
Abstract
Ginsenosides are the major pharmacological components in ginseng. Microorganisms from a ginseng field were isolated to identify transformation of ginsenosides. Based on HPLC and LC–MS analysis, strain LFJ1403 showed strong activities to transform ginsenoside Rb1 to Rd as the sole product. Phylogenetic analysis of 18S rDNA indicated that LFJ1403 belonged to Aspergillus versicolor. Through comparing four systems of transforming Rb1 to Rd, strain LFJ1403 was found to secrete ginsenoside-converting enzymes in the spore production phase of plate culture. This result suggested that the enzyme could be directly obtained from the plate. The spore suspension, which contained the exocrine enzyme, was easy to prepare and efficient for biotransformation of ginsenoside Rb1 to Rd. Further study showed that the maximum bioconversion rate was 96 % (w/w) in shake flasks when a spore suspension system was used with optimized biotransformation conditions. Scale-up of this system to 2L resulted in an 85 % conversion rate. The ginsenoside Rb1 converting enzyme was separated by gradient HPLC with Q-Sepharose column, and its β-glucosidase activity and Rb1-converting ability was assayed by the 4-Nitrophenyl-β-d-glucopyranoside (PNPG) method and HPLC with C18 column, respectively. We obtained 130 U ml−1 enzymatic activity with the purified β-glucosidase. This is the first report on efficiently converting ginsenoside using extracellular enzyme directly from the fungus spore production phase of solid culture.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
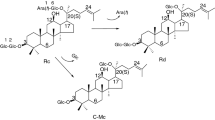
Ginsenoside is the major pharmaceutical component in ginseng (Kim et al. 2007), and accounts for 4 % in ginseng dry root (Oh et al. 2014). According to different glucoside skeletons, ginsenosides are mainly divided into the dammarane type and oleanane type (Quan et al. 2012). The dammarane type can be further classified into protopanoxadiol (PPD) and protopanaxatriol (PPT) by their aglycones (Li et al. 2015). Among these PPD-type ginsenosides, ginsenoside Rd especially exerts many pharmacological activities, such as a promotive effect on proliferation of neural stem cells (Lin et al. 2012), protecting kidney from chemical drugs (Yokozawa and Dong 2001), neutral system protective effects (Li et al. 2011) and preventing contraction of blood vessels (Zeng et al. 2003). However, it is difficult to separate Rd from ginseng because of its low concentration and complex ginseng components (Shi et al. 2007). The amount of the major ginsenoside Rb1 is up to 23.8 % in ginsenosides (Wu et al. 2012a), and it has the same aglycon as ginsenoside Rd; the point of difference is that Rb1 has one more sugar residue at the C-20 position, so ginsenoside Rd can be obtained by removing this residue of ginsenoside Rb1 (Fig. 1).

The conversion pathway of ginsenoside Rb1 to ginsenoside Rd
There are two ways to produce ginsenoside Rd including chemical and biological hydrolysis (Gao et al. 2014; Wu et al. 2012b). Microbial and enzymatic methods are more environmentally compatible and specific than chemical hydrolysis (Hegazy et al. 2015). In the microbial method of fungus biotransformation, ginsenoside Rd has been converted from Rb1 by Acremonium stritum (Chen et al. 2008), and Cladosporium fulvumand (Zhao et al. 2009b) with a conversion rate of 72 % and 86 % respectively. Rd also has been extracted from the ginseng extraction residue by lingzhi (Ganoderma lucidum) (Hsu et al. 2013), in which eight kinds of ginsenosides were detected and the fermentation time was as long as 30 days. Although the microbial approach is feasible, there exist the drawbacks of long conversion time and low biotransformation rate. In the enzymatic manner, ginsenoside Rd has been converted by β-glucosidase from Thermus caldophillus GK24 (Son et al. 2008), Cladosporium fulvum (Zhao et al. 2009a), Penicillium oxalicum sp. 68 (Gao et al. 2012) and Dictyglomus turgidum (Lee et al. 2012). In these reports, the transformation time is much shorter than that of the microbial approaches; however, ginsenoside Rd is sometimes intermediate, and this leads to a relatively low conversion rate. What’s more, the separation and purification processes of β-glucosidase are complicated.
In this study, ginsenoside Rb1-converting fungus was isolated and identified from the soil of a ginseng field, and we investigated the biotransformation of ginsenoside Rb1 to Rd using extracellular enzyme directly from the fungus spore production phase of plate culture. High-performance liquid chromatography (HPLC) analysis showed that Rd was the sole product of this transformation process. The conversion rate increased as high as 96 % at 48 h in shake flasks. Scale-up in a 2L reaction system resulted in an 85 % conversion rate, which indicated that this spore suspension biotransformation system has the potential to be used for Rd production at an industrial scale.
Materials and methods
Materials
Reagent-grade ginsenoside Rb1 and Rd were purchased from Solarbio Co. Ltd (Beijing, China). Total ginsenosides (80 % ginsenoside Rb1, other 20 % compounds are Rb2, Rc, Re, Rf and polysaccharose) were purchased from Hongjiu Biotech Co. Ltd (Jilin, China). 4-Nitrophenyl-β-d-glucopyranoside (PNPG) and 4-nitrophenol (PNP) were purchased from Dingguo Biotech Co. Ltd (Tianjin, China). Super filter tube was purchased from Millipore Co. Ltd (Massachusetts, USA). All chemicals and solvents were of analytical or HPLC grade.
Microorganisms screening and product Rd identification
LFJ1403 was isolated from a ginseng field by a standard dilution plating onto 0.1 g l−1 chloramphenicol PDA plate. Single colonies were purified by transferring onto new plates, and the isolated colony was washed down by 20 mM acetate buffer (PH 5.0) to a sterile shake flask with shaking at 130 rpm and 30 °C for 2 h. After incubation, the spores were filtered with three layers of gauze and were diluted to 5 × 106 spores ml−1. The above spore buffer and 0.5 mg ml−1 total ginsenosides (dissolved in acetate buffer and sterilized through 0.22 μm filter) were reacted with shaking at 130 rpm and 30 °C. The ginsenoside Rb1 conversion ability of isolated strains was tested by HPLC.
The spore buffer of isolate LFJ1403 was mixed with 0.5 mM reagent-grade Rb1 (dissolved in acetate buffer and sterilized through 0.22 μm filter) and cultivated with shaking at 200 rpm and 30 °C. HPLC and LC–MS methods were used to identify the substrate Rb1 and its metabolite.
Strain identification
Isolated strain LFJ1403 was cultured on PDA medium at 30 °C to observe its phenotypic characteristic with a modified Frias-De Leon’s method (Frias-De Leon et al. 2011), and the micromorphological characteristics were analyzed using the microculture method of Ridell (Frias-De Leon et al. 2011). The 18S rDNA gene (1650 bp) was amplified by PCR using genomic DNA isolated from LFJ1403 as a template. Both primers (FR1: 5′-ANC CAT TCA ATC GGT ANT-3′ and NS1: 5′-GTA GTC ATA TGC TTG TCT C-3′) were selected from the fungus universal primers database. Identification and homology search were performed with the BLAST program at the National Center for Biotechnology Information (NCBI) website (http://blast.ncbi.nlm.nih.gov). The phylogenetic tree was constructed using the neighbor-joining method through the MEGA 5.10 program.
Determination of biotransformation system
For obtaining the optimal biotransformation system, spores of Aspergillus sp. LFJ1403 cultured on PDA plate for 2 days were scraped down with 0.1 % Tween80 acetate buffer (pH 5.0) and made into spore buffer (diluted to 5 × 106 spores ml−1). Subsequently, the four biotransformation systems were made as follows: above spore buffer was used as the spore suspension system; the supernatant after spore buffer centrifugation at 25,000 × g for 30 min at 4 °C was used as the supernatant system; the bottom spores after spore buffer centrifugation were again suspended with acetate buffer and used as the centrifugal spore system; the bottom spores were suspended by potato dextrose broth (PDB) and cultured for several days up to spore germination, and the mycelium suspension was used as the mycelium suspension system. Then 0.25 mM (0.277 mg ml−1) reagent-grade ginsenoside Rb1 was added into these four biotransformation systems and reacted with shaking at 200 rpm and 37 °C for 96 h. The conversion abilities of the four systems were detected by comparing the Rb1 conversion rate and enzyme activity.
After the optimal biotransformation system was obtained, to determine the best time of collecting extracellular enzyme, spores on PDA plates were scraped down at 2, 3, 4, 5 and 6 day with 0.1 % Tween80 acetate buffer (pH 5.0) and made into spore suspension systems. These spore suspensions were mixed with 0.277 mg ml−1 reagent-grade ginsenoside Rb1 and cultivated with shaking at 200 rpm and 37 °C for 96 h. The optimum time to form a spore suspension system was identified by HPLC analysis.
Scale-up reaction system
In order to gain the maximum spore yield of plate culture, the spores were transferred from slants and dissolved in sterile water to prepare spore concentration gradients of 104, 105, 106, 107, and 108 ml−1, and 200 μl of each was inoculated to the PDA plate (D90 mm). After 3 days of incubation at 30 °C, spores were scraped down with 10 ml 0.1 % Tween80 acetate buffer (pH 5.0). We diluted the solution to an appropriate concentration so that we could count the spore number through a microscope to determine the spore yield.
The optimal spore concentration resulting from the above procedure was prepared. We inoculated this spore suspension onto larger PDA plates (D150 mm), and put them at 30 °C for 3-day cultivation. The next manipulations were the same as previously described, but the amount of 0.1 % Tween80 acetate buffer was increased proportionally. Then 1L of spore suspension (5 × 106 spores ml−1) was mixed with the same volume of 1.25 mg l−1 reagent-grade ginsenoside Rb1 and this biotransformation system was put in a 5L fermenter (BIOTECH-5JG, Baoxing biological equipment engineering Co. Ltd, Shanghai, China), to proceed under the conditions of 37 °C and 200 rpm.
Analytical methods
The reaction mixture was extracted twice by water-saturated n-butanol and then the n-butanol fraction was evaporated to dryness and methanol was added (Huang et al. 2006). Ginsenosides were assayed by HPLC at 203 nm with a C18 column. The column was eluted at room temperature with acetonitrile (A)/water (B) = 40 : 60 (v/v) at 1.0 ml min−1.
Quantification analysis of ginsenosides was performed by LC–MS system micrOTOF-QII10204. Positive ion mode in electrospray ionization (ESI) was used to analyze ginsenoside Rb1 and its metabolite. The following parameters of positive ion mode were used: scan begin/scan end 200/1500 m/z, set capillary 4500 V, set end plate offset −500 V, set collision cell RF 150 Vpp, set nebulizer 0.8 bar, set dry heater 180 °C, set dry gas 6.0 l min−1.
The β-glucosidase activity was measured by a colorimetric method using PNPG as the substrate. The reaction mixtures were centrifuged at 10,625 × g for 5 min at 4 °C, a crude enzyme was precipitated by adding 60 % saturated (NH4)2SO4 into the supernatant and placed at 4 °C overnight, and the crude enzyme was centrifuged and dialyzed sufficiently in 20 mM acetate buffer (Chang et al. 2014). The hydrolytic reactions containing 400 μl 1 mM PNPG and 100 μl enzyme solution, which were reacted in 20 mM acetate buffer (pH = 5.0), were incubated at 37 °C for 30 min, and the reaction was terminated by the addition of 0.5 ml distilled water and 2.5 ml 0.5 M NaOH (Zhao et al. 2009a). The absorption of released PNP was measured at 405 nm. One unit (U ml−1) of enzyme activity was defined as the amount of enzyme required to release 1 μmol PNP per h under standard conditions (Zhang et al. 2001).
Ginsenoside Rb1-converting enzyme was separated by HPLC at 280 nm with a Q-Sepharose column. The volume of column was 2 ml with 1 ml loading sample. The column was gradient eluted at room temperature with 25 mmol l−1 pH 8.2 Tris–HCL (A)/25 mmol l−1 pH 8.2 Tris–HCl with 0.5 mol l−1 NaCl (B) at 1.0 ml min−1. The volume of B eluent was controlled in the range of 0 and 80 %. The different enzyme fractions were collected to measure the β-glucosidase activities and ginsenoside Rb1-converting abilities by the PNPG method and HPLC with C18 column, respectively. The molecular weight of ginsenoside Rb1-converting enzyme was determined by SDS-PAGE.
Results and discussion
Isolation and identification of Rd-producing fungus
Through repeated transfer to the PDA plate, strain LFJ1403 was purified from the ginseng soil, and the product of its biotransformation activity was identified as the single metabolite ginsenoside Rd by HPLC and LC–MS (Fig. 2a, b).

The HPLC/mass spectrum profiles of ginsenosides in reaction mixtures and the microphotograph of Aspergillus sp. LFJ1403 mycelium growing on the cover slip. a The HPLC profiles of ginsenoside Rb1 and its metabolite Rd. b The mass spectrograms of ginsenoside Rb1 and its metabolite Rd. c The microphotograph of LFJ1403 aerial mycelium. The microculture was performed at 30 °C until the fungus was observed to grow. Subsequently, the cover slip was separated from the agar to be observed under the microscope. The magnification was ×100
The front colour of the colonies was greyish-green and the back was yellow; the texture of the colonies was villiform and dense after 4 days culture. The mycelium shape growing on the cover slip was radial, dense close to the plate bottom and sparse close to the top (Fig. 2c). The phylogenetic tree based on its 18S rDNA sequence (GenBank Accession No. KM925044) was constructed to identify the fungus to the species level (Fig. 3). The phylogenetic tree demonstrated that strain LFJ1403 should be affiliated with the genus Aspergillus. The closest recognized relatives of strain LFJ1403 were identified as Aspergillus versicolor. Strain LFJ1403 had less than 1 % difference in 18S rDNA gene sequence from the corresponding type strain. Morphologic observation and phylogenetic tree analysis both indicated that strain LFJ1403 belonged to A. versicolor. This fungus was stored in the China General Microbiological Culture Collection Center (CGMCC 11038).

Phylogenetic tree based on the 18S rDNA gene sequence showing the relationships between the Aspergillus sp. LFJ1403 strain and related species
Selection of high-efficiency biotransformation system
The four biotransformation systems were mixed with 0.25 mM (0.277 mg ml−1) reagent-grade ginsenoside Rb1, and the conversion rates (w/w) of spore suspension and supernatant were 90 % at 12 h, then remained stable at 94 % at terminal 96 h. In contrast, the conversion rates (w/w) of centrifugal spore and mycelium suspension were 27 and 18 % at 12 h, and then increased slowly to 94 and 62.72 %, respectively (Fig. 4a); this result indicated that LFJ1403 could excrete abundant ginsenoside Rb1-converting enzymes, which were attached to the surface of spores. During the PDA plate culture stage, these enzymes were dissolved into spore buffer and formed a crude enzyme solution. The spore suspension and supernatant systems both contained these enzymes, but the other two systems didn’t. Although the conversion rate was similar, the preparation method of the spore suspension was easier than that of the supernatant, so spore suspension was selected as the final biotransformation system. The conversion rate (w/w) of ginsenoside Rb1 to Rd collecting enzymes at 2 and 3 days was higher than that at 4, 5 and 6 days (Fig. 4b); this result indicated that the activity of extracellular enzyme attached to spores was high in the early spore production stage and slowly decreased with time extension, so spores cultured during 2 and 3 days were washed down to form spore buffer and further made into a spore suspension system.

Effects of different biotransformation systems and extracellular enzyme-collecting time on the production of Rd. a Effects of different biotransformation systems. Circle spore suspension, regular triangle supernatant, square centrifugal spore, inverted triangle mycelium suspension. b Effects of different enzyme-collecting times. Data represent the means of three experiments, and error bars represent the standard deviation
Optimization of biotransformation conditions
Starting with total ginsenosides, ginsenoside Rd yield was higher than that using equal quality of reagent-grade ginsenoside Rb1 (data not shown). This result is from conversion of other PPD-type ginsenosides besides Rb1 in total ginsenosides to Rd, but ginsenoside Rd was mainly from the conversion of ginsenoside Rb1, as confirmed by HPLC (data not shown). In this study, total ginsenosides (80 % ginsenoside Rb1) were used. Up to 1.25 mg ml−1 total ginsenosides, the conversion rate of total ginsenosides to Rd was increased. However, above 1.25 mg ml−1, the conversion rate decreased, but Rd production increased slowly with increasing the concentration of total ginsenosides (Fig. 5a). To achieve a suitable conversion rate and production concentration, 1.25 mg ml−1 total ginsenosides (80 % ginsenoside Rb1) was selected as the substrate concentration. The temperature affected both the reaction rate and the product yield (Eklund et al. 1990), ginsenoside Rb1 can be converted into Rd in various degrees at different temperatures, and the Rd yield was the highest at 37 °C (Fig. 5b). The effect of pH on the biotransformation of Rb1 was also significant. The pH value had no obvious change over time; there was no fungal growth or spore germination in the acetate buffer, so the initial pH value was measured as the pH value of reaction system. The Rd yield was low when the pH was over 7.0 or below 3.0, and was the highest at pH 5.0 (Fig. 5c). Under these optimum conditions, the ginsenoside Rd concentration reached up to 1.2 mg ml−1 with a conversion rate of 96 % (w/w) at 48 h (Fig. 5d).

Effects of biotransformation conditions on the production of Rd. a Effects of different total ginsenosides concentrations. Ginsenoside Rd production (filled square) and conversion rate (open square). Total ginsenosides (concentration was 0.6, 1.25, 1.85, 2.5 and 3.75 mg ml−1 respectively) were mixed with equal volume of spore suspension (1.0 × 106 spores ml−1). The mixtures were incubated with shaking at 200 rpm and 37 °C for 96 h. b Effects of different cultivate temperatures. 20 mM acetate buffer containing 1.25 mg ml−1 total ginsenosides was mixed with equal volume of spore suspension (1.0 × 106 spores ml−1). The mixtures were incubated with shaking at 200 rpm for 96 h at 25, 30, 37 and 40 °C, respectively. c Effects of different pH. 20 mM acetate buffer containing 1.25 mg ml−1 total ginsenosides was mixed with an equal volume of spore suspension (1.0 × 106 spores ml−1). The mixtures were incubated at different pHs with shaking at 200 rpm and 37 °C for 96 h. d Effects of different cultivation times. 20 mM acetate buffer containing 1.25 mg ml−1 total ginsenosides was mixed with equal volume of spore suspension (1.0 × 106 spores ml−1). The mixtures were incubated at pH 5.0 with shaking at 200 rpm and 37 °C for 96 h. Data represent the means of three experiments, and error bars represent the standard deviation
Scale-up biotransformation
The PDA plate (D150 mm) with grown spores is shown in Fig. 6a. As for the optimal spore concentration for inoculation to the plate, the 104 spores ml−1 showed the maximum spore yield (Fig. 6b), so we chose this concentration for plate culture. It was also obvious that high inoculation concentration didn’t lead to high spore yield. This may be caused by too many spores competing for growing space and nutrients, which in turn prevented the production of spores.

Scale-up of Rb1 to Rd biotransformation. a PDA plate (D150 mm) with grown spores. b Effect of different spore concentrations on the spore yield in plate culture. c Picture of the 5L fermenter with a working volume of 2L. d Effect of reaction time on the conversion rate in Scale-up of Rb1 to Rd transformation system. Data represent the means of three experiments, and error bars represent the standard deviation
We expanded the spore suspension biotransformation system in a 5L fermenter with a working volume of 2L (Fig. 6c) under the optimal biotransformation conditions and kept the reaction for 3 days. The HPLC analysis results indicated that the conversion rate reached 80 % (w/w) after 24 h’ transformation, and it trended to be stable during the next 72 h, with an 85 % (w/w) conversion rate (Fig. 6d). This result showed us the potential possibility of applying the spore suspension biotransformation system to industrial transformation of Rb1 to Rd. The process of amplifying the plate culture gradually to get enough spore suspension for use is crucial and this aspect will be studied further.
Enzyme characterization
The specific activity of ginsenoside Rb1-converting enzyme in different biotransformation systems within 96 h were measured by a colorimetric method using PNPG as the substrate. Enzyme activities of spore suspension and supernatant were higher in the range of 80–120 U ml−1 than centrifugal spore and mycelium suspension, which were about 40 and 50 U ml−1 (Fig. 7). This result further demonstrated that β-glucosidase was secreted in the spore production phase of plate culture, and these crude enzymes were collected to form an enzyme solution by washing spores. The enzyme activity of centrifugal spores was low because only spores were reserved by removing the enzyme supernatant, and the activity of mycelium suspension was also low because mycelium produced less β-glucosidase in the growth stage than in the spore production stage. The phenomenon that enzyme activities of supernatant and spore suspension, which both contain the secreted enzyme in the spore production phase of plate culture, were higher than mycelium was caused by different extracellular glucosidase secreting phases. A. versicolor LFJ 1403 secreted more extracellular glucosidase in spore production phase of solid culture than that in mycelium growth phase of liquid culture.

Assay of enzyme activities in different biotransformation systems within 96 h. Circle spore suspension, inverted triangle supernatant, square centrifugal spore, regular triangle mycelium suspension
The crude enzyme solution was concentrated through the process of centrifugation, ammonium sulfate precipitation and ultrafiltration. The individual enzymes in crude enzyme solution were separated by HPLC with a Q-Sepharose column (Fig. 8a), and were collected to analyze β-glucosidase activity and ginsenoside Rb1-converting ability used PNPG method and HPLC detection, respectively. β-glucosidase activities assay indicated that the fourth tube had strong β-glucosidase activity, but others didn’t (Fig. 8b). The enzymatic activity of this purified glucosidase is 130 U ml−1, which is higher than some other glucosidases from fungi, such as 114 U ml−1 by thermophilic fungal strain Melanocarpus sp. MTCC 3922, 52.2 U ml−1 by thermotolerant fungal strain Aspergillus terreus MTCC 6335 (Sonia et al. 2008), and 102.6 U ml−1 by strain Penicillium pulvillorum TUB F-2220, 87 U ml−1 by strain Penicillium cf. simplicissimum TUB F-2378 (Marjamaa et al. 2013), respectively (all the cited data were converted to a unified unit for comparative purposes). HPLC analysis also indicated that the fourth tube had the ability of converting ginsenoside Rb1 to Rd (Fig. 8c). These enzyme activity tests showed that Rb1-converting ability was consistent with the β-glucosidase activity. SDS-PAGE showed that the molecular weight of ginsenoside Rb1-converting enzyme was 97 kDa (Fig. 8d).

Assay of enzyme characterization. a The HPLC profiles of single enzymes separated from crude enzymes with Q-Sepharose column. b Assay of β-glucosidase activity from different collected tubes by PNPG method. c Assay of ginsenoside Rb1-converting ability from different collected tubes by HPLC analysis. d SDS-PAGE analysis of β-glucosidase fraction in the fourth tube. SDS-PAGE was performed on an 8.0 % gel and stained with Coomassie brilliant blue
In summary, a fungus, A. versicolor was isolated and found to be able to convert ginsenoside Rb1 to Rd without further hydrolysis. Through comparing different Rb1 biotransformation systems, A. versicolor in the spore production phase of plate culture was found to excrete ginsenoside Rb1-converting enzyme. The spore suspension system, which containing these exocrine enzyme, was easy to prepare and efficient. The conversion conditions were further optimized and the maximum biotransformation rate (w/w) was up to 96 %. Scale-up in a 2L reaction system resulted in an 85 % conversion rate. Afterwards, the Rb1-converting enzyme was separated by gradient HPLC with Q-Sepharose column, and then the β-glucosidase activity and Rb1-converting ability were assayed, respectively. Consequently, A. versicolor, and its characteristic of excreting highly active enzyme on the solid culture plate, may be suitable for production of ginsenoside Rd.
References
Chang KH, Jo MN, Kim KT, Paik HD (2014) Evaluation of glucosidases of Aspergillus niger strain comparing with other glucosidases in transformation of ginsenoside Rb1 to ginsenosides Rg3. J Ginseng Res 38:47–51
Chen GT et al (2008) Microbial transformation of ginsenoside Rb1 by Acremonium strictum. Appl Microbiol Biotechnol 77:1345–1350
Eklund R, Galbe M, Zacchi G (1990) Optimization of temperature and enzyme concentration in the enzymatic saccharification of steam-pretreated willow. Enzyme Microb Technol 12:225–228
Frias-De Leon MG et al (2011) Phenotypic characteristics of isolates of Aspergillus section Fumigati from different geographic origins and their relationships with genotypic characteristics. BMC Infect Dis 11:116
Gao J et al (2012) Efficient biotransformation for preparation of pharmaceutically active ginsenoside Compound K by Penicillium oxalicum sp. 68. Ann Microbiol 63:139–149
Gao J, Wang J, Cui J, Wang N, Bai Y, Yuan Y, Zhou Y (2014) Purification and characterization of two novel β-glucosidases from Penicillium oxalicum and their application in bioactive ginsenoside production. Biocatal Biotransform 32:199–207
Hegazy ME et al (2015) Microbial biotransformation as a tool for drug development based on natural products from mevalonic acid pathway: a review. J Adv Res 6:17–33
Hsu BY, Lu TJ, Chen CH, Wang SJ, Hwang LS (2013) Biotransformation of ginsenoside Rd in the ginseng extraction residue by fermentation with lingzhi (Ganoderma lucidum). Food Chem 141:4186–4193
Huang C, Wang G, Li H, Xie H, Sun J, Lv H, Lv T (2006) Sensitive and selective liquid chromatography-electrospray ionisation-mass spectrometry analysis of astragaloside-IV in rat plasma. J Pharm Biomed Anal 40:788–793
Kim SN, Ha YW, Shin H, Son SH, Wu SJ, Kim YS (2007) Simultaneous quantification of 14 ginsenosides in Panax ginseng C.A. Meyer (Korean red ginseng) by HPLC-ELSD and its application to quality control. J Pharm Biomed Anal 45:164–170
Lee GW, Kim KR, Oh DK (2012) Production of rare ginsenosides (compound Mc, compound Y and aglycon protopanaxadiol) by β-glucosidase from Dictyoglomus turgidum that hydrolyzes β-linked, but not α-linked, sugars in ginsenosides. Biotechnol Lett 34:1679–1686
Li L et al (2011) Protective effects of ginsenoside Rd against okadaic acid-induced neurotoxicity in vivo and in vitro. J Ethnopharmacol 138:135–141
Li ZY et al (2015) A universal quantitative 1H nuclear magnetic resonance (qNMR) method for assessing the purity of dammarane-type ginsenosides. Phytochem Anal (PCA) 26:8–14
Lin T et al (2012) Promotive effect of ginsenoside Rd on proliferation of neural stem cells in vivo and in vitro. J Ethnopharmacol 142:754–761
Marjamaa K, Toth K, Bromann PA, Szakacs G, Kruus K (2013) Novel Penicillium cellulases for total hydrolysis of lignocellulosics. Enzyme Microb Technol 52(6–7):358–369
Oh JY et al (2014) Investigation of ginsenosides in different tissues after elicitor treatment in Panax ginseng. J Ginseng Res 38:270–277
Quan LH, Min JW, Yang DU, Kim YJ, Yang DC (2012) Enzymatic biotransformation of ginsenoside Rb1 to 20(S)-Rg3 by recombinant β-glucosidase from Microbacterium esteraromaticum. Appl Microbiol Biotechnol 94:377–384
Shi W, Wang Y, Li J, Zhang H, Ding L (2007) Investigation of ginsenosides in different parts and ages of Panax ginseng. Food Chem 102:664–668
Son JW, Kim HJ, Oh DK (2008) Ginsenoside Rd production from the major ginsenoside Rb1 by β-glucosidase from Thermus caldophilus. Biotechnol Lett 30:713–716
Sonia KG, Chadha BS, Badhan AK, Saini HS, Bhat MK (2008) Identification of glucose tolerant acid active β-glucosidases from thermophilic and thermotolerant fungi. World J Microbiol Biotechnol 24:599–604
Wu L, Jin Y, Yin C, Bai L (2012a) Co-transformation of Panax major ginsenosides Rb1 and Rg1 to minor ginsenosides C-K and F1 by Cladosporium cladosporioides. J Ind Microbiol Biotechnol 39:521–527
Wu W, Qin Q, Guo Y, Sun J, Liu S (2012b) Studies on the chemical transformation of 20(S)-protopanaxatriol (PPT)-type ginsenosides Re, Rg2, and Rf using rapid resolution liquid chromatography coupled with quadruple-time-of-flight mass spectrometry (RRLC-Q-TOF-MS). J Agric Food Chem 60:10007–10014
Yokozawa T, Dong E (2001) Role of ginsenoside-Rd in cisplatin-induced renal injury: special reference to DNA fragmentation. Nephron 89(4):433–438
Zeng S et al (2003) Synthesis of 12-epi-ginsenoside Rd and its effects on contractions of rat aortic rings. Chinese Pharmacol Bull 19:282–286
Zhang C, Yu H, Bao Y, An L, Jin F (2001) Purification and characterization of ginsenoside β-glucosidase from ginseng. Chem Pharm Bull 49(7):795–798
Zhao X et al (2009a) A novel ginsenoside Rb1-hydrolyzing β-d-glucosidase from Cladosporium fulvum. Process Biochem 44:612–618
Zhao X et al (2009b) Highly selective biotransformation of ginsenoside Rb1 to Rd by the phytopathogenic fungus Cladosporium fulvum (syn. Fulvia fulva). J Ind Microbiol 36:721–726
Acknowledgments
The authors wish to acknowledge the financial support provided by the National Basic Research Program of China (“973” Program: 2012CB721105) and the Ministry of Science and Technology of China (“863” Program: 2012AA02A701).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lin, F., Guo, X. & Lu, W. Efficient biotransformation of ginsenoside Rb1 to Rd by isolated Aspergillus versicolor, excreting β-glucosidase in the spore production phase of solid culture. Antonie van Leeuwenhoek 108, 1117–1127 (2015). https://doi.org/10.1007/s10482-015-0565-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-015-0565-5