
Abstract
Increasing reports demonstrate that long noncoding RNAs participate in the regulation of numerous malignancies, cervical cancer included. Although lncRNA LOXL1 antisense RNA 1 has been commonly accepted to be an oncogene in many cancers. Here, the role of LOXL1-AS1 in CC still need to be explored. In this study, LOXL1-AS1 was found elevated in CC tissues and cells. LOXL1-AS1 depletion restrained CC cell proliferation, migration, invasion, and angiogenesis in vivo. Furthermore, we found that LOXL1-AS1 upregulated Lysophospholipase 1 expression via sequestering miR‐526b-5p. Rescue assays revealed that overexpression of LYPLA1 reversed the LOXL1-AS1 silencing-induced inhibitory effects on the malignant phenotypes of CC cells. To conclude, this study showed that LOXL1-AS1 facilitates cellular process in CC via functioning as a miR‐526b-5p sponge.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cervical cancer is a common gynecological malignancy, which has become an important public health issue (Marth et al. 2017). It is widely acknowledged that the occurrence of CC results from the interaction between genetic and environmental factors (Niu 2019). The latest statistics show that over 529,800 females are suffering from CC and over 275,100 people die of CC each year, making CC the second fastest growing cancer (Forouzanfar et al. 2011; Kim et al. 2015). Moreover, CC is a leading cause of high mortality in developing countries where access to high-quality healthcare systems that facilitate timely screening and early diagnosis of CC may be limited (Luan and Wang 2018). Despite significant progress made in clinical treatment for CC, the 5-year overall survival of patients with CC is at a poor level (below 40%) (Zhao et al. 2012). Thus, it is exigent to figure out more diagnostic or therapeutic biomarkers for CC.
Long noncoding RNAs (lncRNAs) are transcripts of longer than 200 nucleotides and have limited protein-coding capacity (Bunch 2018). A multitude of studies have revealed the important regulatory functions of lncRNAs in various cellular processes of cancer (Gibb et al. 2011). The differentially expressed lncRNAs have been found in diverse types of cancers (Mercer et al. 2009). For example, MATN1-AS1 suppresses glioblastoma cell proliferation and migration through the modulation of RELA and MAPK signaling (Qiu et al. 2020). LL22NC03-N64E9.1 is a possible prognostic molecular biomarker for lung cancer treatment and promotes cancer cell proliferation (Jing et al. 2018). Additionally, lncRNAs are widely reported to regulate CC progression. OIP5-AS1 increases cell growth in CC by controlling the availability of miR-143-3p to upregulate SMAD3 (Chen et al. 2019). TPT1-AS1 is upregulated in CC and TPT1-AS1 overexpression facilitates CC cell growth, migration, invasion ability by serving as a miR-324-5p sponge (Jiang et al. 2018). Recently, lncRNA LOXL1 antisense RNA 1 (LOXL1-AS1) has been found to exert carcinogenic effects in many cancers. For instance, LOXL1-AS1 contributes to the aggressive behaviors of ovarian cancer cells through the miR-18b-5p/VMA21 axis (Xue 2020). LOXL1-AS1 promotes oncogenic activities in CC cells by miR-28-5p to upregulate RAP1B (Yang 2020). However, the mechanism of LOXL1-AS1 in CC remains largely unexplored, which needs further study.
This study aimed to investigate the expression pattern and biological effects of LOXL1-AS1 in CC. Our findings might provide an innovative idea for CC treatment.
Materials and Methods
Clinical Tissues Collection
Fifty paired CC samples and noncancerous samples were obtained from patients who underwent operation in Binhai County People’s Hospital. The patients were pathologically diagnosed with CC. The specimens were immediately placed in liquid nitrogen after collection and transferred into a refrigerator at −80 °C. The patients and their families signed informed consent, and all the experiments in this study were approved by the Ethics Committee of Binhai County People’s Hospital. None of these patients had undergone antitumor treatment before the operation.
Cell Lines
CC cell lines (HeLa, CaSki, C33A, and SiHa) and the immortalized squamous cells of human cervix Ect1/E6E7 were provided from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS; Gibco, NY, USA) and 1% penicillin/streptomycin (Biochrom, UK) in an incubator with 5% CO2 at 37 °C.
Transfection
To downregulate LOXL1-AS1 expression, short hairpin RNAs (sh-LOXL1-AS1#1 or #2) were purchased from GenePharma (Shanghai, China). Sequence of LYPLA1 was constructed with pcDNA3.1 (Invitrogen, USA) to generate pcDNA3.1/LYPLA1. MiR-526b-5p mimics and NC mimics or miR-526b-5p inhibitor and NC inhibitor were supplied by GenePharma. HeLa and CaSki cells were seeded into 6-well plates at 1 × 10 cells/well and were transfected with the indicated plasmids using Lipofectamine 2000 (Invitrogen, USA). After 48 h of transfection, the transfection efficiency was assessed by RT-qPCR.
RT-qPCR
TRIzol kit (Invitrogen) was utilized to collect total RNA in CC cells. Purified RNA was reverse transcribed into cDNA using the PrimerScript RT Reagent Kit (TaKaRa, Japan), and miRNA from total RNA was reverse transcribed using the Prime-Script miRNA cDNA Synthesis Kit (TaKaRa). The SYBR-Green Real-Time PCR Kit (Takara, Japan) was employed with a Bio-Rad CFX96 system for analysis of RT-qPCR. U6 and GAPDH acted as internal references. Relative quantification was calculated using the 2−ΔΔCt method.
Western Blot
RIPA buffer (Huashun, Shanghai, China) containing protease inhibitor was employed to extract total protein in CC cells. Next, the protein was separated by 10% SDS-PAGE (Epizyme, China, PG110) and transferred onto a PVDF membrane (Millipore, USA). After being blocked with T 5% skim milk for 1 h, the membrane was incubated at 4 °C overnight with the following primary antibodies: FGF2 (Abbkine, PRP, 100009), Ang-2 (Biovision, 7116-50), VEGFA (Abbkine, PRP, 100006) and GAPDH (ProteinTech, USA, 10494). Next, the membrane was incubated with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G secondary antibodies (1:2000, ab6721, Abcam) at room temperature for 2 h. The bands were detected and visualized with the ECL chemiluminescent detection system (Thermo Fisher Scientific, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) Assay
MTT assay was performed with the MTT Cell Proliferation Assay Kit (Roche, USA). HeLa and CaSki cells were cultured in 96-well plates. After incubation for 0 h, 24 h, 48 h, and 72 h, each well was added with 20 μL MTT reagent (Invitrogen; 5 mg/mL). Then the mixture was cultured for 4 h with 5% CO2 at 37 °C. After that, 100 μL dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO, USA) was used to dissolve the formazan. The optical absorbance of each well was measured with an ultraviolet spectrophotometer (Bio-Tek Instruments Inc., USA) at 490 nm.
5-Ethynyl-2′-deoxyuridine (EdU) Assay
EdU assay was carried out to determine cell proliferation as previously described (Sun et al. 2017). HeLa and CaSki cells were seeded in 96-well plates at 4 × 104 cells/well. Afterward, EdU reagent (50 μmol/L; RiboBio Co., Ltd, Guangdong, China) was added to the plates which were then cultured for 1 h with 5% CO2. Next, cells were treated with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA), and then stained with DAPI (Invitrogen). Images of EdU-positive cells were photographed using a fluorescent microscope (CANY, Shanghai, China).
Wound Healing Assay
CC cells after transfection were grown in 6‑well plates (5 × 104 cells/well). When cells reached 90% confluence, an artificial scratch was made in the middle of the cell monolayer using a 200 µL pipette tip. Next, the floating debris was washed away using PBS and serum-free medium was used to replace the old one. The wound closure was observed, and images were captured using an inverted microscope (Nikon, Japan) at 0 h and 24 h. Quantitative analysis was performed using ImageJ version 1.8.0 software.
Transwell Assay
Cell invasion was detected by transwell assay with Matrigel (356234, Millipore, USA). HeLa and CaSki cells (1 × 105) were seeded in the upper chamber with Matrigel-precoated inserts (8 μm in pore size; Corning, NY, USA), which was added with serum-free medium. The lower chamber was added with the medium containing 20% FBS. Next, cells invaded to the lower chamber were fixed with 4% paraformaldehyde, stained with 0.1% crystal violet (Sigma-Aldrich, USA) at room temperature for 15 min, and observed under a light microscope.
Tube Formation Assay
An Angiogenesis Starter Kit (Thermo Fisher Scientific, Shanghai, China) was used for tube formation assay. The Matrigel was dissolved to solidify the matrix at 4 °C overnight. Next, 96-well plates was prechilled and coated with Matrigel. Subsequently, the plates were cultured at room temperature for 30 min. Human umbilical vein endothelial cells were co-cultured with the transfected HeLa and CaSki cells, which were then added to cultured endothelial cell growth medium containing 10% of FBS. After a 24-h incubation at 37 °C in a 5% CO2 atmosphere, the network of tubular structures was observed with an inverted photomicroscope (Olympus, Melville, NY, USA). The data was analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Each experiment was replicated at least 3 times.
Luciferase Reporter Assay
Wild type or mutant type LOXL1-AS1 or LYPLA1 was subcloned into the pmirGLO vector (Promega, WI, USA) to generate LOXL1-AS1-Wt/Mut or LYPLA1 3’UTR-Wt/Mut reporter plasmids. All plasmids were synthesized by GenePharma. The luciferase reporter plasmids (0.1 µg) and miR-526b-5p mimics (40 nM) or NC mimics (40 nM) were transfected into CC cells using Lipofectamine 2000. After48 h, the Luciferase Reporter Assay System (Promega) was used to detect the luciferase activities. Renilla luciferase activities were used as the internal control for the normalization of firefly luciferase activity.
RNA Pull-Down
Bio-NC and Bio- LOXL1-AS1 constructs were synthesized by Shanghai Genepharma Co., Ltd. RIPA lysis buffer containing RNase inhibitors (Thermo Fisher Scientific, Inc.) was utilized to lyse cells. Bio-NC and Bio- LOXL1-AS1 (200 pmol) were added to the supernatants. The Dynabeads™ M-270 Streptavidin (1 mg; cat. no. 65305; Thermo Fisher Scientific, Inc.) and proteinase K (Sigma-Aldrich) were then added and incubated with the supernatants overnight at 4 °C to isolate the RNA. Beads were isolated from the supernatant after centrifugation (2,500 × g, 5 min, 4 °C) and washed with wash buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA, 2 M NaCl and 0.1% Tween-20) followed by another centrifugation step (2,500 × g, 5 min, 4 °C). Finally, the RNA attached to the magnetic beads was washed and eluted. RT-qPCR was used to measure the enrichment of miRNAs.
RNA Immunoprecipitation (RIP) Assay
The RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore, MA, USA) was utilized to validate the interaction between genes. Briefly, CC were seeded into 6-well plates. After 48 h, the cell lysates was obtained after the subsequent treatment of cells with lysis buffer. Magnetic beads coated with Ago2 (1: 50; ab186733) or IgG (1: 100; ab109489) were mixed with the cell lysates and incubated for 6 h at 4 °C. Next, the immunoprecipitated RNA was extracted with the subsequent elution of protein beads. Finally, RT-qPCR was performed to analyze the extracted precipitated RNA.
Fluorescence In Situ Hybridization (FISH)
CC cells were seeded in 24-well plates at a density of 6 × 104 cells/well. Next, the cells were fixed with 4% polyformaldehyde. After treatment with 2 µg/mL protease K (Invitrogen), the cells were incubated in the solution added with 250 µL pre-hybridization, glycine, and ethyl phthalide reagent at 42 °C for 1 h. Next, hybridization solution containing 300 ng/mL LOXL1-AS1 probe (RiboBio Co., Ltd, Guangdong, China) was supplemented and mixed at 42 °C overnight. The cells were then stained with DAPI for 10 min and blocked with the anti-fluorescence quenching agent (Invitrogen). The cells were observed in 5 randomly selected fields of view using a fluorescence microscope.
Statistical Analysis
Data are shown as the mean ± SD. Statistical analyses were conducted using a one-way ANOVA or Student’s t-test. GraphPad Prism software 5.0 was utilized for statistical analysis. All experiments were carried out at least 3 times. The value of p < 0.05 was considered statistically significant.
Results
Inhibition of LOXL1-AS1 Suppresses CC Cell Growth
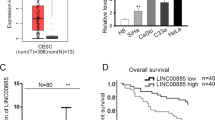
The expression of LOXL1-AS1 in CC tissues and cells was analyzed. In Fig. 1A, LOXL1-AS1 exhibited a higher expression in CC tissues than in normal tissues. Additionally, the high level of LOXL1-AS1 in CC cells was verified (Fig. 1B). These findings suggested that LOXL1-AS1 may be an oncogene in CC. Next, HeLa and CaSki cells were transfected with two LOXL1-AS1-specific shRNAs (sh-LOXL1-AS1#1 or #2) to silence the endogenous expression of LOXL1-AS1. As expected, the LOXL1-AS1 level was decreased in CC cells (Fig. 1C). The specific functions of LOXL1-AS1 in CC were detected by functional assays. As revealed by MTT assay, the transfection of LOXL1-AS1 shRNAs significantly inhibited the viability of HeLa and CaSki cells (Fig. 1D). Similarly, EdU assay also validated that silencing LOXL1-AS1 inhibited CC cell proliferation ability (Fig. 1E). Moreover, we used transwell and wound healing assays to test whether LOXL1-AS1 knockdown affects cell migration and invasion. The knockdown of LOXL1-AS1 notably suppressed the migratory potential of CC cells (Fig. 1F). In addition, the invasive capacity of CC cells was reduced in the sh‐LOXL1-AS1#1/2 group compared to the control group (Fig. 1G). The impact of sh-LOXLA-AS1 on tube formation ability was detected by tube formation assay. We found that LOXLA-AS1 depletion markedly reduced tube formation ability in CC cells (Fig. 1H). Also, the expression of angiogenesis associated proteins (VEGFA, Ang-2, FGF2) in the sh-LOXL1-AS1/2 group was reduced as well (Fig. 1I). Overall, we concluded that LOXL1-AS1 exerts oncogenic effects in CC.

Role of LOXL1-AS1 in CC cells. A LOXL1-AS1 expression in CC tissues was detected by RT-qPCR. n = 50 B LOXL1-AS1 expression in CC cells was measured by RT-qPCR. C The knockdown efficiency of LOXL1-AS1 in HeLa cells and CaSki cells was detected by RT-qPCR. D Cell viability was detected by MTT assay in HeLa and CaSki cells after knockdown of LOXL1-AS1. E EdU assay was carried out to assess cell proliferation after knockdown of LOXL1-AS1. F Cell invasion was evaluated by transwell in cells transfected with sh-LOXL1-AS1#1/2. G Wound healing assay was performed to detect the migratory potential of cells transfected with sh-LOXLA-AS1#1 or #2. H The impact of sh-LOXLA-AS1 on tube formation ability was detected by tube formation assay. I The expression of angiogenesis associated proteins (VEGFA, Ang-2 and FDF2) in HeLa and CaSki cells upon LOXLA-AS1 silencing was measured by western blot. *p < 0.05
LOXL1-AS1 Binds to miR‐526b-5p
As for the regulatory mechanism of LOXL1-AS1, FISH revealed LOXL1-AS1 was mainly distributed in the cytoplasm of HeLa and CaSki cells (Fig. 2A). Studies have showed that cytoplasmic lncRNAs could act as ceRNAs through sequestering miRNAs to release mRNAs (Tay et al. 2014). LOXL1-AS1 was also reported to exert ceRNA functions in other cancers. Consistently, RIP assay revealed that LOXL1-AS1 was enriched in the Ago2 group, demonstrating the presence of LOXL1-AS1 in Ago2-comprised RNA-induced silencing complexes (RISCs) (Fig. 2B). We thus hypothesized LOXL1-AS1 might serve as miRNA sponges in CC. At the starBase website (search category: Pan-Cancer: more than 5 cancer types, AgoExpNum = 1), we found 8 miRNAs possibly binding to LOXL1-AS1. And RNA pull-down assay demonstrated that miR-526b-5p had the strongest binding affinity to LOXL1-AS1 (Fig. 2C). RT-qPCR showed that miR-526b-5p expression was significantly downregulated in CC cells (Fig. 2D). In order to identify whether LOXL1-AS1 and miR-526b-5p interacts with each other, miR-526b-5p was overexpressed by miR-526b-5p mimics (Fig. 2E), and miR‐526b-5p mimics significantly reduced the luciferase activity of wild type LOXL1-AS1 compared to that of the mutant type (Fig. 2F). RIP assay indicated that LOXL1-AS1 and miR-526b-5p were co-immunoprecipitated by Ago2 antibody (Fig. 2G), demonstrating the interaction between LOXL1-AS1 and miR-526b-5p. Moreover, the miR-526b-5p expression level was verified to be downregulated in CC tissues, compared to adjacent normal tissues (Fig. 2H). According to Spearman’s correlation analysis, miR-526b-5p expression was negatively correlated to LOXL1-AS1 expression in CC samples (Fig. 2I). Overall, LOXL1-AS1 could bind to miR‐526b-5p.

LOXL1-AS1 binds to miR‐526b-5p. A FISH assay for the distribution of LOXL1-AS1 in HeLa and CaSki cells. B RIP assay confirmed the existence of LOXL1-AS1 in RISCs. C The binding between LOXL1-AS1 and predicted miRNAs was detected by RNA pull-down assay. D The miR-526b-5p level in CC cells was measured by RT-qPCR. E The overexpression efficiency of miR-526b-5p was detected by RT-qPCR. F The interaction between miR-526b-5p and LOXL1-AS1 was verified by a luciferase reporter assay. G The enrichment of miR-526b-5p and LOXL1-AS1 in the beads conjugated with anti-Ago2 was examined using RIP assay. H The miR-526b-5p level in CC tissues was measured by RT-qPCR. I The expression relationship between miR-526b-5p and LOXL1-AS1 in CC tissues. *p < 0.05, ***p < 0.001
LYPLA1 is Targeted by miR‐526b-5p
To further probe the ceRNA pattern, bioinformatics tools (RNA22, miRmap and PITA) were searched to predict the targets of miR-526b-5p. As a result, seven mRNAs (CTNNB1, KTN1, LYPLA1, ACBD5, NDUFV3, CFI, and SERBP1) were discovered (Fig. 3A). After miR-526b-5p mimics were transfected into CC cells, the miR-526b-5p-bound mRNA expression was detected, and the results showed that LYPLA1 expression had a most significant downregulation in CC cells among all mRNAs (Fig. 3B). The miR-526b-5p binding site in LYPLA1 is predicted and the mutant site was designed for luciferase reporter assay (Fig. 3C). A significantly decreased luciferase activity was detected in cells containing LYPLA1‐Wt reporters after co-transfection with miR‐526b-5p mimics (Fig. 3C). Moreover, we further confirmed that LOXL1-AS1, miR-526b-5p, and LYPLA1 were co-existed in RISCs immunoprecipitated by Ago2 antibody (Fig. 3D). As revealed, the levels of LYPLA1 mRNA and protein were significantly reduced by miR‐526b-5p overexpression and LOXL1-AS1 silencing. Moreover, the levels of LYPLA1 mRNA and protein were significantly increased in the cells silencing miR‐526b-5p (Fig. 3E–F). It suggests that miR‐526b-5p is a negative regulator of LYPLA1. The elevated levels of LYPLA1 mRNA and protein in CC tissues were validated by RT-qPCR and western blotting, respectively (Fig. 3G–H). It was revealed that miR-526b-5p expression was negatively correlated with LYPLA1 or LOXL1-AS1 expression in CC samples (Fig. 3I). To conclude, LOXL1-AS1 positively regulates LYPLA1 expression by sponging miR‐526b-5p.

LYPLA1 is a target of miR-526b-5p. A Potential mRNAs of miR-526b-5p was predicted using online tools. B The expression of mRNAs in HeLa and CaSki cells after overexpressing miR-526b-5p was measured by RT-qPCR. C The binding site between miR-526b-5p and LYPLA1 is predicted in starBase. The interaction between miR-526b-5p and LYPLA1 was measured by luciferase reporter assay. D The interactions among LOXL1-AS1, miR-526b-5p, and LYPLA1 were identified by RIP assay. E–F The effects of miR-526b-5p mimics, sh-LOXL1-AS1#1, or miR-526b-5p inhibitor on the mRNA and protein levels of LYPLA1 were examined by RT-qPCR and western blot. G–H The mRNA and protein levels of LYPLA1 in CC tissues were examined by RT-qPCR and western blot. I The expression relationships among LOXL1-AS1, miR-526b-5p, and LYPLA1 in CC samples. *p < 0.05, ***p < 0.001
LYPLA1 Overexpression Reverses the Regulatory Effect of LOXL1-AS1 Depletion
Finally, we investigated whether LYPLA1 is responsible for the regulation of LOXL1-AS1 in CC. The overexpression efficiency of LYPLA1 in CC cells was examined by RT-qPCR and western blot analyses (Fig. 4A). MTT and EdU assays revealed that LYPLA1 overexpression counteracted the suppressive effect of LOXL1-AS1 inhibition on CC cell proliferation (Fig. 4B and C). Inhibition of LOXL1-AS1 repressed cell migration and invasion, but this effect was reversed upon LYPLA1 overexpression (Fig. 4D and E). Additionally, tube formation assay showed that the co-transfection with pcDNA3.1/LYPLA1 restored the decreased angiogenesis capacity by sh-LOXL1-AS1#1 (Fig. 4F). Also, as shown in Fig. 4G, upregulation of LYPLA1 reduced the sh-LOXL1-AS1-mediated effect on the expression of angiogenesis associated proteins. In summary, LOXL1-AS1 accelerates cellular process in CC by elevating LYPLA1.

LYPLA1 overexpression reverses the regulatory effect of LOXL1-AS1 silencing on cellular progression of CC. A The overexpression efficiency of LYPLA1 in HeLa and CaSki cells was verified by RT-qPCR and western blot. B–C The proliferation of CC cells with indicated transfection was measured using MTT and EdU assays. D–E Transwell and wound healing assays were used to examine the migration and invasion in cells with indicated transfection. F Tube formation capacity was assessed using tube formation assay. G The expression of angiogenesis associated proteins (VEGFA, Ang-2 and FDF2) in cells with indicated transfection was accessed by western blot. *p < 0.05
Discussion
Despite great improvement made in treatment for CC, the overall survival rate remains less than 40% (Jing et al. 2015). It is exigent to elucidate the potential mechanisms in CC progression to develop an effectively targeted therapy. The functional roles of lncRNAs in human cancers has been widely reported (Guttman et al. 2009; Peng et al. 2017; Yang et al. 2014). Numerous lncRNAs are identified to play crucial roles in CC, such as oncogene lncRNAs SOX21-AS1 and SNHG12 (Zhang et al. 2019; Jin et al. 2019), and a tumor suppressor gene lncRNA DGCR5 (Liu et al. 2019). This study focused on the function of LOXL1-AS1 in CC. According to previous research, inhibition of LOXL1-AS1 significantly hampers cell proliferation and migration and facilitates apoptosis in lung adenocarcinoma (Li et al. 2020). LOXL1-AS1 is found overexpressed in lung cancer and aggravates malignant phenotypes of cells (Xie et al. 2019). In our present study, our data revealed that LOXL1-AS1 was upregulated in CC. Downregulation of LOXL1-AS1 inhibited CC cell functions including proliferation, migration, invasion, and angiogenesis ability, suggesting that LOXL1-AS1 performs an oncogenic role in CC.
MicroRNAs (miRNAs) contain 22 nucleotides and exert specific functions post-transcriptionally (He et al. 2019). For instance, miR-126 restrains CC cell migration and invasion by downregulating ZEB1 (Wei 2019). Upregulation of miRNA-494 suppresses the cellular process of CC through binding to Pttg1 (Chen et al. 2015). Additionally, increasing studies showed that lncRNAs could act as miRNA sponges to prevent mRNAs from posttranscriptional degradation under a ceRNA pattern. Moreover, the ceRNA pattern also works in tumor progression. For instance, lncRNA AF147447 represses the aggressive progression of gastric cancer via absorbing miR-34c to modulate MUC2 (Zhou et al. 2016). LncRNA RP4 functions as a ceRNA for miR-7-5p in colorectal cancer (Liu et al. 2018). Here, miR-526b-5p was found to share a binding site for LOXL1-AS1. Additionally, miR-526b-5p expression was downregulated in CC tissues and cells, and negatively correlated with LOXL1-AS1 expression in clinical samples. Collectively, LOXL1-AS1 could bind to miR-526b-5p in CC cells.
To further probe the ceRNA pattern, bioinformatics tools were searched to identify the targets of miR-526b-5p, and lysophospholipase 1 (LYPLA1) was discovered. Previous report indicated that LYPLA1 is overexpressed in non‑small cell lung cancer and its downregulation hampers cell proliferation, migration, and invasion (Mohammed et al. 2019). Here, we confirmed that LYPLA1 was overexpressed in CC tissues. Moreover, overexpression of LYPLA1 reduced the inhibitory effect of LOXL1-AS1 silencing on malignant behaviors of CC cells.
In summary, LOXL1-AS1 was highly expressed in CC and upregulated LYPLA1 to mediate cellular processes in CC by sequestering miR-526b-5p. Our findings may provide a possible theoretical basis for CC treatment.
Data Availability
The datasets used during the current study are available from the corresponding author on reasonable request.
Change history
09 November 2022
A Correction to this paper has been published: https://doi.org/10.1007/s10528-022-10301-9
References
Bunch H (2018) Gene regulation of mammalian long non-coding RNA. Mol Genet Genomics 293(1):1–15
Chen B et al (2015) MiRNA-494 inhibits metastasis of cervical cancer through Pttg1. Tumour Biol 36(9):7143–7149
Chen X et al (2019) Long noncoding RNA OPA-interacting protein 5 antisense transcript 1 upregulated SMAD3 expression to contribute to metastasis of cervical cancer by sponging miR-143-3p. J Cell Physiol 234(4):5264–5275
Forouzanfar MH et al (2011) Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet 378(9801):1461–1484
Gibb EA, Brown CJ, Lam WL (2011) The functional role of long non-coding RNA in human carcinomas. Mol Cancer 10:38
Guttman M et al (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458(7235):223–227
He K et al (2019) Regulatory network reconstruction of five essential microRNAs for survival analysis in breast cancer by integrating miRNA and mRNA expression datasets. Funct Integr Genomics 19(4):645–658
Jiang H et al (2018) Long non-coding RNA TPT1-AS1 promotes cell growth and metastasis in cervical cancer via acting AS a sponge for miR-324-5p. J Exp Clin Cancer Res 37(1):169
Jin XJ et al (2019) Long noncoding RNA SNHG12 promotes the progression of cervical cancer via modulating miR-125b/STAT3 axis. J Cell Physiol 234(5):6624–6632
Jing L et al (2015) HOTAIR enhanced aggressive biological behaviors and induced radio-resistance via inhibiting p21 in cervical cancer. Tumour Biol 36(5):3611–3619
Jing H, et al (2018) A Novel Long Noncoding RNA (lncRNA), LL22NC03-N64E9.1, Promotes the Proliferation of Lung Cancer Cells and is a Potential Prognostic Molecular Biomarker for Lung Cancer. Med Sci Monit 24: 4317–4323.
Kim HJ et al (2015) Long non-coding RNA HOTAIR is associated with human cervical cancer progression. Int J Oncol 46(2):521–530
Li W et al (2020) LncRNA LOXL1-AS1 regulates the tumorigenesis and development of lung adenocarcinoma through sponging miR-423-5p and targeting MYBL2. Cancer Med 9(2):689–699
Liu ML et al (2018) Long noncoding RNA RP4 functions as a competing endogenous RNA through miR-7-5p sponge activity in colorectal cancer. World J Gastroenterol 24(9):1004–1012
Liu Y et al (2019) Downregulation of long noncoding RNA DGCR5 contributes to the proliferation, migration, and invasion of cervical cancer by activating Wnt signaling pathway. J Cell Physiol 234(7):11662–11669
Luan X, Wang Y (2018) LncRNA XLOC_006390 facilitates cervical cancer tumorigenesis and metastasis as a ceRNA against miR-331–3p and miR-338–3p. J Gynecol Oncol 29(6): e95.
Marth C, et al (2017) Cervical cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 28(suppl_4): p. iv72–iv83.
Mercer TR, Dinger ME, Mattick JS (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10(3):155–159
Mohammed A et al (2019) Inhibition of cell proliferation and migration in non-small cell lung cancer cells through the suppression of LYPLA1. Oncol Rep 41(2):973–980
Niu F, et al (2019) The impact of genetic variants in IL1R2 on cervical cancer risk among Uygur females from China: A case-control study. Mol Genet Genomic Med 7(1): e00516.
Peng WX, Koirala P, Mo YY (2017) LncRNA-mediated regulation of cell signaling in cancer. Oncogene 36(41):5661–5667
Qiu S, et al (2020) LncRNA EGOT decreases breast cancer cell viability and migration via inactivation of the Hedgehog pathway. FEBS Open Bio.
Sun Q et al (2017) ER-α36 mediates estrogen-stimulated MAPK/ERK activation and regulates migration, invasion, proliferation in cervical cancer cells. Biochem Biophys Res Commun 487(3):625–632
Tay Y, Rinn J, Pandolfi PP (2014) The multilayered complexity of ceRNA crosstalk and competition. Nature 505(7483):344–352
Wei H, et al (2019) Circular RNA circ_0008450 upregulates CXCL9 expression by targeting miR-577 to regulate cell proliferation and invasion in nasopharyngeal carcinoma. Exp Mol Pathol 110: 104288.
Xie N et al (2019) LncRNA LOXL1-AS1 promotes invasion and proliferation of non-small-cell lung cancer through targeting miR-324-3p. Am J Transl Res 11(10):6403–6412
Xue F, et al (2020) Non-coding RNA LOXL1-AS1 exhibits oncogenic activity in ovarian cancer via regulation of miR-18b-5p/VMA21 axis. Biomed Pharmacother 125: 109568.
Yang G, Lu X, Yuan L (2014) LncRNA: a link between RNA and cancer. Biochim Biophys Acta 1839(11):1097–1109
Yang X, et al (2020) LncRNA LOXL1-AS1 promotes endometrial cancer progression by sponging miR-28–5p to upregulate RAP1B expression. Biomed Pharmacother 125: 109839.
Zhang X et al (2019) Long noncoding RNA SOX21-AS1 promotes cervical cancer progression by competitively sponging miR-7/VDAC1. J Cell Physiol 234(10):17494–17504
Zhao YB et al (2012) Values of three different preoperative regimens in comprehensive treatment for young patients with stage Ib2 cervical cancer. Asian Pac J Cancer Prev 13(4):1487–1489
Zhou X et al (2016) Helicobacter pylori infection related long noncoding RNA (lncRNA) AF147447 inhibits gastric cancer proliferation and invasion by targeting MUC2 and up-regulating miR-34c. Oncotarget 7(50):82770–82782
Acknowledgements
We thank all participators for their help.
Funding
None.
Author information
Authors and Affiliations
Contributions
YZ conceived and designed the experiments. YZ, MZ, LZ, PY, JZ, YW and HW carried out the experiments. YZ and HW analyzed the data. YZ and HW drafted the manuscript. All authors agreed to be accountable for all aspects of the work. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that no conflicts of interest exists.
Ethical Approval
The study was approved from the Ethics Committee of Binhai County People’s Hospital.
Consent to Publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Figure 1 and Figure 4 has been replaced.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Y., Zheng, M., Zhang, L. et al. LncRNA LOXL1-AS1 Facilitates the Oncogenic Character in Cervical Cancer by the miR-526b-5p /LYPLA1 Axis. Biochem Genet 60, 1298–1312 (2022). https://doi.org/10.1007/s10528-021-10182-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-021-10182-4