
Abstract
A microaerophilic, mesophilic, chemoorganoheterotrophic bacterium, designated Y-P2T, was isolated from oil sludge enrichment in China. Cells of the strain were Gram-stain-negative, non-motile, non-spore-forming, rod-shaped or slightly curved with 0.8–3.0 µm in length and 0.4–0.6 µm in diameter. The strain Y-P2T grew optimally at 25 °C (range from 15 to 30 °C) and pH 7.0 (range from pH 6.0 to 7.5) without NaCl. The major cellular fatty acids were C16:0, summed feature 3 (C16:1 ω7c and/or C16:1 ω6c), summed feature 8 (C18:1 ω7c and/or C18:1 ω6c). The main polar liquids of strain Y-P2T comprised phosphatidylethanolamine (PE) and phosphatidylglycerol (PG). The respiratory quinone was Q-10. Acetate and H2 were the fermentation products of glucose. The DNA G + C content was 66.0%. Strain Y-P2T shared the highest 16S rRNA gene sequence similarity (90.3–90.6%) with species within Oceanibaculum of family Thalassobaculaceae in Rhodospirillales. Phylogenetic analyses based on 16S rRNA gene sequences and genomes showed that strain Y-P2T formed a distinct evolutionary lineage within the order Rhodospirillales. On the basis of phenotypic, phylogenetic and phylogenomic data, we propose that strain Y-P2T represents a novel species in a novel genus, for which Shumkonia mesophila gen. nov., sp. nov., within a new family Shumkoniaceae fam. nov. The type strain is Y-P2T (= CCAM 826 T = JCM 34766 T).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The order Rhodospirillales, a member of the class Alphaproteobacteria belonging to the phylum Pseudomonadota, was first proposed by Pfennig and Trüper (Pfennig and Trüper 1971) to replace the name Athiorhodaceae (Molisch 1907), with Rhodospirillum as the type genus (Molisch 1907). It originally comprised three families, named Chlorobiaceae, Chromatiaceae, and Rhodospirillaceae, there are 12 families with validly published names (https://lpsn.dsmz.de/order/rhodospirillales): Acetobacteraceae (Gillis and De Ley 1980), Azospirillaceae (Hördt et al. 2020), Geminicoccaceae (Proença et al. 2018), Kiloniellaceae (Wiese et al. 2009), Reyranellaceae (Hördt et al. 2020), Rhodospirillaceae (Pfennig and Trüper 1971), Rhodovibrionaceae, Stellaceae, Terasakiellaceae, Thalassobaculaceae, Thalassospiraceae and Zavarziniaceae (Hördt et al. 2020). In 2023, Koziaeva et al. (2023) suggested that the order Rhodospirillales should be split into six more new family-level groups according to phylogenomic analyses, including “Magnetospiraceae” and “Magnetovibrionaceae” separated from the family Thalassospiraceae, “Dongiaceae” and “Niveispirillaceae” separated from the family Rhodospirillaceae, “Fodinicurvataceae” from the family Rhodovibrionaceae and “Oceanibaculaceae” from the family Thalassobaculaceae. Rhodospirillales members are widely distributed in freshwater, soil, seawater, plant root and artificial ecosystems. Most members of this order are Gram-stain-negative, spiral or rod-shaped, non-spore-forming, and obligately aerobic or facultatively anaerobic bacteria with ubiquinone Q-10 as the common major respiratory quinone. The order Rhodospirillales is metabolically diverse group, containing chemoorganoheterotrophs, and photoorganoheterotrophs under anoxic conditions in the light (Hördt et al. 2020)
In recent years, it has revealed that Rhodospirillales members present in the oil reservoir related environments, such as oil-contaminated soil and polluted water (Liu et al. 2015; Abbasian et al. 2016; Elumalai et al. 2021). However, just a few of members belonging to Rhodospirillale have the ability to use alkane as energy source (Wu et al. 2021b). The knowledge about ecological roles of Rhodospirillales in oil reservoir is still limited. Elumalai et al. found Rhodospirillales appears in the biofilm on corroded API 5LX carbon steel in produced water of oil reservoir (Elumalai et al. 2021), implying Rhodospirillales probably related with microbially induced corrosion (MIC). Rhodospirillales members including Rhodospirillaceae and Acetobacteraceae were abundant bacteria in the biofilm on corroded steel coupons and Biodiesel Storage Tanks (Procópio 2020; Stamps et al. 2020), populations of these bacteria were accompanied by a continuous corrosion process over the coupons (López et al. 2002; Moura et al. 2018). Corrosion conducted by microorganisms usually influenced through biofilm formation, sulfur metabolism, corrosive metabolites production (such as inorganic or organic acids, and hydrogen sulfide) (Lv and Du 2018; Moura et al. 2018; Elumalai et al. 2021). Several studies have speculated that Rhodospirillales members participate in MIC by producing acetic acid and biofilms, as well as reducing iron (Chen et al. 2019; Chen and Zhang 2019; Procópio 2020; Stamps et al. 2020). Therefore, Rhodospirillales probably link to the corrosion of oil pipelines and storage tanks, suggesting the importance of isolation Rhodospirillales for investigating their ecological functions in oil reservoir environments. Several species have been isolated from the oil production mixture, oil-contaminated soil or oil reservoir water, such as Roseomonas oleicola (Wu et al. 2021a) and Siccirubricoccus phaeus (Li et al. 2021) of the family Acetobacteraceae, Azospirillum oleiclasticum (Wu et al. 2021b) and Azospirillum rugosum (Young et al. 2008) of the family Azospirillaceae, Oleisolibacter albus (Ruan et al. 2019) and Oleiliquidispirillum nitrogeniifigens (Li et al. 2020) of the family Rhodospirillaceae. All these isolates are mesophilic, aerobic or facultatively anaerobic chemoorganoheterotrophs.
In the present study, one strain, designated Y-P2T, was isolated from the oil sludge collected from Shengli oilfield, China. The polyphasic taxonomic analyses revealed strain Y-P2T represents a novel genus in a new family within the order Rhodospirillales.
Materials and methods
Enrichment and isolation
Strain Y-P2T was obtained from the oil sludge of the Shengli oilfield in PR China (37o54’N, 118o33’E). Approximately 10 g of mixture of oil contaminated soil and oily sludge was inoculated into 50 mL of fresh medium for preparation of pre-enrichment culture, which performed as 25 °C. The pre-reduced mineral medium used for enrichment and isolation was prepared with the following components (L−1): 0.5 g NaCl, 0.5 g MgCl2·6H2O, 0.1 g CaCl2·2H2O, 0.3 g NH4Cl, 0.2 g KH2PO4, 0.5 g KCl, 2 ml trace element solution 284 (JCM medium No.284, https://www.jcm.riken.jp/cgi-bin/jcm/jcm_grmd?GRMD=284&MD_NAME =), 1 mg resazurin, 0.5 g cysteine hydrochloride and 1 L distilled water. The strain Y-P2T was isolated with mixed substrate (short-chain fatty acid, glucose, yeast extract and tryptone mixture) by using the extinction dilution method as described previously (Zhang et al. 2018). Unless otherwise stated, R2A (Reasoner’s 2A) liquid medium was used for subculturing and cultivating the strain Y-P2T. R2A medium contained (L−1): 0.25 g tryptone, 0.5 g casein hydrolysate, 0.5 g yeast extract, 0.5 g soluble starch, 0.3 g K2HPO4, 0.1 g MgSO4, 0.3 g sodium pyruvate, 0.25 g peptone, 0.5 g glucose, and 1L distilled water. All media used in this study were prepared and dispensed under 100% N2, and sterilized at 121 °C for 15 min. Strain Y-P2T was deposited in the China Collection of Anaerobic Microorganisms (CCAM 826 T) and Japan Collection of Microorganisms (JCM 34766 T). Oceanibaculum nanhaiense KCTC 52312 T obtained from Korean Collection for Type Cultures (KCTC) was used as the reference strain.
Morphological, physiological, and biochemical tests
The strain Y-P2T incubated at 25 °C for 5 days was used for morphology tests. Gram-staining, flagellum-staining and spore-staining were determined using commercial Gram Staining Kit, Flagellum Staining Kit and Spore Staining Kit (Solarbio, China) according to the manufacturers’ instructions, respectively. The cell shape and size of strain Y-P2T were examined using a scanning electron microscope (JEM-1400 Plus, JEOL, Japan). 0.1% melted agarose gel spread flat on slide and solidified, then take a drop of strain Y-P2T by microscope (Nikon 80i, Japan) for observing motility.
Growth at different temperatures (15, 20, 25, 30 and 37 °C), pH (5.0–8.5, at 0.5-unit intervals), and salinities (0 – 70 g L−1, at 10 g L−1 intervals) were determined in the R2A medium supplemented 10% (v/v) oxygen in the headspace of Hungate tubes (25 ml). The pH values were adjusted using the sterile and HCl or NaOH solution and were buffered with 20 mM MES (pH 5.5, pH 6.0 and pH 6.5), 20 mM PIPES (pH 7.0 and pH 7.5) and 20 mM Tris (pH 8.0 and pH 8.5).The final pH was determined with a pH meter (HORIBA B-712, LAQUAtwin, Japan). Growth was determined by measuring the optical density (OD) at 600 nm using a spectrophotometer (DU730, Beckman, Germany).
For biochemical tests, cells in R2A medium were collected by centrifuging at 13,000 rpm for 5 min at room temperature. Substrate utilization, nitrate reduction, and H2S production were tested with API 20NE, API ZYM and API 20E (bioMérieux, France) and incubated aerobically at 28 °C and 37 °C, respectively, the color change was checked every 18–24 h. Oxidase activity was determined using an oxidase reagent kit (bioMérieux, France) according to the manufacturer’s instructions. Oxygen requirement of strain Y-P2T was tested under 0%, 2%,10% and 20% oxygen (O2:N2, v/v). The fermentation products were analysed by liquid chromatography (Aglient 1200, USA) using AminexHPX-87H column (300 mm × 7.8 mm, 9 µm), H2SO4 (5 mM) was used as the mobile phase at a flow rate of 0.6 ml min−1. H2 in the overhead of the tubes were analysed by gas chromatography (Shimadzu GC2010, Japan).
Chemotaxonomic analysis
For cellular fatty acids, polar lipids and respiratory quinones analyses, cells of strains Y-P2T and O. nanhaiense KCTC 52312 T incubated in R2A medium at 25 °C for 5 days were collected by centrifugation. The fatty acids were separated using gas chromatography (Aglient 8860, USA) and identified with Sherlock software (version 6.3) according to the instructions of Microbial Identification Inc. (MIDI) protocol (Sasser 1990). Respiratory quinones of strain Y-P2T were detected using the protocol described previously (Komagata and Suzuki 1988; Tindall 1990). Polar lipids were extracted using a chloroform / methanol system and were analysed using one- and two-dimensional thin-layer chromatography (TLC) following the method described by Kates et al. (1986).
16S rRNA gene sequencing and phylogenetic analysis
Cells incubated at 25 °C for 5 d was used for extracting genomic DNA using bacteria DNA extraction kit (DP302, TIANGEN, China) following the manufacture’s instruction. 16S rRNA gene fragments were amplified by PCR using the universal primers 27F (5′AGAGTTTGATCMTGGCTCAG3′) and 1492R (5′TACGGYTACCTTGTTACGACTT3′) and were sequenced by ABI 3730XL DNA anazlyzer. The sequence obtained was compared with the available 16S rRNA sequences in EzBioCloud database (https://www.ezbiocloud.net/identify) and NCBI database with nucleotide Basic Local Alignment Search Tool (blastn, https://blast.ncbi.nlm.nih.gov/Blast.cgi), respectively. 16S rRNA gene sequences of closely related species that validly published were derived from genomes from NCBI database or downloaded from EzBioCloud, and then were multiply aligned through MUSCLE v3 (https://drive5.com/muscle/downloads_v3.htm). The phylogenetic analyses were performed by fasttree and MEGA X using the neighbor-joining method and maximum-likelihood method (Felsenstein 1981), respectively. Bootstrap values were calculated based on 1000 replicates. Evolutionary distances were calculated using the Kimura two-parameter method (Kimura 1980). The phylogenetic tree was modified by Itol (https://itol.embl.de/).
Whole–genome sequencing and phylogenomic analysis
The whole genome of strain Y-P2T was sequenced by Novogene Corporation Inc. (Beijing, China) using NovaSeq 6000 System (Illumina, USA). Phylogenomic analysis based on the GTDB taxonomy database (release r95) was performed by using the method described previously (Parks et al. 2018). Genome assembly and annotation were performed using methods described previously (Fan et al. 2023).
Up-to-date bacterial core gene (UBCG) and GTDB phylogenomic trees were reconstructed by UBCG (https://www.ezbiocloud.net/tools) and GTDB-Tk (https://gtdb.ecogenomic.org/), respectively, and were optimized in Evolview website (https://www.evolgenius.info/evolview/#/treeview). The pairwise genomic average nucleotide identity (ANI) and average amino acid identity (AAI) were calculated using OrthoANIu (https://www.ezbiocloud.net/tools/orthoaniu) and Compare M (https://github.com/dparks1134/CompareM), respectively. Percentage of conserved proteins (POCP) between two microbial genomes was calculated as the following formula: (conserved protein number of genome A + conserved protein number of genome B) / (total number of proteins being compared in genome A + total number of proteins being compared in genome B) (Qin et al. 2014).
Results and discussion
Morphological, physiological, and biochemical characteristics
Cells of strain Y-P2T were Gram-stain-negative, non-spore-forming, straightly rod-shaped or slightly curved with 0.8 – 3.0 × 0.4 – 0.6 µm in length and in width (Fig. 1). Growth occurred at 15–30 °C (optimum at 25 °C) and pH 6.0–7.5 (optimum pH 7.0) in absence of NaCl (Fig. S1). Weak growth was observed under anaerobic conditions, but the optimum growth occurred in the presence of oxygen up to approx. 10% (v/v), no growth was observed when oxygen increase to 20% (v/v) (Fig. S2), indicating that strain Y-P2T was microaerophilic.

Scanning electron micrograph of strain Y-P2T. The scale bar is 1 μm
According to API 20NE and API 20E tests (Table 1), strain Y-P2T was positive for urease and gelatinase. The results of API 20NE tests revealed that strain Y-P2T was negative for reduction of nitrate or denitrification, was positive for oxidase activity, but negative for catalase activity, Strain Y-P2T was unable to reduce thiosulfate or ferment tested carbohydrates. In API ZYM test, activities of alkaline phosphatase, esterase (C 4), estase lipase (C 8), acid phosphatase and Naphtol-AS-BI-phosphohydrolase were positive. Acetate and H2 were produced from glucose.
The GenBank accession numbers of the 16S rRNA gene sequence for strain Y-P2T was MZ270534. The GenBank accession number of genome sequence for strain Y-P2T was JAOTID000000000.
Chemotaxonomic characteristics
The predominant fatty acids (> 10%) of strain Y-P2T were C16:0 (34.6%), summed feature 3 (C16:1 ω7c and/or C16:1 ω6c, 23.1%), and summed feature 8 (C18:1 ω7c and/or C18:1 ω6c,17.3%), which accounted for 75.0% of the total fatty acids (Table S1). Differently, O. nanhaiense KCTC 52312 T contained sum in feature 8 (C18:1ω7c and/or C18:1ω6c, 50.0%) and C16:0 (12.6%). The polar liquids of strain Y-P2T comprised one unidentified aminolipid (AL), one phosphatidylethanolamine (PE), one phosphatidylglycerol (PG), three unidentified phospholipids (PLs) and four unidentified polar lipids (Ls) (Fig. S3). The respiratory quinone was Q-10.
Phylogenetic analyses
The complete 16S rRNA gene sequence analysis performed using blastn at NCBI showed that strain Y-P2T shares the highest 16S rRNA gene sequence similarity with species in the genus Oceanibaculum (90.3–90.6%) which was affiliated to “Oceanibaculaceae” (formerly Thalassobaculaceae) within the order Rhodospirillales, followed by Stella humosa DSM 5900 T (90.0%) in family Stellaceae, Thalassobaculum and Nisaea (≤ 89.9%) in Thalassobaculaceae, Varunaivibrio and Magnetovibrio (≤ 89.8%) in “Magnetovibrionaceae” (formerly Thalassospiraceae), Zavarzinia compransoris LMG-5821 T (89.1%) in Zavarziniaceae (Table S2). These 16S rRNA gene sequence similarities were lower than the 94.5% threshold for the delineation of genera and the median sequence identity for the delineation of families (Yarza et al. 2014), suggesting that strain Y-P2T belongs to a new genus or may represent a higher rank taxon.
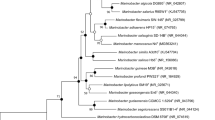
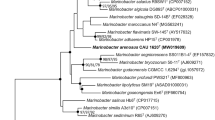
To confirm the phylogenetic relationship between strain Y-P2T and Rhodospirillales members, maximum-likelihood phylogenetic trees based on 16S rRNA gene sequences of strain Y-P2T and the representative genera within order Rhodospirillales were constructed. On the phylogenetic tree, strain Y-P2T was placed in the clade with genera Zavarzinia of the family Zavarziniaceae, but formed an independent evolutionary lineage that is distinguishable among Rhodospirillales families (Fig. S4). The phylogenomic trees reconstructed based on bacterial core gene set (Fig. 2) and 120 single copy genes (Fig. 3) both indicated that strain YP2T clusters with members of “Magnetospiraceae” (Magnetospira), “Magnetovibrionaceae” (Magnetovibrio, Varunaivibrio), and Geminicoccaceae (Defluviicoccus), and formed a clade with Defluviicoccus vanus Ben 114 T.

Phylogenomic tree reconstructed based on up-to-date bacterial core gene set (UBCG, concatenated alignment of 92 core genes) of representative species of genera in the order Rhodospirillales. Bar, 0.1 substitutions per position. Gene support indices (GSIs) are given at branching points. Latin names with quotes have not been validly published

Phylogenomic tree reconstructed based on 120 single copy conservative marker genes of all species with sequenced genomes in the order Rhodospirillales with GTDB pipelines. Bar, 0.1 substitutions per position. Percentage bootstrap values are given at branching points. Latin names with quotes have not been validly published
To further figure out the taxonomic rank of strain Y-P2T, reanalysis of pairwise 16S rRNA gene sequence similarity and indices of pairwise genomic relatedness including ANI, AAI and POCP were performed. Strain Y-P2T had 89.2 ± 0.8% of 16S rRNA sequence similarities to representative species of the family “Oceanibaculaceae” and Thalassobaculaceae (Fig. 4), which were close to the minimum sequence similarity for defining a novel family (Yarza et al. 2014). Meanwhile, the ANI and POCP values between strain Y-P2T and these members were ≤ 69.8 and 43.0% (Table S2), respectively, far lower than the cutoff values for distinguishing genera (83 and 50%, respectively) (Luo et al. 2014; Qin et al. 2014; Jain et al. 2018), and were in the range of relevant inter-family values (ANI 63.7–70.7 and POCP 25.3–49.9%) (Fig. 4); the AAI values were all ≤ 57.8% (Table S2), which were lower the boundary (approximately 60%) for Rhodospirillales families proposed by Koziaeva et al. (2023). In addition, strain Y-P2T was separated from other Rhodospirillales families with the average values of 16S rRNA gene sequence similarity and AAI (86.0 ± 2.0 and 55.3 ± 1.5%, respectively) below the thresholds for family delineation (Fig. 4) (Yarza et al. 2014). All these results suggested strain Y-P2T could be distinguished from families in Rhodospirillales and represents a novel family within Rhodospirillales.

Comparison of 16S rRNA gene identity, AAI, ANI and POCP between strain Y-P2T and representatives of published families within the order Rhodospirillales. The violin plot lines indicate a kernel density of the identity distribution. The black dot indicates the mean and the solid horizontal line indicates the median. *Thalassobaculaceae, members within “Oceanibaculaceae” and Thalassobaculaceae included
Genomic characteristics
The genome of strain Y-P2T had a total length of 5,388,487 bp, and contained 5053 ORFs, 49 tRNA genes, one 5 s rRNA, one 16 s rRNA and one 23 s rRNA (Table S3). The G + C content was 66.0%. 2314, 3540 and 3592 genes were annotated in KEGG, Swiss and GO database, respectively (Table S3).
Glycan biosynthesis pathways
Extracellular polymeric substance (EPS) is a key component of biofilm causing processes of MIC. Strain Y-P2T was able to form EPS when grown under microaerobic condition (Fig. S5). Results based on KEGG annotation showed that 66 genes associated with glycan biosynthesis and metabolism are identified in the genome of strain Y-P2T (Table S4), 20 of which (more than 30%) involved in the biosynthesis of lipopolysaccharide (LPS) which may effect on the Co-Cr and Ti alloys corrosion (Yu et al. 2016). Genes lpxA, lpxCD, lpxI, lpxB and lpxK encoding homologs that consist of pathway synthesizing lipid IVA from UDP-N-acetyl-α-d-glucosamin or a (3R)-3-hydroxytetradecanoyl-[acp] (Table S5). Meanwhile, homologs encoded by genes kdsD, kdsA, kdsC and kdsB formed a pathway for synthesizing CMP-3-deoxy-β-d-manno-octulosonate from d-ribulose 5-phosphate, providing an essential substrate for generating Kdo2-lipid A via the pathway comprised of integral membrane proteins encoded by kdtA, lpxL, IpxK and lpxM (Wang et al. 2015). However, lpxM was not found in genome of strain Y-P2T. In addition, an ADP-l-glycero-β-d-manno-heptose biosynthesis pathway containing enzymes encoded by gmhAC, gmhB and gmhD was employed by Y-P2T to produce the precursor for the inner core region of LPS, but only one gene (gtrB) related to O-antigen repeat unit synthesis was identified. We also found strain Y-P2T had a complete pathway (rmlADBC) to synthesize dTDP-l-rhamnose which is the precursor of l-rhamnose. l-rhamnose and GDP-d-glycero-d-manno-heptose that generated via enzymes encoded by gmhA, hhdA (absent), gmhB, and hhdC are sugar components of bacterial S-layer glycoproteins (Kneidinger et al. 2001; Graninger et al. 2002). It has been demonstrated that S-layer proteins are associated with cells aggregation, bacterial adherence to substrates and surfaces, as well as biofilm formation (Gerbino et al. 2015).
Sulfur metabolism
Sulfur-cycling microorganisms are commonly considered the key players of MIC, and can participate in the process of MIC through sulfate reduction and sulfur disproportionation (Rajala et al. 2022). The genome of strain Y-P2T contained 24 genes for sulfur metabolism (Table S6). Gene sta, and the operons aprAB and dsrAB comprised a complete dissimilatory sulfate reduction and oxidation pathway for the conversion between sulfate and sulfide, which has been described in Dsr-dependent sulphate-reducing bacteria and sulphur-oxidizing bacteria (Neukirchen and Sousa 2021). SOX system oxidizing thiosulfate to sulfate via enzymes encoded by soxDCBAZYX operon was identified in strain Y-P2T. In addition, a flavocytochrome c sulfide dehydrogenase was presented in genome of Y-P2T by fccA and fccB, it has been reported that this enzyme can oxidize self-produced sulfide or exogenous sulfide to sulfite and thiosulfate under aerobic condition in Pseudomonas aeruginosa (Lü et al. 2017).
Conclusion
Physiological comparison revealed that strain Y-P2T shares several common phenotypic features with Rhodospirillales members, such as mesophilic, Gram-stain-negative and major respiratory quinone, but is different to its phylogenetically close relatives in morphological and physiological traits (Table 2). Thalassobaculaceae members are motile, grow with salinity, positive for oxidase activity; “Magnetospiraceae” and “Magnetovibrionaceae” are magnetotactic; while Geminicoccaceae and Stellaceae are aerobic, having different cell shapes, Geminicoccaceae also has abilities to reduce nitrate and hydrolyze gelatin. Meanwhile, values of the 16S rRNA gene sequence similarity, AAI ANI and POCP between strain Y-P2T and published members of Rhodospirillales families were all lower than boundaries for separating Rhodospirillales families. On the basis of the distinct phenotypic and chemotaxonomic characteristics, phylogenetic and genomic evidence mentioned above, strain Y-P2T is proposed as the type strain of a novel species of a new genus, for which the name Shumkonia mesophila gen. nov., sp. nov. is proposed, within a new family Shumkoniaceae fam. nov.
Description of Shumkoniaceae fam. nov.
Shumkoniaceae (Shum.ko.ni.a.ce’ae. N.L. fem. n. Shumkonia a bacterial genus; aceae suffix to denote a family; N.L. fem. pl. n. Shumkoniaceae the Shumkonia family).
The description of Shumkoniaceae is the same as for the genus Shumkonia. The type genus is Shumkonia.
Description of Shumkonia gen.nov.
Shumkonia (Shum.ko’ni.a. N.L. fem. n. Shumkonia named in honour of ShumKo (1031–1095) who found and used oil in the eleventh century in China).
Microaerophilic, mesophilic, chemoorganoheterotrophic bacterium. Gram-stain-negative, non-motile, non-spore-forming, rod-shaped or slightly curved rod. Acetate and H2 are the fermentation products of glucose. The predominant cellular fatty acid is C16:0, and the quinone is Q-10. The type species is Shumkonia mesophila.
Description of Shumkonia mesophila sp. nov.
Shumkonia mesophila (me.sophi.la. N.L. fem. adj. mesophila, middle temperature loving).
Microaerophilic, mesophilic, chemoorganoheterotrophic bacterium. Cells are Gram-stain-negative, non-motile, non-spore-forming, rod-shaped or slightly curved, 0.8–3.0 µm in width and 0.4–0.6 µm in length. Growth occurs optimally under the conditions of 25 °C, pH 7.0 without NaCl. Urease and oxidase positive. Acetate and H2 are the fermentation products of glucose. Catalase, nitrate reduction and denitrification negative. Major cellular fatty acids are C16:0, sum in feature 3 (C16:1 ω7c and/or C16:1 ω6c), sum in feature 8 (C18:1 ω7c and/or C18:1 ω6c). The polar lipids comprise phosphatidylethanolamine (PE), phosphatidylglycerol (PG), one unidentified aminolipid (AL), three unidentified phospholipid (PL) and four unidentified polar lipids (L). Respiratory quinone is Q-10. The genomic DNA G + C content was 66.0%.
The type strain is Y-P2T (= CCAM 826 T = JCM 34766 T) isolated from oil sludge, Shengli, China.
Abbreviations
- AAI:
-
Average amino acid identity
- ANI:
-
Average nucleotide identity
- ANI:
-
Average nucleotide identity
- AL:
-
Aminolipid
- CHES:
-
N-cyclohexyl-2-aminoethanesulfonic acid
- GL:
-
Glycolipids
- L:
-
Lipids
- MES:
-
2-(N-morpholino) ethanesulfonic acid
- PE:
-
Phosphatidylethanolamine
- PG:
-
Phosphatidylglycerol
- PL:
-
Phospholipid
- POCP:
-
Percentage of conserved proteins
- R2A:
-
Reasoner's 2A
References
Abbasian F, Lockington R, Megharaj M et al (2016) The biodiversity changes in the microbial population of soils contaminated with crude oil. Curr Microbiol 72:663–670. https://doi.org/10.1007/s00284-016-1001-4
Anil Kumar P, Aparna P, Srinivas TN et al (2008) Rhodospirillum sulfurexigens sp. nov., a phototrophic alphaproteobacterium requiring a reduced sulfur source for growth. Int J Syst Evol Microbiol 58:2917–2920. https://doi.org/10.1099/ijs.0.65689-0
Bazylinski DA, Williams TJ, Lefevre CT et al (2013) Magnetovibrio blakemorei gen. nov., sp. nov., a magnetotactic bacterium (Alphaproteobacteria: Rhodospirillaceae) isolated from a salt marsh. Int J Syst Evol Microbiol 63:1824–1833. https://doi.org/10.1099/ijs.0.044453-0
Chen S, Zhang D (2019) Corrosion behavior of Q235 carbon steel in air-saturated seawater containing Thalassospira sp. Corros Sci 148:71–82. https://doi.org/10.1016/j.corsci.2018.11.031
Chen RW, Wang KX, Zhou XF et al (2018) Indioceanicola profundi gen. nov., sp. nov., isolated from Indian Ocean sediment. Int J Syst Evol Microbiol 68:3707–3712. https://doi.org/10.1099/ijsem.0.003016
Chen S, Deng H, Liu G et al (2019) Corrosion of Q235 carbon steel in seawater containing Mariprofundus ferrooxydans and Thalassospira sp. Front Microbiol. https://doi.org/10.3389/fmicb.2019.00936
Dar Jean W, Huang SP, Chen JS et al (2016) Tagaea marina gen. nov., sp. nov., a marine bacterium isolated from shallow coastal water. Int J Syst Evol Microbiol 66:592–597. https://doi.org/10.1099/ijsem.0.000756
Dong C, Lai Q, Chen L et al (2010) Oceanibaculum pacificum sp. nov. isolated from hydrothermal field sediment of the southwest Pacific Ocean. Int J Syst Evol Microbiol 60(1):219–222. https://doi.org/10.1099/ijs.0.011932-0
Du Y, Liu X, Lai Q et al (2017) Oceanibaculum nanhaiense sp. nov., isolated from surface seawater. Int J Syst Evol Microbiol 67:4842–4845. https://doi.org/10.1099/ijsem.0.002388
Elumalai P, AlSalhi MS, Mehariya S et al (2021) Bacterial community analysis of biofilm on API 5LX carbon steel in an oil reservoir environment. Bioprocess Biosyst Eng 44:355–368. https://doi.org/10.1007/s00449-020-02447-w
Fan H, Li J, Wu W et al (2023) Description of a moderately acidotolerant and aerotolerant anaerobic bacterium Acidilutibacter cellobiosedens gen. nov., sp. nov. within the family Acidilutibacteraceae fam. nov., and proposal of Sporanaerobacteraceae fam. nov. and Tepidimicrobiaceae fam. nov. Syst Appl Microbiol 46:126376. https://doi.org/10.1016/j.syapm.2022.126376
Felsenstein J (1981) Evolutionary trees from gene frequencies and quantitative characters: finding maximum likelihood estimates. Evolution 35:1229–1242. https://doi.org/10.1111/j.1558-5646.1981.tb04991.x
Foesel BU, Gössner AS, Drake HL et al (2007) Geminicoccus roseus gen. nov., sp. nov., an aerobic phototrophic Alphaproteobacterium isolated from a marine aquaculture biofilter. Syst Appl Microbiol 30:581–586. https://doi.org/10.1016/j.syapm.2007.05.005
Gerbino E, Carasi P, Mobili P et al (2015) Role of S-layer proteins in bacteria. World J Microbiol Biotechnol. https://doi.org/10.1007/s11274-015-1952-9
Gillis M, De Ley J (1980) Intra- and intergeneric similarities of the ribosomal ribonucleic acid cistrons of Acetobacter and Gluconobacter. Int J Syst Evol Microbiol 30:7–27. https://doi.org/10.1099/00207713-30-1-7
Graninger M, Kneidinger B, Fau - Bruno K, Bruno K Fau - Scheberl A, et al (2002) Homologs of the Rml enzymes from Salmonella enterica are responsible for dTDP-beta-L-rhamnose biosynthesis in the gram-positive thermophile Aneurinibacillus thermoaerophilus DSM 10155. Appl Environ Microbiol. https://doi.org/10.1128/AEM.68.8.3708-3715.2002
Hördt A, López MG, Meier-Kolthoff JP et al (2020) Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front Microbiol 11:468. https://doi.org/10.3389/fmicb.2020.00468
Humrighouse BW, Emery BD, Kelly AJ et al (2016) Haematospirillum jordaniae gen. nov., sp. nov., isolated from human blood samples. Antonie Van Leeuwenhoek 109:493–500. https://doi.org/10.1007/s10482-016-0654-0
Hylemon PB, Wells JS, Krieg NR et al (1973) The Genus Spirillum: a Taxonomic Study1. Int J System Evolut Microbiol 23:340–380. https://doi.org/10.1099/00207713-23-4-340
Imhoff JF, Petri R, Suling J (1998) Reclassification of species of the spiral-shaped phototrophic purple non-sulfur bacteria of the alpha-Proteobacteria: description of the new genera Phaeospirillum gen. nov., Rhodovibrio gen. nov., Rhodothalassium gen. nov. and Roseospira gen. nov. as well as transfer of Rhodospirillum fulvum to Phaeospirillum fulvum comb. nov., of Rhodospirillum molischianum to Phaeospirillum molischianum comb. nov., of Rhodospirillum salinarum to Rhodovibrio salexigens. Int J Syst Bacteriol 48(Pt 3):793–8. https://doi.org/10.1099/00207713-48-3-793
Jain C, Rodriguez RL, Phillippy AM et al (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9:5114. https://doi.org/10.1038/s41467-018-07641-9
Kates M (1986) Techniques of Lipidology. 106- 107:241-246.
Kim J, Jeong SE, Khan SA et al (2019) Hwanghaeella grinnelliae gen. nov., sp. nov., isolated from a marine red alga. Int J Syst Evol Microbiol 69:3544–3550. https://doi.org/10.1099/ijsem.0.003656
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. https://doi.org/10.1007/bf01731581
Kneidinger B, Graninger M, Fau-Puchberger M, Puchberger M, Fau-Kosma P et al (2001) Biosynthesis of nucleotide-activated D-glycero-D-manno-heptose. J Biol Chem 276:20935–44. https://doi.org/10.1074/jbc.M100378200
Komagata K, Suzuki K-I (1988) 4 Lipid and cell-wall analysis in bacterial systematics. In: Colwell RR, Grigorova R (eds) Methods in Microbiology. Academic Press, pp 161–207
Koziaeva VV, Sorokin DY, Kolganova TV et al (2023) Magnetospirillum sulfuroxidans sp. Nov., capable of sulfur-dependent lithoautotrophy and a taxonomic reevaluation of the order Rhodospirillales. Syst Appl Microbiol 46:126406. https://doi.org/10.1016/j.syapm.2023.126406
Lai Q, Yuan J, Gu L et al (2009a) Marispirillum indicum gen. nov., sp. nov., isolated from a deep-sea environment. Int J Syst Evol Microbiol 59:1278–1281. https://doi.org/10.1099/ijs.0.003889-0
Lai Q, Yuan J, Wu C et al (2009b) Oceanibaculum indicum gen. nov., sp. nov., isolated from deep seawater of the Indian Ocean. Int J Syst Evol Microbiol 59:1733–1737. https://doi.org/10.1099/ijs.0.004341-0
Lakshmi K, Sasikala C, Ashok Kumar GV et al (2011) Phaeovibrio sulfidiphilus gen. nov., sp. nov., phototrophic alphaproteobacteria isolated from brackish water. Int J Syst Evol Microbiol 61:828–833. https://doi.org/10.1099/ijs.0.018911-0
Lakshmi K, Divyasree B, Ramprasad EVV et al (2014) Reclassification of Rhodospirillum photometricum Molisch 1907, Rhodospirillum sulfurexigens Anil Kumar et al. 2008 and Rhodospirillum oryzae Lakshmi et al. 2013 in a new genus, Pararhodospirillum gen. nov., as Pararhodospirillum photometricum comb. nov., Pararhodospirillum sulfurexigens comb. nov. and Pararhodospirillum oryzae comb. nov., respectively, and emended description of the genus Rhodospirillum. Int J Syst Evol Microbiol 64:1154–1159. https://doi.org/10.1099/ijs.0.059147-0
Lee Y, Park HY, Jeon CO (2019) Zavarzinia aquatilis sp. nov., isolated from a freshwater river. Int J Syst Evol Microbiol 69:727–731. https://doi.org/10.1099/ijsem.0.003214
Li FL, Wang XT, Shan JJ et al (2020) Oleiliquidispirillum nitrogeniifigens gen. nov., sp. nov., a new member of the family Rhodospirillaceae isolated from oil reservoir water. Int J Syst Evol Microbiol 70:3468–3474. https://doi.org/10.1099/ijsem.0.004200
Li FL, Zhang YX, Zhang YK et al (2021) Siccirubricoccus phaeus sp. nov., isolated from oil reservoir water and emended description of the genus Siccirubricoccus. Antonie Van Leeuwenhoek 114:355–364. https://doi.org/10.1007/s10482-021-01516-8
Lin SY, Hameed A, Tsai CF et al (2021) Vineibacter terrae gen nov., sp. Nov., an ammonium-assimilating and nitrate-reducing bacterium isolated from vineyard soil. Int J Syst Evol Microbiol. https://doi.org/10.1099/ijsem.0.005111
Liu JF, Sun XB, Yang GC et al (2015) Analysis of microbial communities in the oil reservoir subjected to CO2-flooding by using functional genes as molecular biomarkers for microbial CO2 sequestration. Front Microbiol 6:236. https://doi.org/10.3389/fmicb.2015.00236
López-López A, Pujalte MJ, Benlloch S et al (2002) Thalassospira lucentensis gen. nov., sp. nov., a new marine member of the alpha-Proteobacteria. Int J Syst Evol Microbiol 52:1277–1283. https://doi.org/10.1099/00207713-52-4-1277
Lü C, Xia Y, Liu D et al (2017) Cupriavidus necator H16 uses flavocytochrome c sulfide dehydrogenase to oxidize self-produced and added sulfide. Appl Environ Microbiol 83(22):e01610-17. https://doi.org/10.1128/AEM.01610-17
Luo C, Rodriguez RL, Konstantinidis KT (2014) MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res 42:e73. https://doi.org/10.1093/nar/gku169
Lv M, Du M (2018) A review: microbiologically influenced corrosion and the effect of cathodic polarization on typical bacteria. Rev Environ Sci Biotechnol 17:431–446. https://doi.org/10.1007/s11157-018-9473-2
Maszenan AM, Seviour RJ, Patel BKC et al (2005) Defluvicoccus vanus gen. nov., sp. nov., a novel Gram-negative coccus/coccobacillus in the 'Alphaproteobacteria’ from activated sludge. Int J Syst Evol Microbiol 55:2105–2111. https://doi.org/10.1099/ijs.0.02332-0
Molisch H (1907) Die Purpurbakterien nach neuen Untersuchungen.
Moura V, Ribeiro I, Moriggi P et al (2018) The influence of surface microbial diversity and succession on microbiologically influenced corrosion of steel in a simulated marine environment. Arch Microbiol 200(10):1447–1456. https://doi.org/10.1007/s00203-018-1559-2
Neukirchen S, Sousa FL (2021) DiSCo: a sequence-based type-specific predictor of Dsr-dependent dissimilatory sulphur metabolism in microbial data. Microb Genom. https://doi.org/10.1099/mgen.0.000603
Parks DH, Chuvochina M, Waite DW et al (2018) A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol 36:996–1004. https://doi.org/10.1038/nbt.4229
Patwardhan S, Vetriani C (2016) Varunaivibrio sulfuroxidans gen. nov., sp. nov., a facultatively chemolithoautotrophic, mesophilic alphaproteobacterium from a shallow-water gas vent at Tor Caldara, Tyrrhenian Sea. Int J Syst Evol Microbiol 66:3579–3584. https://doi.org/10.1099/ijsem.0.001235
Pfennig N, Trüper HG (1971) Type and neotype strains of the species of phototrophic bacteria maintained in pure culture. Int J Syst Evol Microbiol 21:19–24. https://doi.org/10.1099/00207713-21-1-19
Pfennig N, Lunsdorf H, Suling J et al (1997) Rhodospira trueperi gen. nov., spec. nov., a new phototrophic Proteobacterium of the alpha group. Arch Microbiol 168:39–45. https://doi.org/10.1007/s002030050467
Procópio L (2020) Changes in microbial community in the presence of oil and chemical dispersant and their effects on the corrosion of API 5L steel coupons in a marine-simulated microcosm. Appl Microbiol Biotechnol 104:6397–6411. https://doi.org/10.1007/s00253-020-10688-8
Proença DN, Whitman WB, Varghese N et al (2018) Arboriscoccus pini gen. nov., sp. nov., an endophyte from a pine tree of the class Alphaproteobacteria, emended description of Geminicoccus roseus, and proposal of Geminicoccaceae fam. nov. Syst Appl Microbiol 41:94–100. https://doi.org/10.1016/j.syapm.2017.11.006
Qin QL, Xie BB, Zhang XY et al (2014) A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol 196:2210–2215. https://doi.org/10.1128/jb.01688-14
Rajala P, Cheng DQ, Rice SA et al (2022) Sulfate-dependant microbially induced corrosion of mild steel in the deep sea: a 10-year microbiome study. Microbiome. https://doi.org/10.1186/s40168-021-01196-6
Ruan Z, Sun Q, Zhang Y et al (2019) Oleisolibacter albus gen. nov., sp. nov., isolated from an oil-contaminated soil. Int J Syst Evol Microbiol 69:2220–2225. https://doi.org/10.1099/ijsem.0.003428
Sasser M (1990) Identification of bacteria by gas chromatography of cellular fatty acids. USFCC Newsl 20:1–6
Shi BH, Arunpairojana V, Palakawong S et al (2002) Tistrella mobilis gen nov, sp nov, a novel polyhydroxyalkanoate-producing bacterium belonging to alpha-Proteobacteria. J Gen Appl Microbiol 48:335–343. https://doi.org/10.2323/jgam.48.335
Stamps BW, Bojanowski CL, Drake CA et al (2020) In situ linkage of Fungal and Bacterial proliferation to microbiologically influenced corrosion in B20 biodiesel storage tanks. Front Microbiol. https://doi.org/10.3389/fmicb.2020.00167
Su Y, Wang R, Sun C et al (2016) Thalassobaculum fulvum sp. nov., isolated from deep seawater. Int J Syst Evol Microbiol 66:2186–2191. https://doi.org/10.1099/ijsem.0.001008
Tang K, Yang LH, Chen YP et al (2019) Aerophototrophica crusticola gen. nov., sp. nov., isolated from desert biocrusts. Int J Syst Evol Microbiol 71:0. https://doi.org/10.1099/ijsem.0.004677
Tindall BJ (1990) A comparative study of the lipid composition of Halobacterium saccharovorum from various sources. Syst Appl Microbiol 13:128–130. https://doi.org/10.1016/S0723-2020(11)80158-X
Urios L, Michotey V, Intertaglia L et al (2008) Nisaea denitrificans gen. nov., sp. nov. and Nisaea nitritireducens sp. nov., two novel members of the class Alphaproteobacteria from the Mediterranean Sea. Int J Syst Evol Microbiol 58:2336–2341. https://doi.org/10.1099/ijs.0.64592-0
Vasilyeva LV (1985) Stella, a New Genus of Soil Prosthecobacteria, with Proposals for Stella humosa sp. nov. and Stella vacuolata sp. nov. Int J System Evolut Microbiol 35:518–521. https://doi.org/10.1099/00207713-35-4-518
Wang X, Quinn PJ, Fau-Yan A, Yan A (2015) Kdo2 -lipid A: structural diversity and impact on immunopharmacology. Biol Rev Camb Philos Soc. https://doi.org/10.1111/brv.12114
Wang S, Ye ZH, Wang NN et al (2019a) Ferruginivarius sediminum gen. nov., sp. nov., isolated from a marine solar saltern. Int J Syst Evol Microbiol 69:3056–3061. https://doi.org/10.1099/ijsem.0.003589
Wang Z, Zhang Z, Li C et al (2019b) Algihabitans albus gen. nov., sp. nov., isolated from a culture of the green alga Ulva prolifera. Int J Syst Evol Microbiol 69:828–832. https://doi.org/10.1099/ijsem.0.003245
Wang G, Tang D, Li G et al (2020) Denitrobaculum tricleocarpae gen. nov., sp. nov., a marine bacterium from coralline algae Tricleocarpa sp. Int J Syst Evol Microbiol 70:3335–3339. https://doi.org/10.1099/ijsem.0.004173
Wiese J, Thiel V, Gärtner A et al (2009) Kiloniella laminariae gen. nov., sp. nov., an Alphaproteobacterium from the marine macroalga Laminaria saccharina. Int J Syst Evol Microbiol 59:350–356. https://doi.org/10.1099/ijs.0.001651-0
Williams TJ, Lefèvre CT, Zhao W et al (2012) Magnetospira thiophila gen. nov., sp. nov., a marine magnetotactic bacterium that represents a novel lineage within the Rhodospirillaceae (Alphaproteobacteria). Int J Syst Evol Microbiol 62:2443–2450. https://doi.org/10.1099/ijs.0.037697-0
Wu D, Liu H, Zhou Y et al (2021) Roseomonas oleicola sp. nov., isolated from an oil production mixture in Yumen Oilfield, and emended description of Roseomonas frigidaquae. Int J Syst Evol Microbiol. https://doi.org/10.1099/ijsem.0.005064
Wu D, Zhang XJ, Liu HC et al (2021) Azospirillum oleiclasticum sp. nov., a nitrogen-fixing and heavy oil degrading bacterium isolated from an oil production mixture of Yumen Oilfield. Syst Appl Microbiol 44:126171. https://doi.org/10.1016/j.syapm.2020.126171
Yamada K, Fukuda W, Kondo Y et al (2011) Constrictibacter antarcticus gen. nov., sp. nov., a cryptoendolithic micro-organism from Antarctic white rock. Int J Syst Evol Microbiol 61:1973–1980. https://doi.org/10.1099/ijs.0.026625-0
Yarza P, Yilmaz P, Pruesse E et al (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635–645. https://doi.org/10.1038/nrmicro3330
Yoon JH, Kang SJ, Park S et al (2007) Caenispirillum bisanense gen. nov., sp. nov., isolated from sludge of a dye works. Int J Syst Evol Microbiol 57:1217–1221. https://doi.org/10.1099/ijs.0.64910-0
Young CC, Hupfer H, Siering C et al (2008) Azospirillum rugosum sp. nov., isolated from oil-contaminated soil. Int J Syst Evol Microbiol 58:959–963. https://doi.org/10.1099/ijs.0.65065-0
Yu W, Qian C, Weng W et al (2016) Effects of lipopolysaccharides on the corrosion behavior of Ni-Cr and Co-Cr alloys. J Prosthet Dent. https://doi.org/10.1016/j.prosdent.2016.01.002
Zhang GI, Hwang CY, Cho BC (2008) Thalassobaculum litoreum gen. nov., sp. nov., a member of the family Rhodospirillaceae isolated from coastal seawater. Int J Syst Evol Microbiol 58:479–485. https://doi.org/10.1099/ijs.0.65344-0
Zhang X, Tu B, Dai L-r et al (2018) Petroclostridium xylanilyticum gen. nov., sp. nov., a xylan-degrading bacterium isolated from an oilfield, and reclassification of clostridial cluster III members into four novel genera in a new Hungateiclostridiaceae fam. nov. Int J Syst Evol Microbiol 68:3197–3211. https://doi.org/10.1099/ijsem.0.002966
Acknowledgements
We thank Professor Paul A. Lawson at University of Oklahoma (Norman, OK 73019, USA) and Professor Aharon Oren at the Hebrew University of Jerusalem for help with checking the derivation of the names.
Funding
This study was supported by Agricultural Science and Technology Innovation Project of the Chinese Academy of Agriculture Science (CAASASTIP-2016-BIOMA), Central Public-interest Scientific Institution Basal Research Fund (1610012023002, 1610012023003 and 1610012023004), and National Natural Science Foundation of China (no. 92051108).
Author information
Authors and Affiliations
Contributions
Min Yang: Physiological and Biochemical experiment, data analysis and article writing ; Xue Zhang: Enrichment and isolation; Shichun Ma: Project guidance and promotion, data analysis and article revision; Qiumei Zhang: Genomic Data Analysis and phylogenetic analysis; Chenghui Peng: Physiological data collection; Hui Fan: Electron acceptor and substrate experimen; Jiang Li: Genomic Data Analysis; Lirong Dai: Strain preservation and activation; Lei Cheng: Funding support and guidance. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, M., Zhang, X., Ma, S. et al. Shumkonia mesophila gen. nov., sp. nov., a novel representative of Shumkoniaceae fam. nov. and its potentials for extracellular polymeric substances formation and sulfur metabolism revealed by genomic analysis. Antonie van Leeuwenhoek 116, 1359–1374 (2023). https://doi.org/10.1007/s10482-023-01878-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-023-01878-1