F The effect of aloperine on infarct size, expressed as a percentage of the area at risk (AAR), in the control (CON) and aloperine (ALO) groups. n = 6–7 per group. ##P < 0.01 vs. CON (by unpaired t test). G Bar graph showing the area at risk from the left ventricle (AAR/LV). NS: nonsignificant difference between groups (P > 0.05, by unpaired t test). n = 6–7 per group. PostI/R mean serum LDH (H), CK-MB (I) and α-HBDH (J) levels in rats subjected to 45 min of myocardial ischemia and 3 h of reperfusion. n = 6–7 per group. ***P < 0.001 vs. sham, #P < 0.05, ##P < 0.01 vs. CON (by one-way ANOVA). LDH: lactate dehydrogenase; CK-MB: creatine kinase-MB; α-HBDH: α-hydroxybutyrate dehydrogenase. K Representative H&E-stained heart sections post-I/R. n = 5 rats per group. CON, control; ALO, aloperine. The arrow points at haemorrhage (red), edema (blue), or cytoplasmic vacuolation (black). L Histological evaluation of myocardial damage post-I/R injury using a grading system (see Methods section). n = 5 rats per group. ###P < 0.001 vs. CON (by unpaired t test)
Hemodynamic Measurements
Hemodynamic data are presented in Table 1. Repeated measures ANOVA was used to detect differences in LVSP (P < 0.001), LVEDP (P < 0.001), dP/dtmax (P < 0.01), and -dP/dtmax (P < 0.001) among the sham, CON, and ALO groups during the 3 h of reperfusion. Hemodynamic parameters were similar at baseline in each group. There was a significant interaction effect between the groups and time for LVSP (P < 0.001), LVEDP (P < 0.001), dP/dtmax (P < 0.01), and -dP/dtmax (P < 0.001). Myocardial I/R impaired cardiac function, as reflected by the decreases in LVSP and dP/dtmax and the increases in LVEDP and -dP/dtmax (P < 0.001 vs. sham). However, compared with CON, aloperine significantly improved cardiac hemodynamics, as measured by LVSP, LVEDP, dP/dtmax, and -dP/dtmax (P < 0.01 for all).
Serum Enzymes
LDH, CK-MB, and α-HBDH are serum biomarkers associated with cardiac damage. We tested the serum concentrations of the abovementioned enzymes and found that myocardial reperfusion injury caused significant elevations in the serum LDH, CK-MB and α-HBDH levels, which are indicative of myocardial damage post-I/R. However, the levels of these enzymes were markedly lower in the aloperine group than in the control group, suggesting that aloperine effectively ameliorated cardiac damage after reperfusion injury (P < 0.05 or P < 0.01; Fig. 2H–J).
Histological Evaluation
Heart sections were stained with H&E, and the severity of myocardial damage was evaluated. After reperfusion, different types of myocardial injury, including vacuolization, hemorrhage, edema and steatosis, were observed in control hearts. However, treatment with aloperine resulted in minimal myocardial degradation (Fig. 2K). The pathological score of the control rats was 2.6 ± 0.2, which was significantly greater than that of the aloperine-treated rats (1.1 ± 0.1, P < 0.001; Fig. 2L). In addition, we did not observe any necrosis in either group.
Apoptotic Measurement
TUNEL-positive cardiac myocytes were observed in all the experimental groups (Fig. 3A). However, after myocardial I/R, more apoptotic nuclei were observed in the control group (30.0 ± 4.1%) than in the aloperine group (17.8 ± 2.1%) (P < 0.001; Fig. 3B). The typical changes in Bcl-2 and Bax protein expression are displayed in Fig. 3C and D. The expression level of Bcl-2 (an antiapoptotic protein) was decreased upon reperfusion in the control and aloperine groups compared to that in the sham-operated group (P < 0.001). In contrast, the expression of Bax (a proapoptotic protein) was significantly increased in the CON and ALO groups post-I/R (P < 0.05 or P < 0.001 vs. sham). However, aloperine significantly increased the Bcl-2-to-Bax ratio (P < 0.01 vs. CON) by increasing the cardiac expression of Bcl-2 (P < 0.001 vs. CON) and decreasing Bax expression (P < 0.001 vs. CON), suggesting that aloperine treatment effectively prevents apoptosis (Fig. 3E–G).

Aloperine inhibits cardiac myocyte apoptosis. A Representative images of TUNEL-stained heart sections. Green indicates TUNEL-positive nuclei. Blue indicates total nuclei (DAPI). CON, control; ALO, aloperine. B The average percentage of apoptotic cells in the ischemic regions in each group. n = 5 per group. ###P < 0.001 vs. CON (by unpaired t test). Representative images of immunostaining for Bcl-2 (C) and Bax (D) proteins in heart sections post-I/R. n = 5 per group. sham: sham-operated; CON, control; ALO, aloperine. E–G Bar graph showing the analysis of Bcl-2 (E) and Bax (F) protein expression and the Bcl-2/Bax ratio (G) in heart sections after 3 h of myocardial reperfusion. n = 5 per group. *P < 0.005, ***P < 0.001 vs. sham, ##P < 0.01,###P < 0.001 vs. CON (by one-way ANOVA)
Aloperine Enhanced Ventricular ERK1/2 Phosphorylation post-I/R
We examined whether aloperine affects cell survival pathways after reperfusion. The expression of signaling molecules, including P38 MAPK, ERK1/2, GSK-3β, AKT, STAT-3 and STAT-5, was examined in this study. We checked the phosphorylation levels of each protein by normalization to the total protein. P38 MAPK was phosphorylated after I/R compared to that in the sham group (P < 0.01); however, aloperine did not further increase its phosphorylation level (P > 0.05 vs. CON, Fig. 4A). Strikingly, we previously found that ERK1/2 participates in aloperine-induced antiarrhythmic activities [10]. Here, we also showed that, compared with control treatment, aloperine treatment (1.5 ± 0.1) significantly enhanced ERK1/2 phosphorylation by 1.7-fold (0.9 ± 0.1, P < 0.001; Fig. 4B). We used an IHC assay to further evaluate ERK1/2 expression levels. Consistent with our western blotting results, we detected phospho-ERK1/2-immunoreactive cardiac myocytes in the ventricles of the mice in each group. Although the expression levels of p-ERK1/2 were increased in the CON and ALO groups compared with those in the sham-operated group (P < 0.001), aloperine significantly enhanced ERK1/2 phosphorylation by 2.5-fold compared to the control group (P < 0.001, Fig. 4C). These results indicate that the ERK1/2 pathway is associated with aloperine-induced cardioprotection. Rats in both the control and aloperine groups exhibited increased AKT phosphorylation (P < 0.01 vs. sham); however, the aloperine treatment did not further alter the phosphorylation status of AKT (P > 0.05; Fig. 4D) or its downstream target GSK-3β (P > 0.05; Fig. 4E). In addition, no differences were detected in the phosphorylation levels of STAT-3 (P > 0.05, Fig. 4F) or STAT-5 (P > 0.05, Fig. 4G) between the aloperine and control groups. Thus, the beneficial effect of aloperine was unlikely to be associated with the P38 MAPK, AKT, GSK-3β, STAT-3 and STAT-5 signaling pathways.

Aloperine enhances ventricular ERK1/2 phosphorylation post-I/R. A Representative western blot (left) and band densities (right) of ventricular p-P38 MAPK and t-P38 MAPK after I/R injury. n = 4–5 per group. **P < 0.01 vs. sham (one-way ANOVA). sham: sham-operated; CON, control; ALO, aloperine. B Representative western blot (left) and band densities (right) of ventricular p-ERK1/2 and t-ERK1/2. n = 4–5 per group. **P < 0.01 or ***P < 0.001 vs. sham. ###P < 0.001 vs. CON (by one-way ANOVA). C Representative images (left) and analysis (right) of immunostaining for p-ERK1/2. n = 5 per group. ***P < 0.001 vs. sham. ###P < 0.001 vs. CON (by one-way ANOVA). Representative western blot (left) and band densities (right) of ventricular p-AKT and t-AKT (D), p-GSK-3β and t-GSK-3β (E), p-STAT-3 and t-STAT-3 (F), and p-STAT-5 and t-STAT-5 (G). n = 4–5 per group. **P < 0.01 or ***P < 0.001 vs. sham (by one-way ANOVA)
The Effect of Aloperine on Ventricular β-catenin Expression
The β-catenin signaling pathway is an established pathway that participates in the pathophysiological process of I/R injury [13]. We therefore investigated the involvement of the β-catenin pathway in our experimental model. Western blotting revealed that after 3 h of reperfusion, the expression level of β-catenin was lower in control hearts (0.6 ± 0.2) than in sham-operated hearts (1.5 ± 0.1, P < 0.001). However, aloperine significantly upregulated β-catenin expression (1.1 ± 0.1, P < 0.001 vs. CON; Fig. 5A). Moreover, our immunohistochemistry results confirmed our western blotting results by showing that aloperine treatment significantly altered β-catenin expression compared with that in the control group (P < 0.01; Fig. 5B). Our data indicated that β-catenin may also play an important role in aloperine-mediated cardioprotection.

Aloperine alters ventricular β-catenin expression post-I/R. A Representative western blot (left) and band densities (right) of ventricular β-catenin after I/R injury. n = 4–5 per group. *P < 0.05 or ***P < 0.001 vs. sham, ###P < 0.001 vs. CON (by one-way ANOVA). sham: sham-operated; CON, control; ALO, aloperine. B Representative images (left) and analysis (right) of immunostaining for β-catenin. n = 5 per group. **P < 0.01 or ***P < 0.001 vs. sham. ##P < 0.01 vs. CON (by one-way ANOVA)
Inhibition of ERK1/2 Blocks ERK1/2/β-catenin Signaling and Recreates Myocardial Injury
To investigate the role of the ERK1/2-catenin signaling pathway in aloperine-induced cardioprotection against myocardial I/R injury, rats in the CON and ALO groups were treated with a specific ERK1/2 inhibitor (U0126) prior to cardiac reperfusion. After 3 h of reperfusion, the phosphorylation of ERK1/2 was decreased in the aloperine-treated hearts and in the hearts treated with U0126 (P < 0.001 vs. the non-U0126-treated ALO group; Fig. 6A). The results from the IHC assay also confirmed that aloperine-induced ERK1/2 phosphorylation was abolished by U0126 (P < 0.001, ALO vs. ALO + U0126; Fig. 6B). We then investigated the relationship between the ERK1/2 and β-catenin pathways and the associated cardioprotection of aloperine. Importantly, the results of western blot analysis indicated that the expression level of β-catenin was lower in the ALO + U0126 group than in the ALO group (P < 0.001; Fig. 7A). IHC assays also showed that U0126 decreased the increase in β-catenin in the aloperine-treated hearts (P < 0.01 vs. ALO; Fig. 7B). These data indicate that the ERK1/2-β-catenin signaling axis may participate in aloperine-mediated cardioprotection.

U0126 abolishes aloperine-induced ERK1/2 phosphorylation. A Representative western blot (left) and band densities (right) of ventricular phospho-ERK1/2 after I/R injury. n = 4–5 per group. †††P < 0.001 vs. ALO (by one-way ANOVA). sham: sham-operated; CON, control; ALO, aloperine. B Representative images (left) and analysis (right) of immunostaining for phospho-ERK1/2. n = 5 per group. U: U0126. †††P < 0.001 vs. ALO (by one-way ANOVA). Values for sham, CON and ALO were repeated from Fig. 4C for comparison

Effect of pharmacological inhibition of ERK1/2 phosphorylation on β-catenin expression post-I/R. A Representative western blot (left) and band densities (right) of ventricular β-catenin after I/R injury. n = 4–5 per group. ††P < 0.01 or †††P < 0.001 vs. ALO (by one-way ANOVA). sham: sham-operated; CON, control; ALO, aloperine. B Representative images (left) and analysis (right) of immunostaining for β-catenin. n = 5 per group. U: U0126. ††P < 0.01 vs. ALO (by one-way ANOVA). Values for sham, CON and ALO were repeated from Fig. 5B for comparison
We then examined whether pharmacological inhibition of ERK1/2 phosphorylation affects the beneficial effects of aloperine. As shown in Table 2, the effect of U0126 on systemic hemodynamics was examined. As expected, compared with those in the ALO group, the rats in the ALO + U0126 group exhibited impaired systolic and diastolic function, suggesting that pharmacological inhibition of ERK1/2 activity effectively abolished the hemodynamic benefits of aloperine. Typical images of TTC-stained heart sections with or without U0126 are shown in Fig. 8A. As shown in Fig. 8B, U0126 robustly disrupted the cardiac benefits induced by alloperine, as the infarct area ratio in the ALO + U0126 group was significantly greater than that in the ALO group after cardiac reperfusion (50.7 ± 6.3% versus 36.1 ± 7.4%, P < 0.01). There was no difference among the groups regarding the AAR/LV ratio (P > 0.05; Fig. 8C). In addition, we found that rats in the ALO + U0126 group exhibited higher serum LDH (Fig. 8D), CK-MB (Fig. 8E) and α-HBDH (Fig. 8F) levels than did those in the ALO group (P < 0.05), similar to those in the CON group. Moreover, aloperine-treated rats exhibited severe myocardial damage post-I/R in the presence of U0126, as evidenced by impaired cardiac structure and integrity (Fig. 8G) and increased pathological scores (P < 0.001 vs. ALO; Fig. 8H). We found that aloperine effectively inhibited cardiac apoptosis in our study (Fig. 3). However, aloperine-treated hearts treated with U0126 (30.6 ± 2.2%) exhibited more TUNEL-positive nuclei than did untreated hearts (17.8 ± 2.1%, P < 0.001; Fig. 9A). Furthermore, we also evaluated the expression levels of apoptosis-related proteins (Fig. 9B). Compared with those in the ALO group, the rats in the ALO + U0126 group had reduced ventricular expression of the Bcl-2 protein (P < 0.001; Fig. 9C), increased Bax expression (P < 0.001; Fig. 9D), and a decreased Bcl-2/Bax ratio (P < 0.01; Fig. 9E).

U0126 abolishes aloperine-induced cardioprotection post-I/R. A Representative images of TTC-stained infarcted rat hearts from each group treated with or without the inhibitor. CON, control; ALO, aloperine. B Effect of U0126 on infarct size after reperfusion. n = 6–7 per group. ††P < 0.01 vs. ALO (by one-way ANOVA). Values for CON and ALO were repeated from Fig. 2F for comparison. C AAR/LV ratio. n = 6–7 per group. Values for CON and ALO were repeated from Fig. 2G for comparison. Post-I/R mean serum levels of LDH (D), CK-MB (E) and α-HBDH (F). n = 6–7 per group. †P < 0.05 vs. ALO (by one-way ANOVA). LDH: lactate dehydrogenase; CK-MB: creatine kinase-MB; α-HBDH: α-hydroxybutyrate dehydrogenase. Values for sham, CON and ALO were repeated from Fig. 2H–J for comparison. G Representative images of H&E-stained heart sections from the CON and ALO groups treated with or without U0126. n = 5 rats per group. CON, control; ALO, aloperine. The arrow points at haemorrhage (red), edema (blue), or cytoplasmic vacuolation (black). H Histological evaluation. n = 5 rats per group. †††P < 0.001 vs. ALO (by one-way ANOVA). Values for CON and ALO were repeated from Fig. 2L for comparison

Effect of U0126 on myocardial apoptosis. A Left: Representative images of TUNEL-stained heart sections in the presence or absence of U0126. CON, control; ALO, aloperine. Right: Apoptotic nuclei/total nuclei ratio. n = 5 per group. †††P < 0.001 vs. ALO (by one-way ANOVA). Values for CON and ALO were repeated from Fig. 3B for comparison. B Representative images of immunostaining for the Bcl-2 (upper) and Bax (lower) proteins in heart sections. n = 5 per group. Analysis of Bcl-2 (C) and Bax (D) protein expression and the Bcl-2/Bax ratio (E) post-I/R. n = 5 per group. ††P < 0.01 or †††P < 0.001 vs. ALO (by one-way ANOVA). Values for sham, CON and ALO were repeated from Fig. 3E–G for comparison
Discussion
The results of the present study revealed the beneficial effects of aloperine on the prevention of myocardial I/R injury. Specifically, it limited infarct size, improved hemodynamics, decreased pathological scores and inhibited cardiac apoptosis. Our data also showed that enhanced ventricular ERK1/2 phosphorylation post-I/R may play a role in aloperine-induced cardioprotection. To the best of our knowledge, this is the first study showing the infarct-sparing effect of aloperine in a murine model of myocardial I/R injury.
Sophora alopecuroides Linn (Ku-Dou-Zi) has several active components, among which quinolizidine alkaloids serve as the main active ingredients. Evidence has shown that aloperine-type alkaloids have many significant therapeutic functions, including organ protection [14]. It can suppress pulmonary necroptosis in lipopolysaccharide-induced acute lung injury [15], regulate inflammatory responses in dextran sodium sulfate-induced colitis [16], protect pancreatic β-cells against streptozotocin-induced injury [17], protect kidneys against ischemia‒reperfusion-induced acute renal injury [18], alleviate brain damage against cerebral I/R injury [19], and protect lungs against bleomycin-induced pulmonary fibrosis [20]. In addition, aloperine is beneficial for the vascular system. Aloperine was shown to have KCNQ-dependent vasorelaxant effects [7] and could ameliorate monocrotaline-induced pulmonary artery remodeling [8]. In terms of the heart, the effect of aloperine is largely unknown. Few studies have been conducted to evaluate the cardioprotective effect of aloperine. Aloperine was first identified as a cardioprotective drug target in a rat model of coronary microembolization-induced myocardial injury [9]. We next found that it exerted strong antiarrhythmic effects against severe lethal ventricular arrhythmia and sudden cardiac death [10]. Here, we further demonstrated the occurrence of an aloperine-induced infarct-sparing effect. We showed that aloperine administered at the onset of reperfusion effectively attenuated myocardial damage post-I/R, as evidenced by a reduced infarct size, inhibited cardiac apoptosis and ameliorated hemodynamics. Our results are in agreement with those of others showing that sophocarpine, another active ingredient compound of Sophora alopecuroides L., could significantly limit infarct size and improve cardiac function against myocardial I/R injury [11].
Cell death is one of the major pathophysiological characteristics of myocardial I/R injury. Regulated cell death pathways, such as ferroptosis and necroptosis, play important physiological roles during ischemia–reperfusion conditions [21]. Ferroptosis is characterized by lipid peroxidation and is triggered by excessive iron [22]. Uncontrolled accumulation of iron causes ROS accumulation, lipid peroxidation, and cell death. Ferroptosis inhibitors, such as liproxstatin-1, can ameliorate myocardial damage after myocardial I/R injury [23]. In addition, several traditional Chinese herbal medicines or natural compounds have ferroptosis-inhibiting abilities. For example, baicalin, a natural flavonoid isolated from Scutellaria root, protects the heart against myocardial I/R injury by suppressing ACSL4-mediated ferroptosis [24]. Naringenin, which is widely distributed in citrus fruits, was shown to have an infarct-sparing effect by inhibiting ferroptosis by regulating the Nrf2-GPX4 axis [25]. Furthermore, resveratrol, a kind of natural polyphenol, inhibits ferroptosis and ameliorates myocardial damage after myocardial I/R injury [26]. In addition, necroptosis, another pathway of regulated necrosis, plays an important role in myocardial I/R injury. Activation of necrosomes, including RIP3, RIP1, and MLKL, is needed, leading to cell swelling, inflammation, cell membrane disruption and cell death [27]. The expression levels of RIP1, RIP3 and MLKL were elevated in mouse hearts after myocardial I/R injury [28]. The application of necrostatin-1, a RIP1-targeted inhibitor of necroptosis, resulted in alleviated cell death [29]. All of these results support the theory that ferroptosis and necroptosis may serve as potential therapeutic targets for myocardial ischemia–reperfusion injury. Apoptosis, another form of programmed cell death, has been extensively studied in various animal models. It can be initiated through the extrinsic or death receptor pathway and the intrinsic or mitochondrial pathway. In the present study, we showed that the aloperine group had fewer TUNEL-positive cells after reperfusion than did the control group. Moreover, aloperine treatment caused an increase in Bcl-2 and a reduction in Bax, resulting in an increase in the Bcl-2/Bax ratio. These data revealed that aloperine may protect hearts against myocardial I/R injury by inhibiting cardiac apoptosis. Ferroptosis, necroptosis and apoptosis may have complementary effects on myocardial I/R injury. The underlying mechanism through which aloperine could confer a protective signal to the heart is not completely known. Although the beneficial effect of aloperine was confirmed in the present study, the regulatory role of aloperine in necroptosis and ferroptosis needs further attention. Future experimental or clinical studies are needed to confirm its efficacy and mechanism against myocardial I/R injury.
This study is the first to demonstrate the infarct-sparing effect of aloperine against myocardial I/R injury, despite the established knowledge of its existing protective role in other organs. Furthermore, we explored the underlying protective mechanism associated with survival signaling pathways (Fig. 10). The extracellular signal-regulated kinase (ERK) pathway and the p38 MAPK pathway are vital signaling components of the mitogen-activated protein kinase (MAPK) pathway that can modulate various biological activities, including cell survival, apoptosis, death, proliferation, and differentiation [30]. ERK1/2 is widely expressed and can be activated by both extracellular and intracellular stimuli. It has been reported that protective stimuli or reagents, such as ischemic preconditioning [31] or postconditioning [32] and SGLT2 inhibitors [33, 34], can cause ERK1/2 activation; therefore, pharmacological inhibition of ERK1/2 phosphorylation could abolish their protective effects. The current study revealed that, compared with the control treatment, aloperine significantly enhanced ventricular ERK1/2 phosphorylation, while U0126, a specific ERK1/2 inhibitor, inhibited ERK1/2 phosphorylation. Further analysis demonstrated that U0126 nullified the aloperine-induced infarct-sparing effect, accompanied by the persistence of cardiac damage in the aloperine-treated hearts, as evidenced by increased apoptosis, elevated serum cardiac enzyme levels, and deteriorated myocardial histology. These results indicate that the ERK1/2 signaling pathway may contribute to the aloperine-mediated anti-myocardial infarction effect after myocardial I/R injury. Our current study confirms the vital role of ERK1/2 in the aloperine-induced infarct-sparing effect against myocardial I/R injury and is in agreement with our previous study in which activation of the ERK1/2 pathway was related to an antiarrhythmic effect of aloperine against lethal ventricular arrhythmias and sudden cardiac death post-I/R injury [10].

Schematic illustration of the potential mechanisms of aloperine-induced cardioprotection in myocardial ischemia and reperfusion injury
Reactive oxygen species (ROS), which include superoxide anions, hydrogen peroxide and hydroxyl radicals, are often associated with oxidative stress. However, elevated ROS also serve as signaling molecules to regulate physiological functions [35]. Increasing evidence has shown that ERK1/2 is a redox-regulated kinase. An increase in ERK1/2 phosphorylation is associated with activation of the redox cascade. Redox-induced ERK activation has been detected in different cell types. For example, in lung tumor cells, ERK activity was linked to redox-induced cell proliferation [36]. Furthermore, Miyamoto et al. reported that ERK activation was dependent on the redox state in PC12 cells [37]. Redox-induced ERK activation was also shown in cardiomyocytes [38]. In addition, the redox system was shown to participate in cardioprotective signal transduction. It acts as a trigger of IPC and can posttranslationally modify redox-sensitive proteins, including ERK1/2 [39]. Evidence has shown that oxidative stress indirectly regulates ERK. It triggers other cytokines and growth factors, such as EGFR, upstream of the MAPK/ERK pathway and subsequently activates the ERK molecule [40]. It has been indicated that morphine treatment protects the heart against myocardial I/R injury. The underlying mechanism may involve promoting intracellular ROS generation and activation of ERK via ROS-EGFR signaling [41]. Notably, Penna et al. showed that acidic early reperfusion protected hearts against myocardial I/R injury and activated prosurvival kinases, possibly by modulating the redox environment [42]. Notably, aloperine has been shown to have antioxidative effects in other animal models [43], and it can attenuate hydrogen peroxide-induced injury [44]. The compartmentalization of ROS determines whether cell death or redox-related signaling occurs. Aloperine can mediate the balance between ROS production and scavenging in response to deleterious stimuli and may affect redox homeostasis, thus triggering cellular redox signaling and resulting in cardioprotection. We found that aloperine activated ERK1/2, a redox-sensitive kinase. However, our current study design cannot elucidate how aloperine mediates ROS production and therefore activates ERK1/2 during myocardial I/R injury. It is likely that ROS may act as triggers and mediators of aloperine-mediated cardioprotection and ERK1/2 activation.
The p38 MAPK pathway is a stress-response kinase. The p38 pathway is activated upon ischemia‒reperfusion [45]. AKT is a powerful survival signal that serves as an important modulator of cardiac function [46]. As a downstream signaling target of AKT, phosphorylation at serine 9 of GSK-3β leads to its deactivation (inhibition) and thus may be a contributing mechanism to the benefits observed in ischemic conditioning [47] Here, we tested the ventricular phosphorylation levels of these abovementioned proteins and found that aloperine-treated hearts exhibited no further enhancement of their phosphorylation levels when compared with control hearts. These findings indicate that the P38 MAPK, AKT and GSK-3β pathways are not involved in aloperine-induced cardioprotection against myocardial I/R injury. Moreover, STAT-3 and STAT-5 belong to the signal transducer and activator of transcription (STAT) family. STAT-3 can be activated through phosphorylation of its tyrosine 705 residue (Y705) [48]. We previously found that the aloperine-induced antiarrhythmic effect was associated with the activation of STAT-3; furthermore, the beneficial effect was diminished by the administration of a STAT-3 inhibitor [10]. In contrast, in our current study, we found that the cardioprotective effect of aloperine was not accompanied by the upregulation of STAT-3 phosphorylation. We also tested whether STAT5, another established cardioprotective protein in the STAT family, is activated upon aloperine treatment. We previously found that the phosphorylation level of STAT-5 was increased in ischemic, conditioned diabetic and nondiabetic hearts after I/R injury in rats [49]. In contrast, in a model of myocardial I/R-induced lethal arrhythmia, we failed to detect alterations in STAT-5 phosphorylation [10]. In the present study, using a rat model of 45 min of LAD ligation followed by 3 h of reperfusion, we did not observe any difference in STAT-5 activation between groups treated with or without aloperine. Taken together, the results of the present study suggest that aloperine exerts a cardioprotective effect against myocardial I/R injury through activation of the ERK1/2 signaling pathway but not via other signaling pathways, such as the p38 MAPK, AKT, GSK-3β, STAT-3 or STAT-5 pathways.
The β-catenin pathway is a vital regulator of cell survival, proliferation and development [50]. It is associated with the pathophysiological process of myocardial I/R injury. Previous studies have demonstrated that myocardial I/R injury is associated with the downregulation of β-catenin [51], and its overexpression may reduce myocardial infarct size after MI [52]. Moreover, knockdown of HIF-1α and β-catenin exacerbates oxygen–glucose deprivation/reperfusion-induced cardiac myocyte injury [53]. However, protective stimuli, such as ischemic preconditioning, can cause β-catenin accumulation in cardiac myocytes [54]. In agreement with these findings, in the present study, we observed that, under ischemic conditions, the expression of β-catenin was significantly attenuated. Furthermore, we showed that aloperine treatment protected the heart, and this effect was accompanied by an increase in the level of β-catenin, which may serve as a downstream target of GSK-3β. The inhibition of GSK-3β activity using GSK-3β inhibitors or protective stimuli may cause GSK-3β phosphorylation and lead to an increase in β-catenin levels [55]. In the present study, we did not observe any significant evidence that GSK-3β is involved in aloperine-mediated cardioprotection; thus, it is logical to deduce that the increase in ventricular β-catenin levels in the aloperine group was unlikely to be associated with GSK-3β inhibition. Notably, in the present study, ERK1/2 phosphorylation was enhanced in the aloperine-treated group. The ERK1/2 and β-catenin pathways may participate in cross-talk interactions with other cell types that may modulate cell differentiation [56]. However, the relationship between these two pathways and aloeperine-associated cardioprotection against myocardial I/R injury remains obscure. With the use of U-126, a specific ERK1/2 inhibitor, we found that the inhibition of ERK1/2 activity blunted aloperine-induced ERK1/2 phosphorylation while abrogating the increase in β-catenin in aloperine-treated hearts, indicating that aloperine-mediated cardioprotection may be associated with the ERK1/2/β-catenin signaling axis. Notably, Funato et al. showed that treating cells with a low dose of H2O2 enabled β-catenin activation and further initiated the expression of specific genes relevant to cell survival and proliferation [57]. These findings indicate that redox signaling may interact with β-catenin signaling. We found that aloperine activated ERK1/2, a redox-sensitive kinase, while U0126 affected the aloperine-induced increase in β-catenin in our study. Moreover, the EGFR-ERK signaling cascade can mediate β-catenin transactivation [58]. EGFR is the downstream target of ROS. This result supports the possible interactions of ROS-EGFR-ERK and β-catenin signaling. However, based on the current study design, we cannot explain what caused the increase in β-catenin expression in mice exposed to aloperine. It is possible that aloperine may mediate ROS and redox signaling, to some extent, by regulating the β-catenin pathway.
Our study has several limitations. First, aloperine was given at the onset of myocardial reperfusion (postconditioning). Although this approach is the most clinically feasible strategy, it remains unknown whether other timeframes, such as before myocardial ischemia (preconditioning) or during ischemia (perconditioning), would confer cardioprotection. Moreover, the dose‒response effect and dose‒limiting toxicity of aloperine should be further determined. Second, our current study was performed with 45 min of ischemia and 3 h of reperfusion, and our results indicated only the AMI and early reperfusion stages. However, whether aloperine affects long-term survival after myocardial infarction or cardiac fibrosis deserves further investigation. Third, aloperine was intravenously injected into the experimental animals via the femoral vein in our study, and it will be of great interest to test the beneficial effect of aloperine via other noninvasive drug delivery routes. Fourth, we evaluated several major survival signaling pathways associated with reperfusion injury and found that the cardioprotective effect of aloperine is related to the ERK1/2/β-catenin signaling pathway. However, how aloperine regulates the ERK1/2/β-catenin signaling cascade in cardiomyocytes requires further investigation. Furthermore, identification and characterization of other signaling molecules and pathways are necessary in the future.
In summary, our study demonstrated that aloperine exerts a strong cardioprotective effect against myocardial I/R injury, as evidenced by reduced infarct size, ameliorated cardiac damage and improved cardiac function. This aloperine-induced cardioprotection is associated with an ERK1/2/β-catenin-dependent signaling pathway. These data may suggest potential clinical applications of aloperine in the treatment of cardiac damage after reperfusion injury in clinical settings.
Data Availability
All the data in this study are available upon reasonable request from the corresponding author.
References
Collaborators GBDCoD. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1151–210. https://doi.org/10.1016/S0140-6736(17)32152-9.
Heusch G. Myocardial ischemia: lack of coronary blood flow, myocardial oxygen supply-demand imbalance, or what? Am J Physiol Heart Circ Physiol. 2019;316(6):H1439–46. https://doi.org/10.1152/ajpheart.00139.2019.
El Nasasra A, Zeymer U. Current clinical management of acute myocardial infarction complicated by cardiogenic shock. Expert Rev Cardiovasc Ther. 2021;19(1):41–6. https://doi.org/10.1080/14779072.2021.1854733.
Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357(11):1121–35. https://doi.org/10.1056/NEJMra071667.
Tahir M, Ali S, Zhang W, et al. Aloperine: a potent modulator of crucial biological mechanisms in multiple diseases. Biomedicines. 2022;10(4). https://doi.org/10.3390/biomedicines10040905.
Yang C, Yu Y, Wu F, et al. Vasodilatory effects of aloperine in rat aorta and its possible mechanisms. Chin J Physiol. 2018;61(5):293–301. https://doi.org/10.4077/CJP.2018.BAH609.
Manville RW, van der Horst J, Redford KE, et al. KCNQ5 activation is a unifying molecular mechanism shared by genetically and culturally diverse botanical hypotensive folk medicines. Proc Natl Acad Sci USA. 2019;116(42):21236–45. https://doi.org/10.1073/pnas.1907511116.
Wu F, Yao W, Yang J, et al. Protective effects of aloperin on monocroline-induced pulmonary hypertension via regulation of Rho A/Rho kinsase pathway in rats. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;95:1161–8. https://doi.org/10.1016/j.biopha.2017.08.126.
Mao Q, Guo F, Liang X, Wu Y, Lu Y. Aloperine activates the PI3K/Akt pathway and protects against coronary microembolisation-induced myocardial injury in rats. Pharmacology. 2019;104(1–2):90–7. https://doi.org/10.1159/000500761.
Hu Z, Li J, Liu Q, Manville RW, Abbott GW. The plant-derived alkaloid aloperine prevents ischemia/reperfusion injury-induced sudden cardiac death. FASEB J: Off Publ Fed Am Soc Exp Biol. 2023;37(7):e22999. https://doi.org/10.1096/fj.202300253R.
Li C, Gao Y, Tian J, et al. Sophocarpine administration preserves myocardial function from ischemia-reperfusion in rats via NF-kappaB inactivation. J Ethnopharmacol. 2011;135(3):620–5. https://doi.org/10.1016/j.jep.2011.03.052.
Hu Z, Crump SM, Zhang P, Abbott GW. Kcne2 deletion attenuates acute postischemia/reperfusion myocardial infarction. Cardiovasc Res. 2016;110(2):227–37. https://doi.org/10.1093/cvr/cvw048.
Schunk SJ, Floege J, Fliser D, Speer T. WNT-beta-catenin signaling - a versatile player in kidney injury and repair. Nat Rev Nephrol. 2021;17(3):172–84. https://doi.org/10.1038/s41581-020-00343-w.
Li Y, Wang G, Liu J, Ouyang L. Quinolizidine alkaloids derivatives from Sophora alopecuroides Linn: bioactivities, structure-activity relationships and preliminary molecular mechanisms. Eur J Med Chem. 2020;188:111972. https://doi.org/10.1016/j.ejmech.2019.111972.
Cui YR, Qu F, Zhong WJ, et al. Beneficial effects of aloperine on inflammation and oxidative stress by suppressing necroptosis in lipopolysaccharide-induced acute lung injury mouse model. Phytomed : Int J Phytother Phytopharmacol. 2022;100:154074. https://doi.org/10.1016/j.phymed.2022.154074.
Fu X, Sun F, Wang F, et al. Aloperine protects mice against DSS-induced colitis by PP2A-mediated PI3K/Akt/mTOR signaling suppression. Mediat Inflamm. 2017;2017:5706152. https://doi.org/10.1155/2017/5706152.
He W, Zhou H, He X. Aloperine protects beta-cells against streptozocin-induced injury to attenuate diabetes by targeting NOS1. Eur J Pharmacol. 2022;916:174721. https://doi.org/10.1016/j.ejphar.2021.174721.
Hu S, Zhang Y, Zhang M, et al. Aloperine protects mice against Ischemia-Reperfusion (IR)-Induced renal injury by regulating PI3K/AKT/mTOR signaling and AP-1 activity. Mol Med. 2016;21(1):912–23. https://doi.org/10.2119/molmed.2015.00056.
Li Z, Cao X, Xiao L, Zhou R. Aloperine protects against cerebral ischemia/reperfusion injury by activating the PI3K/AKT signaling pathway in rats. Exp Ther Med. 2021;22(4):1045. https://doi.org/10.3892/etm.2021.10478.
Yin W, Han J, Zhang Z, Han Z, Wang S. Aloperine protects mice against bleomycin-induced pulmonary fibrosis by attenuating fibroblast proliferation and differentiation. Sci Rep. 2018;8(1):6265. https://doi.org/10.1038/s41598-018-24565-y.
Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99(4):1765–817. https://doi.org/10.1152/physrev.00022.2018.
Guo Y, Zhang W, Zhou X, et al. Roles of ferroptosis in cardiovascular diseases. Front Cardiovasc Med. 2022;9:911564. https://doi.org/10.3389/fcvm.2022.911564.
Feng Y, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun. 2019;520(3):606–11. https://doi.org/10.1016/j.bbrc.2019.10.006.
Fan Z, Cai L, Wang S, Wang J, Chen B. Baicalin prevents myocardial ischemia/reperfusion injury through inhibiting ACSL4 mediated ferroptosis. Front Pharmacol. 2021;12:628988. https://doi.org/10.3389/fphar.2021.628988.
Xu S, Wu B, Zhong B, et al. Naringenin alleviates myocardial ischemia/reperfusion injury by regulating the nuclear factor-erythroid factor 2-related factor 2 (Nrf2)/System xc-/glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis. Bioengineered. 2021;12(2):10924–34. https://doi.org/10.1080/21655979.2021.1995994.
Li T, Tan Y, Ouyang S, He J, Liu L. Resveratrol protects against myocardial ischemia-reperfusion injury by attenuating ferroptosis. Gene. 2022;808:145968. https://doi.org/10.1016/j.gene.2021.145968.
Guo X, Chen Y, Liu Q. Necroptosis in heart disease: molecular mechanisms and therapeutic implications. J Mol Cell Cardiol. 2022;169:74–83. https://doi.org/10.1016/j.yjmcc.2022.05.006.
Oerlemans MI, Liu J, Arslan F, et al. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol. 2012;107(4):270. https://doi.org/10.1007/s00395-012-0270-8.
Cao L, Mu W. Necrostatin-1 and necroptosis inhibition: pathophysiology and therapeutic implications. Pharmacol Res. 2021;163:105297. https://doi.org/10.1016/j.phrs.2020.105297.
Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90(4):1507–46. https://doi.org/10.1152/physrev.00054.2009.
Rossello X, Yellon DM. The RISK pathway and beyond. Basic Res Cardiol. 2018;113(1):2. https://doi.org/10.1007/s00395-017-0662-x.
Cheng X, Li H, Yan Z, Liu J, Hu Z. Ischemic limb preconditioning-induced anti-arrhythmic effect in reperfusion-induced myocardial injury: is it mediated by the RISK or SAFE pathway? Pflugers Arch. 2022;474(9):979–91. https://doi.org/10.1007/s00424-022-02716-5.
Hu Z, Ju F, Du L, Abbott GW. Empagliflozin protects the heart against ischemia/reperfusion-induced sudden cardiac death. Cardiovasc Diabetol. 2021;20(1):199. https://doi.org/10.1186/s12933-021-01392-6.
Huang D, Ju F, Du L, et al. Empagliflozin protects against pulmonary ischemia/reperfusion injury via an extracellular signal-regulated kinases 1 and 2-Dependent mechanism. J Pharmacol Exp Ther. 2022;380(3):230–41. https://doi.org/10.1124/jpet.121.000956.
Sies H, Belousov VV, Chandel NS, et al. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat Rev Mol Cell Biol. 2022;23(7):499–515. https://doi.org/10.1038/s41580-022-00456-z.
Galli S, Antico Arciuch VG, Poderoso C, et al. Tumor cell phenotype is sustained by selective MAPK oxidation in mitochondria. PLoS ONE. 2008;3(6):e2379. https://doi.org/10.1371/journal.pone.0002379.
Miyamoto Y, Sakai R, Maeda C, et al. Nitric oxide promotes nicotine-triggered ERK signaling via redox reactions in PC12 cells. Nitric Oxide Biol Chem. 2011;25(3):344–9. https://doi.org/10.1016/j.niox.2011.06.006.
Xiao L, Pimentel DR, Wang J, et al. Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol. 2002;282(4):C926–34. https://doi.org/10.1152/ajpcell.00254.2001.
Otani H. Reactive oxygen species as mediators of signal transduction in ischemic preconditioning. Antioxid Redox Signal. 2004;6(2):449–69. https://doi.org/10.1089/152308604322899521.
Jaenen V, Fraguas S, Bijnens K, et al. Reactive oxygen species rescue regeneration after silencing the MAPK-ERK signaling pathway in Schmidtea mediterranea. Sci Rep. 2021;11(1):881. https://doi.org/10.1038/s41598-020-79588-1.
Xu J, Bian X, Zhao H, et al. Morphine prevents ischemia/reperfusion-induced myocardial mitochondrial damage by activating delta-opioid Receptor/EGFR/ROS pathway. Cardiovasc Drugs Ther. 2022;36(5):841–57. https://doi.org/10.1007/s10557-021-07215-w.
Penna C, Perrelli MG, Tullio F, Angotti C, Pagliaro P. Acidic infusion in early reperfusion affects the activity of antioxidant enzymes in postischemic isolated rat heart. J Surg Res. 2013;183(1):111–8. https://doi.org/10.1016/j.jss.2012.12.029.
Zhou H, Li J, Sun F, et al. A review on recent advances in aloperine research: pharmacological activities and underlying biological mechanisms. Front Pharmacol. 2020;11:538137. https://doi.org/10.3389/fphar.2020.538137.
Ren D, Ma W, Guo B, Wang S. Aloperine attenuates hydrogen peroxide-induced injury via anti-apoptotic activity and suppression of the nuclear factor-kappaB signaling pathway. Exp Ther Med. 2017;13(1):315–20. https://doi.org/10.3892/etm.2016.3962.
Steenbergen C. The role of p38 mitogen-activated protein kinase in myocardial ischemia/reperfusion injury; relationship to ischemic preconditioning. Basic Res Cardiol. 2002;97(4):276–85. https://doi.org/10.1007/s00395-002-0364-9.
Mullonkal CJ, Toledo-Pereyra LH. Akt in ischemia and reperfusion. J Invest Surg: Off J Acad Surg Res. 2007;20(3):195–203. https://doi.org/10.1080/08941930701366471.
Yang S, Abbott GW, Gao WD, et al. Involvement of glycogen synthase kinase-3beta in liver ischemic conditioning induced cardioprotection against myocardial ischemia and reperfusion injury in rats. J Appl Physiol. 2017;122(5):1095–105. https://doi.org/10.1152/japplphysiol.00862.2016.
Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19(21):2628–37. https://doi.org/10.1038/sj.onc.1203481.
Liu X, Chen H, Yan Z, et al. Remote liver ischemic preconditioning attenuates myocardial ischemia/reperfusion injury in streptozotocin-induced diabetic rats. Sci Rep. 2021;11(1):1903. https://doi.org/10.1038/s41598-021-81422-1.
Stamos JL, Weis WI. The beta-catenin destruction complex. Cold Spring Harb Perspect Biol. 2013;5(1):a007898. https://doi.org/10.1101/cshperspect.a007898.
Ban Q, Qiao L, Xia H, et al. beta-catenin regulates myocardial ischemia/reperfusion injury following heterotopic heart transplantation in mice by modulating PTEN pathways. Am J Transl Res. 2020;12(8):4757–71.
Hahn JY, Cho HJ, Bae JW, et al. Beta-catenin overexpression reduces myocardial infarct size through differential effects on cardiomyocytes and cardiac fibroblasts. J Biol Chem. 2006;281(41):30979–89. https://doi.org/10.1074/jbc.M603916200.
Dong W, Weng JF, Zhu JB, et al. CREB-binding protein and HIF-1alpha/beta-catenin to upregulate miR-322 and alleviate myocardial ischemia-reperfusion injury. FASEB J: Off Publ Fed Am Soc Exp Biol. 2023;37(9):e22996. https://doi.org/10.1096/fj.202200596RRRRRR.
Kaga S, Zhan L, Altaf E, Maulik N. Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic preconditioned myocardium. J Mol Cell Cardiol. 2006;40(1):138–47. https://doi.org/10.1016/j.yjmcc.2005.09.009.
Ni B, Sun M, Zhao J, Wang J, Cao Z. The role of beta-catenin in cardiac diseases. Front Pharmacol. 2023;14:1157043. https://doi.org/10.3389/fphar.2023.1157043.
Tian L, Xiao H, Li M, et al. A novel Sprouty4-ERK1/2-Wnt/beta-catenin regulatory loop in marrow stromal progenitor cells controls osteogenic and adipogenic differentiation. Metabolism: Clin Exp. 2020;105:154189. https://doi.org/10.1016/j.metabol.2020.154189.
Funato Y, Michiue T, Asashima M, Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signaling through dishevelled. Nat Cell Biol. 2006;8(5):501–8. https://doi.org/10.1038/ncb1405.
Ji H, Wang J, Nika H, et al. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol Cell. 2009;36(4):547–59. https://doi.org/10.1016/j.molcel.2009.09.034.
Funding
This study was supported by Grant No. 82270326 (to ZH) from the National Natural Science Foundation of China.
Author information
Authors and Affiliations
Contributions
Participated in the research design: Zhaoyang Hu
Conducted experiments and the study: Shichao Wei, Feng Ju, Junshen Xiao, Jiaxue Li, Ting Liu, Zhaoyang Hu
Performed the data analysis or interpreted the data: Shichao Wei, Feng Ju, Junshen Xiao, Jiaxue Li, Ting Liu, and Zhaoyang Hu
Wrote or contributed to the writing of the manuscript: Zhaoyang Hu
Corresponding author
Ethics declarations
Ethics Approval
All the experimental procedures complied with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH Publication 8th edition, 2011) and were approved by the Institutional Animal Care and Use Committee of Sichuan University (approval number: 20211709A).
Consent to Participate
Not applicable.
Consent to Publish
Not applicable.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wei, S., Ju, F., Xiao, J. et al. Aloperine Alleviates Myocardial Injury Induced by Myocardial Ischemia and Reperfusion by Activating the ERK1/2/β-catenin Signaling Pathway. Cardiovasc Drugs Ther (2024). https://doi.org/10.1007/s10557-024-07566-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s10557-024-07566-0